0022-538X/81/0400441-09$02.00/0
Method for Induction of
Mutations in
Physically
Defined
Regions of the
Herpes Simplex Virus Genome
ROZANNE M.
SANDRI-GOLDIN,'
MYRONLEVINE,I* ANDJOSEPH C.GLORIOSO2 Departmentof HumanGenetics'
andUnitforLaboratory AnimalMedicine,'
University of MichiganMedicalSchool, Ann Arbor, Michigan 48109 Received10November 1980/Accepted 29 December1980
A
procedure
wasdeveloped for inducing
mutations in isolated restriction enzymefragments
ofherpes simplex virus
type 1(HSV-1)
DNAwith nitrous acid.The mutations
werethen transferred
tothe viral
genome bygenetic recombina-tionduring cotransfection
ofrabbit
kidney cells with
themutagenized
fragmentsand
intact HSV-1 DNA. The HpaI restriction
enzymefragments
LD, B, LG, I,and J
weremutagenized.
Temperature-sensitive
mutants werefound
atfrequen-cies of
1 to5%
amongthe
progenyof
the transfections. Syncytial
mutants also werefound
athigh
frequency when fragment
B orLD
wasusedfor
mutagenesis.Fifteen of these
mutants, 11 temperaturesensitive and
4syncytial,
wereused for
further studies,
including complementation analysis, DNA synthesis, and marker
rescue.
Marker
rescuedata
presented here and in the accompanying publication
(A.
L.
Goldin, R. M.
Sandri-Goldin, M. Levine, and J. C. Glorioso, J. Virol.
38: 50-58,1981)
confirm the
mapposition of
someof the
newly
isolated
mutants.The
genomeof herpes
simplex virus
type 1(HSV-1) is
adouble-stranded DNA molecule of
about
100 x10c
daltons
(3,
11,16). DNA
mole-cules isolated from virions consist of
twocova-lently linked
componentsdesignated
long
(L)
and short
(S), each of which consists
ofunique
sequences
(UL
and
Us) flanked
by
inverted
re-peats(27). The
Land S
componentsinvert with
respect to
each other
sothat four isomeric
ar-rangementsdesignated
P
(prototype),
IL
(inver-sion of
L), Is
(inversion
of
S),
and
ISL (inversion
of S and
L)
occur inequimolar
amounts(5, 6,
13,28).
The size and
complexity of the
genomethus
present
problems
in
elucidating
the
processesinvolved in viral
replication,
latency,
and
trans-formation. The
availability
of
conditionally
le-thal
mutantsin
all partsof
the viral
genomewould
greatly
aid in
defining
the mechanis of
viral
biosynthesis
and virus-host interactions.
Anumber of
laboratories have isolated
tempera-ture-sensitive
(ts)
mutants,and
acomplemen-tation
analysis
of these
mutantshas
led
tothe
identification of
23complementation
groupsof
HSV-1
(25).
Still the genome is far fromsatu-rated
withmutants, andthere
arelarge
regions
such as
Us
whicharemutationally
silent.Many
of
thecurrently
availablemutants wereisolated
after
randommutagenesis
of the genome withthymine-specific
mutagens, oftenresulting
inthe isolationof
mutantsin thesamecomplementa-tion group
by
several
different laboratories(25).
Chu
etal.
(4)
developed
aprocedure
for the
invitro induction of mutations in preselected
re-gions of
the HSV-1 genome by using thecyto-sine-specific
mutagenhydroxylamine.
Thismethod
resulted
inthe identification of
three newcomplementation
groups (4).In
this
report, wedescribe
asimilar
approachfor the isolation of
mutants,using nitrous acid
as
the
mutagen.The
procedure involves the
invitro
mutagenesis ofspecific
restriction enzymefragments which
aresubsequently allowed
torecombine with the viral
genomeduring
cotrans-fection with intact viral DNA.
Inthis
waythe
mutation is induced within
adefined
region and
then transferred
tothe
viral
genome.MATERIALS AND
METHODS
Cells and virus.
Primary
rabbitkidney (RK)
cells
wereprepared from3-to5-day-old rabbits and were
passed no morethan two times before use. African greenmonkeykidney (Vero) cells and RK cellswere passed routinely inEagleminimal essential medium (MEM; GIBCOLaboratories) supplementedwith non-essential aminoacids, 10%heat-inactivated fetal calf serum (GIBCO), 100
pg
ofstreptomycin per ml, and 100U ofpenicillinperml. Stocks ofwild-typeHSV-1 strain KOS and ts mutant virusweregrownby infec-tion of Vero cellsat lowmultiplicityand titered by plaque assayonVero cellsasdescribedpreviously(1). KOS stocks were grown at 37°C. ts mutant stocks weregrownat340C,
thepermissive temperature. Thenonpermissivetemperature forts mutants was
390C.
