Vol.46, No.2 JOURNALOFVIROLOGY,May1983, p. 424-433
0022-538X/83/050424-10$02.00/0
Copyright01983, American SocietyforMicrobiology
RNA
Encoded
by the IR1-U2
Region of Epstein-Barr
Virus
DNA
in Latently Infected,
Growth-Transformed
Cells
VICKY VANSANTEN, ANDREW CHEUNG, MARY HUMMEL, ANDELLIOTT KIEFF* KovlerViral Oncology Laboratories, The University of Chicago, Chicago, Illinois 60637
Received 22 October1982/Accepted 28 January 1983
The IR1-U2region ofEpstein-Barr virus DNA consists of multiple copies ofa
3.1-kilobase (kb) repeatsequence,IR1, whichmaps totheleft ofa3.3-kb unique region, U2. Although hybridizations of cytoplasmic polyadenylated RNA from latently infected cells to viral DNA indicate that the IR1-U2 region encodes a
substantial fractionof the viral RNA in thesecells,onlyasinglelow-abundance 3-kb cytoplasmic polyadenylated RNA has been identified on Northern blots. Furtheranalysis of thecytoplasmicpolyadenylated RNAencoded by the IR1-U2 regionindicates that(i)the RNAis transcribed from lefttoright; (ii) thereareonly three copies ofthe 3-kb RNAper cell; and (iii) the RNA is spliced. The RNA
hybridizesto possibly contiguous 0.56- and 1.3-kb U2 domains. These domains andpartof IR1 hybridizetothe3-kbcytoplasmic RNA. DNAbetween IR1 and the 0.56-kb U2 domain does not hybridize to the 3-kb RNA. The CCAAT-34 nucleotide-TATAA sequence in IR1 may be part of the promotorfor the 3-kb cytoplasmic polyadenylated RNAsince(i)it enables left-to-right transcriptionof
IR1byaHeLacellextract, and(ii) latently infected cells contain giant
polyade-nylated nuclear RNAs which differ in size by 3 kb, as would be expected if transcription initiates in any copy of IR1 and continues through the rightward remaining copies into U2.
Infection of normal human B lymphocytes withEpstein-Barr virus (EBV) results in
persis-tentlatent infection and cellulargrowth transfor-mation (14, 36). Latentgrowth-transforming in-fection is characterized by the presence of the complete viralgenome(21, 39,50)ascovalently closedcircularepisomes(28, 34); by the
expres-sion of polyadenylated polyribosomal RNA
from the IR1-U2, IR3, and U5regions (19, 22, 24, 25, 37, 48); and by the expression ofnew
intranuclear (42) and cell membrane (45)
anti-gens. A similar stateof virus infection
character-izes latently infected B lymphocytes in normal
seropositive humans (33) and malignant African Burkitttumorcells (10).
Probes which include IR1 or U2 (BamHI fragments V, X, and H; Fig. 1) identify a
3-kilobase (kb) RNA incytoplasmicextractsofa
latently infected cell line, IB4 (24, 48). The
importance of theEBV IR1-U2regionin induc-ing growth transformation is also suggested by studies oftheP3HR-1 EBVstrain. TheP3HR-1
genome is deleted forthe entire U2-1R2 region
(7, 18, 23, 38, 40). The virus produced by
P3HR-1cellscaninfectlymphocytesbutcannotinduce growthtransformation (32, 41).
cDNA madefromthe 3' end of IB4 cell RNA hybridizes more tothe EBV DNABamHI
frag-mentH,which contains therightpartof U2 and IR2, than to BamHI fragments V or X, which contain IR1 and the IR1-U2juncture,
respec-tively, suggesting that the 3' end of the 3-kb
RNAisin U2orIR2(48). Since the cDNA also hybridizes to BamHI fragments V and X, the data are also compatible with the existence of several RNAs with different 3' ends. The
se-quence of IR1 revealsa CCAAT-34
nucleotide-TATAAA sequence and a TATA sequence
which could bepartofpromotersforrightward transcription from IR1 intoU2(8). Other
impor-tant features of IR1 include multiple potential splice donor and acceptor sites, a 1.1-kb open
reading frame, and an unusual 500-base pair
dyad symmetry which would have a profound
effect on the secondary structure ofRNA
en-codedby IR1 (8).