DNApreparation. HSV-1 (KOS) DNAwas pre-pared from both extracellularvirionsand infectedcell lysatesasfollows. Verocellmonolayersinroller bot-tleswereinfected with KOSat amultiplicityof infec-41on November 10, 2019 by guest
http://jvi.asm.org/
SANDRI-GOLDIN, LEVINE, AND GLORIOSO tionof0.05 to 0.1and incubatedat37°C. When cyto-pathic effectwasgeneralized, cellswerescraped into the medium. The suspensionwascentrifuged atlow speed to pellet the cells. The supernatant fluidwas removed andcentrifugedat7,500xg inaGSArotor at4°C for4htopellet extracellular virus. The viral pelletwassuspended inasmall volume of TE buffer (0.01 M Tris-hydrochloride, pH 8.0, and 0.001 M EDTA). Thecellularpelletwassuspended inasmall volume of 0.01 M Tris-hydrochloride and 0.01 M EDTA, pH 8.0, and frozen. The cellular suspension was thawed and mixed with the extracellular virus. Sodiumdodecyl sulfatewasaddedto afinal concen-tration of 1%, and N-lauroyl sarcosinate (Sarkosyl) wasaddedtoafinal concentration of 0.5%. RNase A (Sigma ChemicalCo.)wasaddedto afinal concentra-tionof500ug/ml. Solutions of RNaseA at 10mg/ml were heatedat83°C for20minbeforeusetoinactivate any contaminating DNase activity. The lysate was incubatedat 37°C for60min. Atthattime, Pronase wasaddedto afinal concentration of2mg/ml. Pronase solutions of20mg/mlwereheatedat83°Cfor20min before use also toinactivate DNase. The lysatewas incubatedat37°C for 12 to 16h. Thevolume of the lysatewasthenadjustedto25ml with TEbuffer, and 32.5 gofCsClwasadded. TheDNA suspensionwas mixed very gently, and the refractive index of the suspension was adjusted to 1.4005 to 1.4010. DNA-CsCl solutionswerecentrifugedat40,000 rpm for20h in aBeckman VTi5O rotorat 20°C. The tubes were
puncturedat the bottom, and 1.0-ml fractionswere collected. The refractive indexwasreadfor each frac-tion, and those fractions comprising the viral DNA peak (densities of 1.740 to 1.720) were pooled and centrifuged twosuccessive timesasdescribed above. The viral DNAwasdialyzed extensively against TE buffer and then ethanolprecipitatedby the addition of'/o volume ofa3M sodiumacetate-100 mM mag-nesium acetatesolution and2.5 volumes of absolute ethanol. Thesuspensionwasheldat-20°Covernight and thencentrifugedat20,000 rpm inanSW27rotor at0°C for60min. ViralDNA wasgently suspended in 1.0mlof TE buffer.
ts mutant DNA used in marker rescue mapping experimentswasisolated from HSV-1 mutant-infected cell lysates. Verocell monolayers were infected with various ts mutants at amultiplicity of infection of 0.05 to 0.1 and then incubated at 34°C until cytopathic effectwasgeneralized. Extracellular virus waspelleted andsuspendedasdescribed above andthen combined withthecellularpellet.Theprocedures for lysis and digestionof thelysate with RNase and Pronase were asdescribed above. After incubation with Pronase, the DNAwasmixed with CsCl(initial refractive index of 1.4005 to1.4010) and centrifuged in a VTi5O rotor at 40,000 rpm for20 hat 20°C. Fractions in the density range of 1.740 to 1.715 were pooled and dialyzed against TE andthenprecipitated with ethanol. The DNA wasresuspended in TE buffer. After this pro-cedure, the viral DNA was still contaminated with cellular DNA. The viral DNA was not purified further, however,sincethecontaminatingcellularDNA served asthecarrierDNA inthetransfection so that no other carrier DNAneeded to beadded.
Salmon spermDNA (lyophilized; Sigma Chemical
Co.) usedascarrierDNAin themutagenesis proce-dure was suspended in TE buffer at a concentration of 5 mg/ml. The DNA was extracted with an equal volume of TE-saturated phenol four to six successive times followed by two extractions with chloroform-isoamyl alcohol (49:1) and then extensively dialyzed against TE buffer. The DNAwas precipitated with ethanol as described above and resuspended in TE buffer.
Restriction endonuclease cleavage of DNA and purification of DNA fragments. Viral DNA (10 to 15
,ug)
wasdigested with HpaI (Bethesda Re-search Laboratories)at 1U ofenzyme perjig
of DNA in 20 mMTris-hydrochloride, pH 7.2, 20 mM MgCl2, 20mMKCl, and1mMdithiothreitol for2h at37°C. The reactionwasstopped by the addition of '/lo volume of0.5MEDTA, pH 8.0, and 'Avolume ofasolution containing 5%bromophenol blue and 25% Ficoll in 30 mMNaH2PO4,36mMTris, and1mMEDTA, pH 8.0. Fragmentswereseparated by electrophoresis in 0.8% agarose. Electrophoresis was carried out on vertical slabgels (13.5 by20by0.3cm) in30mMNaH2PO4,36 mMTris, and1mMEDTA, pH 8,at80 Vfor15 to 18 h, in tubegels (15 by0.6cm)at 40V for15 to18h,or inhorizontalgels(8.5by22by0.6cm)at80V for20 to 24 h. Gels were stained with 1 ,ug of ethidium bromide permlandvisualized under long-waveUV light. Bandswere excised from thegelandstoredat 4°C overnight in0.5ml of TE buffer. DNA was ex-tracted from the agarosebytwosuccessive freeze-thaw stepsin which theDNA-agarose bandwasfrozenat -70°C and then thawed at 37°C. The agarose was pelleted bycentrifugationat15,000xg for5min inan Eppendorf microfuge. The supernatantwascarefully removed, and 1.0ml of TE bufferwasaddedto the agarose pellet and mixed. The freeze-thaw step was repeated, and the agarose was again pelleted. The supernatantwascombined withthefirst supernatant. One-tenth volume ofa3 Msodium acetate-100mM magnesiumacetatesolution and about10volumes of absolute ethanol were added to the DNA solution,whichwasthenkeptat-20°C overnighttoprecipitate the DNA andto remove theethidium bromide. The DNAwaspelleted bycentrifugationinanSW27rotor at24,000 rpm for90minat0°C.Controlexperiments using32P-labeled KOS DNA showed that60 to80% of the32pcountscould be recoveredfrom each band in the agarosegelsby this extractiontechnique.
Mutagenesisof HSV-1 DNAfragments.Purified restriction enzymefragments extracted from agarose gelsasoutlinedabove wereresuspended in 100jl of TEbuffer after ethanolprecipitation. Foreach muta-genesis treatment, 10jig of HSV-1 KOS DNA was digested with HpaI and electrophoresed in agarose. The amountof each DNAfragment recovered from thegelwasthen addedto a10-foldvolume(1.0ml)of nitrous acid reaction mixture (0.1M sodium acetate buffer, pH 4.6, containing0.05 MNaNO2 and2x
10'
Mspermine).DNA was incubated in thepresenceof nitrous acid at37°Cforthe times indicatedin individ-ualexperiments. The reactionwasstoppedby raising thepHto 7.0(32) by theaddition of 9.0ml of ice-cold transfection buffer(8 g ofNaCl,0.37gofKCl,0.1gof Na2HPO4, 1.0g ofdextrose,and5g of HEPES [N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid] per J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
liter, pH 7.1) (12) containing 10 ug of salmon sperm DNA perml. The mutagenized DNA fragments were used immediatelytocotransfect RK cells.
Transfection procedure.Transfections using mu-tagenizedfragmentswerecarriedoutby a procedure similar to that describedby Grahametal. (12). Intact KOS DNA was added at0.5,ug/ml to mutagenized DNA fragments in transfection buffer as described above.Calcium chloridewasadded to a final concen-trationof 125 mM. The DNA solutions were allowed tostandat roomtemperaturefor30minfor precipi-tates toform. Two milliliters of this solutionwasadded to a 60-mm petri dish containing a monolayer of subconfluent RK cells which had been seeded into the dish4to 5hearlier. Thegrowth mediumwasremoved from thecells before the addition of the transfection solution. DNAwasallowedtoadsorbtothe RK cells at340C for60min.Thesolution wasthenremoved, and5.0ml of MEMsupplementedwith5%fetal calf serum wasaddedtothe dishes. The cellswere incu-batedat340C for4h,atwhich time the mediumwas removed and replaced with 5 ml of MEM supple-mented with 5% fetal calfserum.Incubationat340C wascontinued until generalized cytopathic effect was observed in themonolayers.Disheswerethenfrozen untilassayed.
Mutant isolation.Progenyvirus from the cotrans-fectionsusingmutagenized fragmentswereassayedon Verocell monolayersin100-mmpetri dishesat
340C.
Approximately 100 to 200well-isolatedplaqueswere picked from each transfection with each fragment.
The virus in each plaquewastested for temperature-sensitive growth by assay at 34 and 390C. Those isolates whichplatedatefficienciesatleast 103 times lowerat39°Cthanat34°Cwereplaque purifiedthree times and used for furtherstudy.
Complementationtests.Thets mutantsisolated werecomplemented with each otherbyaprocedure
similar to the quantitative complementation test of Schafferetal.(24, 25) asdescribedpreviously(9).
Measurementof DNAsynthesis.Vero cell mono-layers in 25-cm2 flasks (2 x 106cells) were infected withamultiplicityof infection of10of eachmutantor wild-type KOS and then incubated at370C for 1 h. The inoculum was removed and monolayers were washed twice with phosphate-buffered saline, after which5 mlof MEM containing5%fetal calfserum wasadded. Theflaskswereincubatedat
390C
for4h, atwhich time10,uCiof[methyyl-3H]thymidine
perml(specific activity,6.7Ci/mmol;NewEnglandNuclear Corp.)wasaddedtoeachflask,and incubationat
390C
was continued for an additional 20 h. Cells were scrapedinto the medium and thenpelleted
bylow-speed centrifugation. The cellular pellets were sus-pendedin1.0ml of0.01MTris-hydrochloride, pH8.0, and 0.01 M EDTA. Sodiumdodecyl sulfate and Sar-kosylwereadded tofinal concentrationsof 1%each, and RNase A was added to afinal concentration of 500ug/ml. Thelysateswereincubated at
370C
for1h, atwhichtime 2mg ofPronaseper mlwasadded, and incubationat370Cwascontinuedfor anadditional12 h. Solutions ofRNase and Pronase were heated at830C
for20min before being usedto inactivate any DNase. Thevolume ofeachlysatewasadjustedto 5.0 mlwithTEbuffer,and6.5g ofCsClwasaddedtoeachtube. The refractive index was adjusted to 1.4005 to 1.4010, and DNA-CsClsolutions were centrifuged at 35,000 rpmin a type 50 fixed-angle rotor at
200C
for 72h. Fractions (200pl)
werecollected by puncturing the bottoms of the tubes. The refractive index of each fraction was read, and then samples were precipitated ontofilter paper with 10% trichloroacetic acid. Filters werewashedwith 5%trichloroacetic acid followed by distilledwaterandthen dehydrated with acetone, all at4C. Filtersweredriedand counted.RESULTS
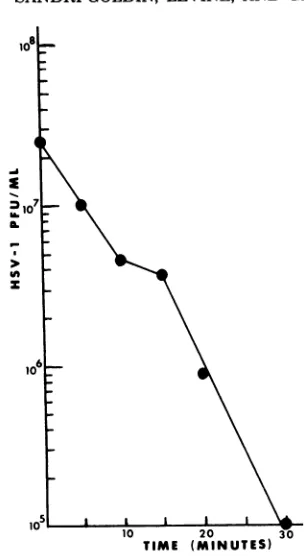
Nitrous acid inactivation
of intact,infec-tious HSV-1 DNA. Mutagenesis
of nucleicacids by
nitrousacid
occursprimarily byoxida-tive
deaminationof
adenine to hypoxanthineand of
cytosine
touracil(26,
31,33), resulting
inbase substitutions, although
a number of otherreactions have been
shown to occur (for review, see reference 34). Nitrous acid mutagenesis ofduplex
DNA hasbeen
shown to beless efficient
than
mutagenesis
ofsingle-stranded
DNA (15, 19, 20,32).