Theobjectiveof thisreportistodefine further (i) the sites for initiation oftranscription from
IR1 in vitro; (ii) the direction of transcription
from IR1 and U2 in latently infected
growth-transformedIB4cells; (iii) thenumber and size ofRNAs transcribed from IR1 and U2 in IB4
424
on November 10, 2019 by guest
http://jvi.asm.org/
IR1-U2-ENCODED EBV RNA IN CELLS
A.
IR1OD) TR U1
,R
,IR
-ulI. U3 ,R3 K4
small segments
totalling 0.9 Kb (1.9kb) TATA" 06kb 1.3kb
IRI IRI1(D) U2
Bam V BmHI X
mature 3kb nRNA
IR2,,i U3 BamMIH
I I I
-I 0 1 2 3 4 5 6 7 8 9 i0 11
Kb
OBmHI
if P3HR- Ideletion
FIG. 1. (A) Schematic diagram of the EBV genome. The genome is divided into five regions of unique
sequencecomplexity (Ut through U5) bounded by direct tandem copies of DNAsequencesatthe termini (TR) andatinternal siteswithin the molecule (IR1through IR4). Thediscontinuity in themapbetween U4 and U5
reflects the fact that the B95-8 isolate of EBV is deleted foraregion ofthe DNA which containsafourth internal
repeat,IR4.Thedeletion hasnoknowneffectonthebiological properties of the virus. IR1(D) indicatesthefinal incompletecopyofIRL. (B) Model for the 3-kb mRNA transcribed from the EBV IR1-U2 region inIB4cells.A BamHIrestrictionenzyme mapofthe IR1, U2, and IR2regions is shown. TATAAA indicates the position of the promoter-likesequenceatnucleotide 1137. RNAs transcribed from this regionareshown above themap.Some
oftheprimary transcripts begin in copies of BamHI fragmentVtothe left of theoneshownonthemap.In this
model,splicing of long primary transcriptsgeneratesa3-kb mRNA. The 0.6- and 1.3-kb RNAsegmentscould be
acontinuous1.9-kbsegment.The uncertaincontinuityof thesesegmentsisindicatedby the dotted line in the 1.9-kbsegment.Other RNAsegmentssplicedontothe 1.9-kbsegmenthavenotbeendemonstrated andareindicated
inparentheses. Thepolyadenylic acid 3' end (not shown) probably constitutes the remaining 0.2 kb of the3-kb maturemRNA.
cells;and (iv) whether the 3-kb polyadenylated
RNA previously identified in the cytoplasm of
IB4cells is spliced.
MATERIALSANDMETHODS
Ceil culture.TheIB4cell linewasestablishedbyin
vitro transformation of human cord blood lympho-cytesbythe B95-8 isolate of EBV and isnegativefor antigens expressed earlyandlate in EBVreplication (24).Cellsweremaintainedby addinganequalvolume ofRPMI1640medium(GIBCOLaboratories) supple-mented withl1ofetalcalfserum(K.C. Biologicals)
every 3 days. RNA was prepared from cells 3 days afterfeeding.
Recombinant DNA. Recombinant EBV (B95-8) DNAswereusedforhybridizationtoRNAblots and invitrotranscription. PlasmidspDK14,pDF322, and pDK286areclones of BamHIfragmentsV,X,andH, respectively (9). ForS1 nucleaseanalysis, restriction fragments from the library were subcloned into the vector plasmid pKH47 (6). The recombinant EBV-pBR322 plasmids were cut with BamHI to free the EBV DNA. Before ligation into the BamHI site of
pKH47,either the EBV DNAwasisolatedby electro-phoresis in agarose gels or the pBR322 vector was
destroyed by cuttingwithEcoRI,anenzymethat does not cut within the EBV DNA. Ligated DNA(ligase
was obtained from Bethesda Research Laboratories)
was transfected into Escherichia coliHB-101 bythe calcium phosphate technique (15). The subclones of BamHIfragments V, X,and HwerenamedpKH47-V, pKH47-X,andpKH47-H, respectively.
PlasmidDNAwaspreparedfrom bulk cultures(12).
After ethanol precipitation, DNA to be used for S1 nuclease analysiswas suspended in 10mM Tris (pH
8.0)-i mM EDTA andwasstored at-70°C.
Restric-tionenzymes werepurchasedfromBethesdaResearch LaboratoriesorNewEngland Biolabs andwere used asrecommendedbythemanufacturers.