However, duplex
DNA isreadily
mutated
by
nitrous acid in the presence ofvar-ious alcohols, phenols,
orprimary amines (32).Of
theabove-mentioned compounds, spermine
has been
found
to causeappreciable
stimulation
of the
production
of mutants(32).
Therefore,
we added 2 x10-4
Mspermine
to areaction mixconsisting
of 0.1 Msodium
acetatebuffer
and 0.05 MNaNO2
atpH
4.6. In an attempt todetermine conditions for
mutagenesis,inactiva-tion of the
infectivity of
whole viral DNA innitrous acid
wasmeasured.
Intact,HSV-1
(KOS) DNA wasadded
to the nitrous acid reactionmixture and
incubated at37°C.
Aliquots
wereremoved
atvarious
times,
and the
infectivity
of the DNA was testedby transfection
ofRK cells.
By
25min, about 99% of the
transfecting activity
of KOS
DNAhad been
destroyed (Fig.
1).Al-though
this
was not adirect
measureof the
mutagenic activity of
nitrous acid onKOS DNA,
25min was
chosen
asthe
tirne
of treatment inmutagenesis
experiments,
since a 1% survival oftransfecting
activity
indicated
thatthe
majority
of DNA
molecules
hadreceived
atleast
onelethal hit.
Mutagenesis
of DNA
fragments.
Formu-tagenesis of
fragments,
HSV-1
(KOS)
DNAwasdigested with the restriction endonuclease
HpaI.
Fragments L1D, L2D (13.8
megadaltons [Md]),
B(12.4
Md),
LiG,
L2G (10.5 Md),
1(5.5Md),
and J (4.6Md) (Fig.
2) were selected because thesefragments
represented
avariety
of sizes and becausethey
spanned regions
intheUL
segment, thejoint
region,
and theUs
segment.Therefore,
the
general
usefulness of thetechnique
through-outthegenome could be tested.
Fragments
L1D andL2D
have the same molecularweight
andon November 10, 2019 by guest
http://jvi.asm.org/
LEVINE, GLORIOSO
IA-zA
10 20 30 TIME (MINUTES)
FIG. 1. Nitrous acidinactivation of the transfect-ing activity of HSV-1 (KOS) DNA. Ten-microliter samplescontaining2.1pgof KOS DNAwereadded to aseriesof tubescontaining200 ilof nitrous acid reaction mixture (see MaterialsandMethods). Each samplewasincubatedat37°Cfor the times indicated; then2.0mlof ice-cold transfection buffer containing salmon sperm DNA (10 Lg/ml)wasaddedto inacti-vatethe nitrous acid.CaCl2(1.25M) was addedtoa final concentrationof125mM, and the suspensions wereheldat roomtemperaturefor30min.Aliquots (0.8ml) of theDNA-CaCl2suspension were added to each ofthree 60-mmpetri dishes containing1 x 10' to 2 x 10; RK cells.After removal of the DNA solu-tions and addition of fresh growth medium as de-scribed in Materials andMethods, the plates were incubated at34°C for8daysandthen frozenuntil assayed. Infectious virusfirst appeared in the mono-layers atabout 5days, with maximum titers being achieved in thezerotimepointsat 7to 8days.
thereforecould not be
separated
onagarose
gels. This isalsotrue forfragmentsLIG
and L2G. For this reason, these fragmentswill
bereferred to as LDand LG. DNAwasextracted from bands in agarose gels asdescribed
inMaterials
and Methods. Thefragmentswere mutagenized sep-arately with nitrous acid for 25min
at37°C andthen
mixed with intact KOS DNA, and the mixture was used to cotransfect RK cells at340C.
Well-isolated plaques resultingfrom these transfections werepicked and tested for temper-ature-sensitivegrowth. Table 1 shows thenum-ber of plaques tested from each
mutagenized
fragment and the number of mutants found. ts mutants were isolated at
frequencies
of about 1 to 5%. Eleven ts mutants wereplaque purified three times and used in further studies. Nine of these mutants had titers 104 timeshigher
at 34°C than at390C,
and two had titers103
timeshigher
at 340C than at 390C. Inaddition,
a number of mutantshaving syncytial
plaque
mor-phology
(syn)
were also isolated(Table
1).
Four syn mutants wereplaque
purified
three times for furtherstudy.
Mutantswerenamed accord-ing to the HpaI fragment which wasmutagen-ized
for theirisolation,
forexample,
tsJ17 andsynLD70.
Complementation of
mutants.To
deter-mine whether
mutantsisolated from
transfec-tions using
the samefragment
werein different
complementation
groups,standard
quantitative
complementation
tests(24, 25)
wereperformed
between
these mutants.Mutants isolated with
fragment J,
I,
orLG
complemented
(Table 2),
demonstrating
thatit
waspossible
toisolate
mutants in
different
complementation
groupsfrom the
samefragment.
However, three of the
mutants
isolated with
fragment
Bdid
notcom-plement. These
mutantsmaybe
clonally related;
14 L~~~1. vii 41.1 .
0 10 20 30 40 30 60 70 80 90 100
FIG. 2. HpaI restriction endonuclease map of
HSV-1 (KOS)DNA (21). The map represents theP orientationofthe genome. Themolecularweights (in megadaltons)ofthefragmentswhichwere mutagen-izedareindicated above thefragments.
TABLE 1. Mutant isolationbynitrous acid mutagenesisofHSV- 1HpaI fragments
No.of No. ofmutants HpaI fragment plaques
tested ts syn
LID,L2D 120 1 3
B(1)a 150 7
B(2)a 100 >10%
L,G,L2G 120 2
I 240 2
J 215 2
aB(1)andB(2)representtwodifferenttransfection experiments.
p
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.494.76.228.67.346.2] [image:4.494.262.457.358.509.2]TABLE 2. Complementation of temperature-sensitive mutants
Mixed infections Complementationindex'
J17xJ176 7.0
I27 x I232 5.9
LG4 x LG15 6.2
B50xB54 0.6
B50xB100 4.8
B50xB124 0.5
I27xJ17 13.5
I27 xJ176 4.9
127xB50 5.7
J17xB50 6.2
J176xB50 4.2
LD84xB50 4.0
LD84xLG4 4.3
LD84xLG15 7.6
LG4xB50 6.4
a
Determined
astiter ofmutants 1 + 2 at39°C/ (titer ofmutant1at390C
+titerofmutant2at3900).
Acomplementation index of2 orgreater isconsidered significant (24).
this will be tested
by genetic recombination todetermine whether that is the
case orwhether
they
representmutations atdifferent allelic sites
in the
samegene.Some of the
mutantsisolated
by mutagenizing
different
fragments
werealso
tested, and all of
these
mutantscomplemented (Table 2).
Inad-dition,
twoof the
mutants(tsLG4
andtsLD84)
were
complemented
with
tsmutantsrepresent-ing
10of the
20complementation
groups in ourlaboratory collection and
werefound
tocomple-ment
with
all 10 groups(data
notshown). We
do
notknow
atthis
time whether any ofthe
mutantsrepresent new
complementation
groups inHSV-1.
However,
astandard
set ofHSV-1
complementation
groups(25) has
recently
be-come
available
tous, and we arein the
processof
testing the
new mutants with the standard set.Viral DNA
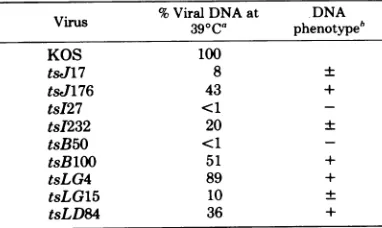
synthesis phenotypes
of
tsmutants.
The
amountof viral
DNAsynthesized
at390C
by each
ts mutant was measured andcompared
withthe
amountsynthesized by
wild-typeKOS virus
at390C.
MutantstsJ176, tsB100,
tsLG4,
and
tsLD84synthesized
greater than 20%of the
wild-type
KOS DNA
level,
whereas
tsJ17,
tsI232, and
tsLG15
synthesized
20% or less of theKOS
level(Table
3).
Mutants tsI27 and tsB50 were DNAnegative,
sincenoviral DNA was detected at390C.
DNAphenotypes
based on the percentage ofwild-type
level wereas-signed
asdesignated by
Aronetal.(2).
Confirmation of
mappositions
of
mu-tants. A
major
advantage
ofinducing
mutations inspecific regions
of the genome is that the mappositions of
the mutants should be delineated from the map positions of the fragments usedfor
mutagenesis. Two approaches were used toconfirm
the locations of some of the newlyiso-lated
mutants: complementation with a mutantwhich has been
mapped previously andmarker rescueby cotransfection of RKcells
withmutant DNA andwild-type
restriction enzyme frag-ments.The mapposition of mutant
synLD70,
which wasisolated
byusing fragment LD for mutagen-esis (seeFig.
2), was confirmed bycomplemen-tation with
a previously mappedmutant.
synLD70 had
asyncytial
plaquemorphology
on Verocells different from
that of syn mutantswhich
wereisolated
by using fragment B or from mutants synLD105 and synLD42. In addition,unlike the other newly
isolated syn mutants, synLD70 does not produce glycoprotein C(T. C.Holland,
J.C.
Glorioso,
R. M. Sandri-Goldin, and M.Levine,
manuscript in preparation). TheHSV-1
strain mP mutant MP (14) is syncytial and also does notsynthesize glycoprotein C (16, 29). The defect has been shown to mapbetween 0.7and
0.83units
onthe HSV-1 map by markertransfer
experiments (23).
Todetermine
whether MPand
synLD70
are in the samecomplemen-tation
group, Verocells
were mixedly infectedwith
MPand
synLD70 and
wereanalyzed
forcomplementation of the
synphenotype and for
the
synthesis
ofglycoprotein C. Control
infec-tionsof
synLD70
andwild-type
KOS and of MPand
KOS
wereperformed
atthe same time tobe
sure that the synphenotype
and theglyco-protein C defect of both
mutants wererecessive andcould be
complemented.
It wasfound
that theKOS
phenotype
wasdominant for both mu-tants.However,
in mixed infection ofsynLD70
TABLE 3. DNAsynthesisphenotypesofts mutants inducedby nitrous acid
Virus %ViralDNAat DNA
Virus
390C0
phenotypehKOS 100
ts,J17 8 +
tsJ176 43 +
tsI27 <1
-tsI232 20 +
tsB50 <1
-tsBlOO 51 +
tsLG4 89 +
tsLG15 10 +
tsLD84 36 +
'Determined aspercentviralDNA in
mutant-in-fected cellsat 39°C/percent
wild-type
KOS DNA at390C.
b+, 20%ofwild-type KOS DNAlevel; +, -20% of
wild-type KOS DNA level; -, no detectable viral DNA.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.494.252.443.481.595.2]46
and
MP, the
monolayers
werestillfound
tofuse,
indicating
nocomplementation
of the synphe-notype. In
addition,
noglycoprotein C
was pro-duced in the mixedinfection,
indicating
thatsynLD70
wasmutated at the samelocus
asMP(Holland
et al.,manuscript
inpreparation).