PreparationofRNA.Cytoplasmicandnuclear frac-tions were prepared from IB-4 cells (48). DNA was
sheared withaTekmar Tissuemizerfor the
prepara-tionofnuclear RNA(5).Vanadylribonucleoside
com-plex (10mM)wasusedas anRNaseinhibitor (4). RNA
wasstored in 20 mM Tris (pH7.4)-2mMEDTA-0.2%
sodiumdodecyl sulfateat -70°C.
Forsome experiments, nuclear RNA was further
freed of DNAbypelietingincesiumchloride.RNAin 1 ml of 10mM Tris(pH 7.4)-2 mM vanadylribonucleo-sidecomplex with CsCladdedtoarefractive indexof 1.4017 was overlaid with a CsCl solution with a
refractive index of1.4003 andwas sedimented inan
SW41 rotorat34,000rpmfor6h.The RNAwasnot
degradedduring cesium chloride centrifugation since the45S ribosomal RNAprecursor wasstillvisible in gelsofCsCl-purifiednuclear RNA.
In vitro transcription. BamHI fragment V DNA (2 Fg) was incubated at30°C for 60min with 30 ,ul of HeLacellextract(Bethesda ResearchLaboratories)in 50 p.l ofa solution consisting of 5 mM ATP, 5 mM
CTP, 5 mM GTP, 0.05 mM UTP, 10 ,uCi of
[a-32P]UTP, 10 mM creatine phosphate, and 0.14 mM EDTA (29, as specified bythe manufacturer). After completion ofthe reaction, protein was removed by
phenol andchloroform isoamyl alcohol extraction in the presence of 0.5% sodium dodecyl sulfate-7 M
urea. Nucleic acidwas precipitated, suspended, and B.
U5 TR
primary transcripts
46,
To a a m
3-0
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.490.107.393.75.227.2]426 VAN SANTEN ET AL.
subjected to electrophoresis in 1.4% agarose methyl mercury gels (3).
Preparation andhybridization of RNA blots.Nuclear polyadenylated and non-polyadenylated RNAs were separated by one cycleof oligodeoxythymidylic acid [oligo(dT)]-cellulose column chromatography (2, 35). Cytoplasmic polyadenylated RNAwaspurified by one or twochromatography cycles. The yield of cytoplas-micpolyadenylated RNA was between 1 and 2 mg per 8x 109 cells.Denaturation of RNA, electrophoresis in formaldehyde-agarose gels (27), and blotting onto ni-trocellulose (46) were as previously described (48). Thesize of nuclear RNAs wasdetermined by plotting the square root of the size against the log of the electrophoretic mobility (27). Ribosomal precursor RNAsizes of 6.9 and 14 kb and ribosomal RNA sizes of5.2and 2kbwereusedas standards(49). Electro-phoresis ofcytoplasmic and nuclear RNA wasin 0.8 and 0.6% agarose gels, respectively. Specific DNA restriction fragments were recovered from agarose (HGTP, Seakem) gels by electrophoresis against a dialysis membrane. Fragmentswerethenlabeled with 32Pby nick translation (43) and hybridized to RNA blots as previously described (48) without dextran sulfate.
The number of IR1-U2 3-kb cytoplasmic polyade-nylated RNA molecules per cellwasdeterminedfrom the relative levels ofhybridization of the RNA and known amounts ofDNA to recombinant viral DNA probes and from the RNAyieldper cell.
Si nuclease analysis. S1 nuclease analysis (6) of RNAwasperformed byusing strand-separated DNA. Individual strands of EBV restriction fragments sub-cloned into the vector plasmid pKH47were purified
by binding to oligo(dT) or oligodeoxyadenylic acid
[oligo(dA)]-cellulose (16). Nonradioactive
strand-sep-arated DNA(25to100ng)washybridizedto50 ,ugof
IB-4 cytoplasmicpolyadenylatedRNAoryeast RNA
in 10 ,ulof50%'o (vol/vol)recrystallizedformamide-0.4
M NaCl-40 mM PIPES
[piperazine-N,N'-bis(2-ethanesulfonicacid),pH6.5]-1mMEDTA-0.1% sodi-um dodecyl sulfate at 50°C for 3 h. Nonhybridized DNAsequences wereremovedbydigestionin 0.3 ml of 0.28 M NaCl-30 mMNaC2H302 (pH 4.4)-4.5mM
ZnSO4-20
p.g
each of native and denatured salmon sperm DNA per ml, with S1 nuclease (Boehringer-MannheimCorp.)at1,000to2,000U/ml,for 30 minat37°C. After ethanolprecipitation,RNAwasdestroyed by incubation in 0.1 M NaOH at 70°C. The
RNA-cDNAwaselectrophoresedon1.5% alkaline agarose
gels(31), transferredtonitrocellulose(44), and
detect-ed by hybridization to nick-translated probes made
from EBV DNA restriction fragments purified from recombinant DNAplasmids.AHinfldigestofplasmid pDK286orpKH47-HDNA wasincludedin eachgel and blot to provide size markers. In some experi-ments, the RNA-cDNA was alsoelectrophoresed in formaldehyde agarosegelsasdescribed forRNA(48). Bothtypes ofdenaturing gelsyieldedidentical results for the sizeof the protectedDNA.
RESULTS
Nuclear RNAs encoded by the IR1-U2BamHI
fragments V, H, and H. BamHI fragmentV, X,
and H probes hybridized almost exclusivelyto
the samenuclear RNAs (Fig. 2). Atraceof the
3.0-kb cytoplasmic RNA was present in the polyadenylated nuclearRNAfraction(identified
as 2.9kb in theautoradiogram shown in Fig. 2).
Two classesofRNAs larger than3 kbcouldbe distinguished. The largest nuclear RNAs were
polyadenylated and range in size from approxi-mately 22 to 14 kb in decrements of
approxi-mately 3kb.ManyRNAspecies between 5 and
13kbwerein boththepolyadenylated and
non-polyadenylated fractions. Itwas, therefore, not
possible to discern whether the stepladder of polyadenylated RNAs continues through the
rangeof 5to 13 kb.
BamHIfragment H includesnot only theright
partof U2 but also IR2and theleft2.7 kbofU3
(Fig. 1). In addition to those nuclear RNAs in
common with BamHI fragments V and X,
BamHI fragment H identifies a 2-kb nuclear polyadenylatedRNA anda moreabundant
1.1-kbnuclear non-polyadenylated RNA.Theselast RNAsare encodedby the U3 region ofBamHI fragment H, whereas the RNAs incommon with
BamHI fragments V and X donot extend into IR2 orU3. Thus, probes which include mostly the U2part ofBamHIfragmentH hybridizeto
the same RNAsasBamHI fragmentsV andX,
whereas probes from BamHI fragment H U3
hybridizetothe 1.1- and 2.0-kb RNAs butnot to
theotherRNAs (Fig. 2;other datanot shown). IR2 does not hybridize to any ofthe nuclear
RNAs of latently infected cells (11). All ofthe
nuclear RNAs were confirmed to be RNA and
not DNA by their density in neutral cesium
chloride.
Direction of transcription of the 3-kb
cytoplas-mic polyadenylated RNA. To determine the
di-rection of transcription of the cytoplasmic polyadenylated RNA, BamHIfragments V, X, and Hwere cloned intoplasmid pKH47 (16)so
that the strands could be separated. Plasmid pKH47 is a derivative of pBR322 with adenine and thymine homopolymers inserted into the opposite strands so that the strands can be separated byusingoligo(dT) and oligo(dA)
col-umns(16). To relate the separated EBV DNA
strands which were cloned into pKH47 to the
strandsofthe EBVgenome, it was necessary to
determine the relative orientation of the EBV
andpKH47 restriction endonuclease sites andto
identify which pKH47 strands contain polydeox-yadenylic acid or polydeoxythymidylic acid.
The strategy used to identify the
polydeoxya-denylic acid-containing strand of pKH47 is
shown inFig.3. Theorientation ofEBV DNA in
pKH47-V, pKH47-X, and pKH47-H was then
determined by restriction endonuclease
analy-sis.
Each of the separated strands of plasmids pKH47-V, pKH47-X, and pKH47-H was then
hybridized tocytoplasmic polyadenylatedRNA J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
IR1-U2-ENCODED EBV RNA IN INFECTED CELLS
PA(+)
PA(-)
V
X H
* - 22.1
69.11
- 9.14it --
13.7
6
9& &*
5.3tg|6
20. -2.0
H(R)
V
X H
H(L)
H(R)
-1
-13
6.9k
-6.5
5.3
g1|-4.6
-2.0
2.0.