Fragment
LD spans 0.78 to 0.92mapunits
and coverspart of theregion
in which MPhas
beenmapped.
Therefore,
thelocation
infragment
LDof
thedefect
insynLD70 is consistent with the
mapping of
MP.Three other
mutants weremapped by
marker rescue. Toisolate the
mutants, restriction en-zymefragments
wereextracted from bands
in agaroseafter
onegel
purification.
Subsequent
marker
rescuemapping experiments showed,
however,
thatfragments isolated
inthis
way werefrequently
contaminated with otherfrag-ments,
resulting
inhigh
background levels
of rescueof
a mutantby
more than onefragment.
Although
asecond
gel purification
minimized
this
problem,
recovery of DNA after twogel
purifications
wasusually
veryinefficient. We
used
twomethods
to increasethe
purity
andyield of
DNArestriction
fragments for marker
rescue.
First,
wedeveloped
atwo-dim-ensional
gel
electrophoresis procedure which resulted
inincreased
purification
ofthe
fragments
overthat
achieved by
onesingle
gel
purification and
stillyielded
DNA recoveriesof 50to70%.
Figure
3A showsthe
gel pattern of
HpaI fragments
which
were
electrophoresed
vertically
in a1% agarose tubegel.
The tubegel
wasthen
placed
horizon-tally
2 cmfrom
the end of aglass
plate,
and a 6-mm0.5%
agaroseslab
gel
waspoured around
the tube
gel. After
asecond
horizontal
electro-phoresis (Fig.
3B), bands
wereexcised,
and the DNA wasextracted
asdescribed earlier.
Table
4shows
theresults
of amarker
rescueexperiment
using
tsLD84. tsLD84 was rescuedby fragment B,
notby fragment
LD.Fragment
B
(12.4
Md)
migrated slightly faster
than LD (13.8Md)
(seeFig.
3), sothat
the LDband on agel
presumably
wouldbe contaminated
withfragment
Bafter
onegel purification.
Itis likely,
then, that it was the
contaminating
Bfragment
which wasmutagenized in the
induction
of mu-tant tsLD84. Since this mutant is actually lo-cated in B and notLD, it has been renamed ts B84.The
two-dimensional
gelprocedure, although
increasing
thepurity and yield of specific DNAfragments,
didnoteliminate
theproblem
ofcon-taminating fragments
entirely. The second method we used to enable us to obtain large amounts ofspecific DNAfragments
wasto clone the HSV-1 genome. Theaccompanying
paper (10) describes thecloning
ofEcoRIfragments ofHSV-1
(KOS)
inaplasmid
vector.These cloned EcoRI fragments were used in cotransfectionexperiments
with intact DNA from mutantstsB84, tsLG4,
andtsJ176. The results from the marker rescue experiments are presented in the accompanying paper (10). Those data confirm that the location of the mutation in tsB84 is in the region of HpaI fragment B. The positions of tsLG4 in fragment LG and tsJ176 in fragment J arealso confirmed in themapping
experimentswith cloned
fragments (10).
DISCUSSION
Thisreport describes a method for the induc-tion of mutainduc-tions in
specific regions
of the viral genome.The procedure involves in vitro muta-genesis ofpurified restrictionenzyme fragments by nitrous acid. The mutations arethen
trans-ferred to the viral genome by cotransfection of RK cells with the mutagenized fragments and intact HSV-1 DNA so that a recombination eventbetween thefragment
and theviral DNA molecule can occur. The efficient marker rescue of mutantsby cotransfection ofwild-type
DNA fragments and mutant DNA(18,
22, 30) sug-gested to us that mutants could be isolated by thisapproach at reasonablefrequencies.
Infact,
this proved to be the case, for 1 to
5%
of the plaques tested in eachmutagenesis
treatment weretemperature sensitive forgrowth (Table 1).
Syncytial
mutantsalso
wereisolated with
high
frequency by
thisprocedure (Table
1).Chu et al.
(4)
have described asimilar
ap-proachfor
theisolation
of HSV-1 ts mutants that useshydroxylamine
asthe
mutagen. We chose nitrous acid as the mutagenbecause
it acts oncytosine
and adenineresidues (26,
31,33).
HSV-1 hasaguanine-plus-cytosine
content of67%
(17);
therefore,
amutagenwhich
interacts
with cytosine would be
advantageous
in theinduction of mutations
inguanine-plus-cytosine-richregions of
the
genome.Hydroxylamine also
interacts
withcytosine
(8).However,
mutagen-esis with nitrous acidmay have other
advantages
over
mutagenesis
withhydroxylamine.
Hydrox-ylamine ismutagenic primarily
on single-stranded DNA(8), so that mutagenesis of HSV-1 DNAfragments
must be conducted underpartially denaturing
conditions (4). Becausecomplete
denaturation results in loss ofinfectiv-ity,
conditions must beadjusted
suchthat only asmallpercentage
of the HSV-1 DNA is dena-tured (4). Therefore the mutagenic action ofhydroxylamine
may belimited
tocytosine resi-dues inadenine-plus-thymine-rich
regions of the genome. Nitrous acid alsomutagenizes
duplex DNA lessefficiently
thansingle-stranded
DNA (15, 19, 20, 32), but theefficiency
ofmutagenesis J. VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
LG
B
K
[image:7.494.104.394.78.529.2]LDI DE
F
G,H
I J RSjL
M
FIG. 3. Two-dimensionalagarosegelpurification of HpaI restrictionfragments ofHSV-1 (KOS) DNA.(A)
Tenmicrograms ofHSV-1(KOS)DNAwasdigestedtocompletion withHpaI. Thesamplewasloadedonto
a1.0%agarosetubegel (0.6 by15cm)andelectrophoresed verticallyat40Vfor18h.Thegelwasstained with
ethidium bromidetovisualizethe bands. Theoriginisontheleft-handsideof thefigure. (B)The tubegelwas
placed horizontallyabout 2cmfromtheedgeofaglassplate,anda0.5%agaroseslab gel (8.5 by22by0.6cm) waspouredaround it.Thegelwaselectrophoresed horizontallyat80Vfor22h. Theoriginisatthetopofthe
gel.DNAbands visualizedbyethidium bromidestainingwerecutfromthegel, and DNAwasextractedas
described in Materials andMethods.