2.... . 1
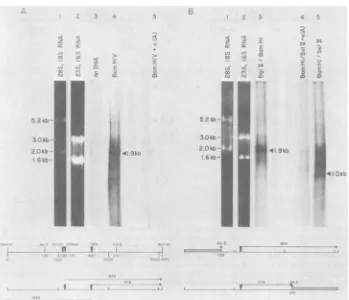
FIG. 2. Sizes ofIB4nuclear RNAs transcribedfrom EBV BamHIfragments V, X,and H.32P-labeledprobes
of BamHIfragmentsV, X,and Hwerehybridizedtoblots ofpolyadenylated[PA(+)]andnon-polyadenylated [PA(-)] nuclear RNA. The BamHIfragmentV probeisPstIfragmentb(see Fig. 5). H(L)andH(R)indicate
probesmade from the SstI-a and SstI-bfragmentsofBamHIfragmentH(see Fig. 7).Ribosomal and precursor ribosomal RNAs fromIB-4cells(A)wereusedassize standards(49). RNAs detectedby hybridizationtothe DNAfragment probes areindicatedbylines. RNAsidentified in bothpolyadenylatedandnon-polyadenylated RNAarenotindicatedonthe blotsofpolyadenylatedRNA.
and treated with S1 nuclease. RNA protected
parts of the EBV DNA R strand of all three
clones, indicating thatRNAis transcribedfrom
lefttoright from BamHI fragments V, X,and H
(see Fig.5through 7). CytoplasmicRNA did not
protectanyof the L strand from SI digestion. Invitrotranscription from IR1. The
observa-tions that cytoplasmic polyadenylated RNA is
transcribed from the R strand of BamHI
frag-mentsV, X, and H and that thesamefragments
encodelarge nuclear RNAs differing in size by
thelength of IR1 suggestthattranscription may
be initiated in any copy ofIRL. A CCAAT-34
nucleotide-TATAAA sequence similarto many
RNApolymerase II promoter sequences and a
less similar TATA sequence have been defined
in IR1 and are in the correct orientation to
promote transcription from left to right (8).
Utilization of these promoter-like sequences
wastestedby usingaHeLacellextract(29) and
BamHIfragment V DNA. BamHI fragment V is
onecompletecopyofIRL.Therelevantmapsof
BamHI fragment V are shown in Fig. 4. The
CCAAT-34 nucleotide-TATAAA sequence
would promote transcription ofa1.9-kb runoff
product, whereas the TATA sequence would
promotetranscription ofa1.4-kb RNA(ora
1.6-kb RNA if the promoterfunctions in the
oppo-sitedirection).Transcripts of approximately 1.9
kbweredetected(Fig. 4A).Thesynthesis of this
RNAwas inhibited by 0.8 ,ug ofalpha-amanitin
perml, suggesting that the 1.9-kb runoff product
is transcribed by RNA polymerase II. As
ex-H
[image:4.490.84.417.73.290.2].../
FIG. 3. Determination of the oligo(dA)- or
oligo(dT)-bindingstrand ofplasmid pKH47. (A) Sche-matic diagram of the pKH47-X DNA used in the experiment. The plasmid DNA was cleaved with HindIII (H), labeled with 32P at the 5' ends (-) (30),andthen cleaved with BamHI(B).The oligo(dA)-and oligo(dT)-binding sites are indicated by closed rectangles.Onlythe strand withacounterclockwise 5' to3' directioncontainsalabeled end which would be
covalently attached to oligo(dA) or oligo(dT). This plasmidDNAwasdenatured and strandseparated by
binding to oligo(dA)- and oligo(dT)-cellulose. (B) Autoradiogramofanagarosegel containingthe mate-rial recovered from oligo(dA)-cellulose (A) and oligo(dT)-cellulose (T). The data indicate that the 5' labeled end isonthepoly(A)-containing strand.
-1.1 VOL.46, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.490.287.409.429.527.2]428 VAN SANTEN ET AL.
.~
_:i.~,~ ..- i-.
_
.w
-4 . FXt X 4 ',.w
[image:5.490.76.425.71.371.2]I
FIG. 4. Transcriptionfrom EBVBamHI fragmentVinvitro.BamHI fragmentVDNApurified from plasmid
pDK14 (9) was transcribed bya HeLa cell extract. The labeledtranscripts were electrophoresed in methyl
mercury agarose gelsand detectedbyautoradiography. Marker human andE.coli ribosomalRNAs are shown in
lanes 1 and 2. InA, the products of transcription from BamHI fragmentVDNA are shownin lane4.Negative controls in whichnoBamHI fragmentVDNA was added(lane 3) orinwhichRNApolymeraseIIwasinhibited
by0.8 jigofalpha-amanitin perml (lane 5) are shown. Promoter-like sequences identified in the nucleotide
sequence (8)areshown in themapofBamHI fragmentV(A). The numbers indicatenucleotides. The map below
shows the expected length of runoff products if transcription begins near each promoter-like sequence.
Transcriptionin either direction from thepromoter-like sequence atnucleotide 1663 is shown. In B, products of
invitrotranscriptionaftercleavageof the BamHIfragmentVtemplate DNA withBglII (lane 3) orSstII(lane 5) are shown. Lane 4 shows the products oftranscriptionfrom SstIl-cleaved BamHI fragment V DNA in the
presenceof 0.8jigofalpha-amanitin perml.Adiagramoftheexpectedeffect of cleavage of the template DNA on
the size of thetranscript initiated nearthepromoter-like sequence atnucleotide 1137 is also shown (B).
pected from the restriction endonuclease sites, cleavage ofBamHIfragment V with BgIII had no effect on the length ofthe runoff product, whereascleavage withSstII shortened the
run-offproductto1 kb(Fig.4B). This indicates that
the CCAAT-34 nucleotide-TATAAA sequence
is partofafunctionalpromoter.A small amount
of1.9-kb RNAwas madewithout BamHI
frag-mentV. Thisendogenous templateactivity was
unaffected by0.8 ,ug ofalpha-amanitinperml.
Si nuclease analysis of cytoplasmic RNA
en-coded by BamHJ fragments V, X, and H. The
DNA sequencesofBamHIfragmentsV, X, and
H which encode cytoplasmic polyadenylated
RNAweremapped by Si nucleaseanalysiswith the strand-separatedDNAsofplasmids
pKH47-V, pKH47-X, and pKH47-H. The R strand of each fragment was hybridized with IB4
cyto-plasmic polyadenylated RNA, andunhybridized
DNAwasdegraded with Si nuclease. The
RNA-protectedDNAwaselectrophoresedin
denatur-ingagarosegels,blottedontonitrocellulose,and
detected by hybridization to 32P-labeled EBV
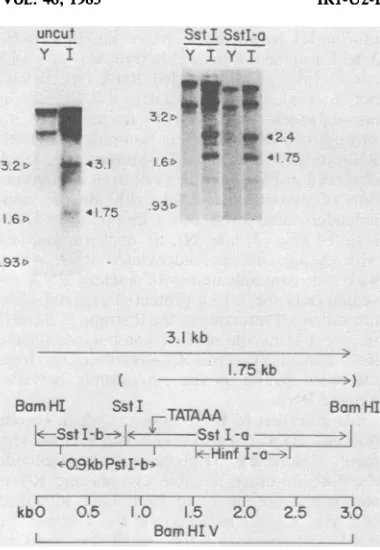
DNAfragments. The RNA protecteda 1.75-kb
segment of BamHI fragment V and the
full-length 3.1-kbBamHIfragmentV(Fig.5). Minor
protected DNAs of 1.75 to 3.1 kb were also
detected. When only DNA to the right of the
Sstl site (see map in Fig. 5) was used in the
hybridization, the full-length protected DNA
and the slightly smaller minor species were
reduced in size to 2.4 kb, and the RNA still
J.VIROL.
"I
;..1
'. I
on November 10, 2019 by guest
http://jvi.asm.org/
429
Sstl SstI-o Y I Y I
32N #*
*< P 424
16> - 4* *4175
93-.93>
3.1 kb
1.75 kb
... ... BmHI SstI -TATAAA
'-09kb PstI-bT ifI-£- >~
r -...T T "".- s .... ,__
kbO 0.5 i.5 2_(0 2.5 3.0
Bam HTV
extends tothe rightwardBamHI site.
Further-more, the data indicate that the minor species
and thefull-length protected DNAdiffer onlyat
their 5' end, which istobe left of the SstI site.
RNAprotecteda0.56-kb segmentofBamHI fragment X and a family of DNA segments
ranginginsize from 1.1 kbtofull-lengthBamHI fragment X (Fig. 6). Cleavage ofBamHI
frag-ment X DNA with restriction endonucleases
XhoI andHpaI, which cutprogressively closer
toitsright end,progressively removed the
high-er-molecular-weight components of the family
and had no effect on the size of the 0.56-kb
protected segment. Identical results were
ob-tained by using only the parts ofBamHI
frag-mentXtothe right of the XhoI and HpaI sites.