of double-stranded DNA can be increased by possible to mutagenize guanine-plus-cytosine-adding spermineto the reaction (32), although richregionsof thegenomemoreefficiently with
the mechanism of action of spermine has not nitrous acid. Finally, the procedure described
beenfully elucidated (7). Therefore, it should be herefornitrous acid mutagenesisissimpleand
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 4. Markerrescueof tsLD84by HpaI fragments
HpaI Titer
fragment 34°C 39°C Rescue'
None 2.52x106 1.3x 10 0.5 LD 7.8 x 106 1.0 x 104 0.2 B 3.65x106 1.95x 105 5.0 LG 1.98 x 106 2.0 x 103 0.1 D, E 6.3 x106 1.8 x 103 0.03 I 1.55 x 106 1.17 x 104 0.75 RS 2.45 x105 7.85 x 102 0.3
Determinedas(titerat39°C/titerat34°C)x 100.
rapid, which should facilitate the isolation of large numbers ofmutants.
Another advantage ofinducing mutations by in vitro mutagenesis of specific restriction en-zyme fragments is that in theory the mutants canbeplacedonthephysicalmapofthegenome
by virtue of theirinduction.However,theuseof
fragments purified fromagarose gelscan result
in mutation ofcontaminating DNA fragments instead of thefragment intended. As described
above,mutanttsB84wasisolatedby usingDNA
extracted fromband LD but wassubsequently
foundtomapinfragmentB(Table 4). Although additionalgel purificationscanreduce
contami-nationoffragments, the problemcanbe
circum-vented entirely by using cloned HSV-1
frag-ments. Forthis reason, as well asfor the easy
isolation oflarge quantities of specific fragments of HSV-1 DNA, we cloned the HSV-1 genome
by using HSV-1 EcoRI fragments (10).
Addi-tional mapping experiments using some of the
mutantsisolated in thisreportand cloned EcoRI fragmentsarereported in the accompanying pa-per (10). We are also mutagenizing specific
cloned EcoRI fragments by the procedure
re-ported here in an attempt to saturate certain
definedregions of thegenomewithmutants.
In summary, the procedure described here
should be generallyuseful for the induction of
mutations in defined regions of any viral
ge-nome. This is especially useful for viruses like
HSV-1, which have a large genome, since the mutagenesis can be directed to specific frag-ments.
ACKNOWLEDGMENTS
We thank Alan L. Goldin for valuable discussions and
advice andJoseph Bellestrifor excellent technicalassistance.
This workwassupported bygrants07711and05860from
theNationalScienceFoundationand by Public Health Ser-vice grant 5P40-RROO200 from the National Institutes of
Health.R.M.S-G. is the recipient of Public Health Service individual postdoctoral research service fellowship F32-A105653 fromtheNational Institutes of Health.
LITERATURE CITED
1. Alder, R., J. C. Glorioso, and M. Levine. 1978. Infection by herpes simplex virus andcells of nervous system origin: characterization of anonpermissive interaction. J. Gen. Virol. 39:9-20.
2.Aron, G. M., D.J. M.Purifoy, and P. A. Schaffer. 1975.DNAsynthesis and DNA polymerase activity of herpes simplex virus type 1 temperature-sensitive mu-tants.J. Virol. 16:498-507.
3.Becker, Y., H.Dym,and I.Sarov.1968.Herpes simplex virusDNA. Virology 36:184-192.
4. Chu, C.-T.,D. S.Parris, R.A. F.Dixon, F. E. Farber, and P. A. Schaffer. 1979.Hydroxylamine mutagenesis ofHSV DNA and DNA fragments: introduction of mutations into selected regions ofthe viral genome. Virology 98:168-181.
5. Clements,J.B., R.Cortini, and N. M. Wilkie. 1976.
Analysis of herpesvirus DNA substructure by means of restrictionendonucleases. J. Gen. Virol. 30:243-256. 6. Delius, H., and J. B. Clements.1976.Apartial
denatur-ation map ofherpes simplex virus type1DNA:evidence for inversions of theunique DNAregions. J. Gen. Virol. 33:125-134.
7. Frankel, A. D.,B. K.Duncan, and P. E. Hartman.
1980.Nitrous aciddamagetoduplex deoxyribonucleic acid:distinction between deamination of cytosine resi-dues andanovelmutational lesion. J. Bacteriol.142: 335-338.
8. Freese,E., and H. B. Strack.1962.Induction of
muta-tions in transforming DNAby hydroxylamine. Proc. Natl. Acad. Sci. U.S.A.48:1796-1803.
9. Glorioso,J.C.,M.Levine,T. C.Holland,andM.S. Szczesiul. 1980. Mutant analysis ofherpessimplex virus-induced cell surfaceantigens: resistanceto
com-plement-mediated immune cytolysis. J. Virol.
35:672-681.
10. Goldin, A. L., R. M.Sandri-Goldin,M.Levine,and J. C. Glorioso.1981.Cloningofherpes simplexvirustype
1sequencesrepresenting the whole genome. J. Virol. 38:50-58.
11. Grafstrom, R. H., J. C.Alwine, W. L. Steinhart, C. W.Hill,and R. W.Hyman.1975.The terminal repe-tition ofherpessimplexvirus DNA.Virology 67:144-157.
12. Graham,F.L.,G.Veldhuisen,and N. M. Wilkie.1973.
Infectious herpesvirus DNA. Nature (London) New Biol.245:265-266.
13. Hayward, G. S., R. J. Jacob, S. C. Wadsworth, and B. Roizman. 1975. Anatomy ofherpessimplexvirus
DNA:evidence for fourpopulations of moleculesthat
differintherelative orientations of theirlong and short segments.Proc. Natl. Acad. Sci.U.S.A. 72:4243-4247.
14. Hoggan, M. D., and B. Roizman.1959.Theisolation andproperties ofavariant ofherpes simplex producing multinucleated giantcellsinmonolayercultures inthe presence of antibody. Am. J.Hyg. 70:208-219.
15. Horn,E.E., andR. M.Herriott. 1962.Themutagenic action on "single-stranded" (denatured) hemophilus transforming DNA. Proc. Natl. Acad. Sci. U.S.A.48: 1409-1416.
16.Keller, J. M., P. G. Spear, and B. Roizman. 1970. Proteinsspecified by herpessimplexvirus. III. Viruses differingin their effects on thesocial behavior of
in-fected cellsspecify different membraneglycoproteins.
Proc.Natl. Acad. Sci. U.S.A.65:865-871.
17. Kieff, E.D., S. L. Bachenheimer, andB.Roizman.
1971. Size, composition, andstructureof the
deoxyri-bonucleic acid ofherpes simplexvirussubtypes1and2.
J. Virol.8:125-132.
18.Knipe, D. M., W. T.Ruyechan,R.W.Honess, and B. Roizman.1979.Moleculargenetics ofherpes simplex
virus: theterminalasequencesoftheLand S
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.494.59.253.70.207.2]nentsareobligatorily identical andconstitute apartof astructuralgenemappingpredominantlyin the S com-ponent.Proc.Natl. Acad. Sci. U.S.A. 76:4534-4538.
19. Kotaka, T., and R.L. Baldwin. 1964.Effects ofnitrous
acid on the dAT copolymer asa template forDNA polymerase.J.Mol.Biol.9:323-339.
20. Litman, R. M. 1961.Geneticandchemical alterations in
the transforming DNA of Pneumococcus caused by
ultraviolet light and bynitrousacid.J. Chem. Phys. 58: 997-1003.
21. Morse, L.S., T. G. Buchman, B. Roizman,andP.A.
Schaffer.1977.Anatomyofherpessimplex virus DNA. IX.Apparentexclusion ofsomeparentalDNA
arrange-mentsin the generation ofintertypic (HSV-1xHSV-2) recombinants.J.Virol. 24:231-248.
22. Parris, D. S., R. A. F. Dixon, andP.A. Schaffer. 1980. Physical mapping of herpes simplex virus type 1 ts
mutantsbymarkerrescue: correlation ofthephysical and geneticmaps.Virology 100:275-287.
23. Ruyechan, W. T., L. S. Morse, D. M. Knipe, and B. Roizman. 1979.Molecular genetics of herpessimplex virus. II.Mapping of themajorviralglycoproteinsand
of the genetic loci specifying the social behavior of infected cells. J. Virol. 29:677-697.
24. Schaffer,P.A.,G. M.Aron,N.Biswal,and M. Ben-yesh-Melnick.1973.Temperature-sensitivemutantsof
herpes simplexvirus type 1:isolation,complementation andpartial characterization. Virology52:57-71. 25. Schaffer, P. A., V. C. Carter, and M. C. Timbury.
1978.Collaborative complementation study of
temper-ature-sensitive mutants ofherpessimplexvirus types 1 and 2. J. Virol. 27:490-504.
26. Schuster, H. 1960. The reaction ofnitrous acid with
deoxyribonucleic acid. Biochem. Biophys. Res.
Com-mun. 2:320-323.
27. Sheldrick, P., and N. Berthelot.1974.Inverted
repeti-tions in thechromosome ofherpessimplexvirus. Cold
Spring HarborSymp. Quant.Biol. 39:667-678. 28. Skare,J., and W. C. Summers. 1977. Structure and
function ofherpesvirusgenomes.II.EcoRI, XbaI,and
HindIIIendonucleasecleavagesites onherpes simplex virustype 1 DNA.Virology76:581-595.
29. Spear, P.G.1976.Membraneproteinsspecified by herpes simplex viruses. I. Identification of fourglycoprotein
precursors and theirproductsintype 1-infected cells. J.
Virol.17:991-1008.
30. Stowe, N. D., J.H.Subak-Sharpe, and N.M. Wilkie. 1978.Physicalmapping of herpessimplexvirus type 1 mutationsbymarker rescue. J. Virol.38:182-192. 31. Tessman,I. 1959.Mutagenesisinphages4x174and T4
andproperties of thegenetic material.Virology 9:375-385.
32. Thomas, H. F., P. E.Hartman,M.Mudryj,and D.L.
Brown. 1979. Nitrous acidmutagenesisofduplexDNA as athree-componentsystem.Mutat. Res.61:129-151. 33. Vielmetter, W.,and H. Schuster. 1960.Thebase
spec-ificity ofmutation inducedbynitrous acid inphageT2. Biochem.Biophys.Res.Commun.2:324-328. 34. Zimmerman, F.K. 1977.Genetic effects of nitrous acid.
Mutat. Res.39:127-148.