Cleavage of pKH47 with HindlllremovesDNA
tothe left of the
HindIll
site in BamHIfragmentBonmHI X (Fig. 6) while leaving the HindIII-bfragment
...,h..._.-..;..j.-7;
1^X Y- - Y r I Y -:
FIG. 5. S1 nuclease analysis of IB-4 cytoplasmic polyadenylated RNA transcribed from the R strand of EBV BamHI fragment V. Hybridizations were done
with yeastRNA ascontrols (Y) andwith IB-4 cyto-plasmic polyadenylated RNA (I). In SstI hybridiza-tions, plasmid pKH47-Vwascleaved with SstI before
strandseparation and hybridizationtoRNA.The SstI site is shown in themapof BamHIfragmentVbelow. After SstIrestriction, EBV DNA sequences onboth
sides of the restriction site remained covalently
at-tachedtopolydeoxyadenylic acid,wererecoveredby
bindingtooligo(dT)-cellulose, andwerehybridizedto
RNA. In SstI fragment a hybridizations, the DNA was also cleaved with a restriction endonuclease
whichcleaves within thevectorsothat only the SstI
fragment a part of BamHI fragment V is recovered andhybridizedtoRNA.The blots shownweremade fromalkaline gels. The positions of marker DNAs (A) andDNAs protectedby IB-4 RNA and notbyyeast RNA (A) are shown. Sizes are indicated in kb. A
restrictionenzyme mapof BamHIfragmentV indicat-ing themap position and size of protected DNAs is shown below. TATAAAindicates the promoter-like
sequenceatposition 1137 in Fig. 3. The possiblerange
ofposition of the1.75-kb protectedDNAsegmentis indicated by parentheses. The directionof transcrip-tionofthe RNA is indicated byarrows. The 0.9-kb
PstI fragment b and Hinfl fragment a, used for
hybridization to blots of cytoplasmicRNA in Fig. 7,
arealso shown.
protected a 1.75-kb segment. Thus, the RNA
protecting the 1.75-kb segment is transcribed
entirely fromDNAtotheright of the SstI site,as
would be expected if the RNA begins nearthe
CCAAT-34 nucleotide-TATAAA sequence and
;
93
--i14
-~. 6 -56 .5
0.56kb -1.9 kb11het
...__.__... ... .. ~~~~~~~~~~~~~~~~o
Bam HI XhoT HpaI Hindm
,I I~~~~~~~~~~~~~~~~
Barn HIT
H,r I-b---h - Hird
kb 05 0 .5 2 0
[image:6.490.48.238.64.339.2]BormHI X
FIG. 6. S1 nuclease analysis of IB-4 cytoplasmic polyadenylatedRNAtranscribed from the R strand of EBVBamHI fragment X. In the lanes labeled XhoI andHpaI, DNAonbothsides of the restriction sites
was used in the hybridization to RNA. In the lanes labeledXhoIL andXhoIR, only DNA to the left or
right, respectively, was used. Since HindlIl also
cleaves thevector,only HindlIl fragmentbwasused
in the lanes labeled HindlIl. The brackets indicatea
family ofprotectedDNAsegmentsof different sizes. Theblots shownweremadefromformaldehyde
agar-osegels.BamHIfragmentXcontainsapartialcopyof IR1, shownasIR1(D),andpartofU2.ThepartofIR1 PstIfragmentbwhich is within BamHIfragmentXis indicated below the BamHI fragment X restriction endonuclease map. Thetransition from IR1 to U2is 123basepairstotheright of the XhoIsite shown. The HindIIIfragmentsaand bofBamHIfragmentXused for hybridization to blots ofcytoplasmic RNA (see Fig.7)areshown. For furtherexplanation,seelegend
toFig.4and5. uncut
Y I
3 2 p-
P
43.141.'75
VOL.
lo
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.490.252.445.289.483.2]430 VAN SANTEN ET AL.
uncut
Y I
41 N
3 2
>1-6.> E
1
413
93-< ., 4
.93>z,Io
13 kb BamHI
-...
S...
sta Hinf I c dgfkbO 2
Bamr U2
:2
FIG. 7. SI nuclease a] ized to the R strand of Hybridizations with IB4 RNA (N) are included. 1 from alkaline gels. TheSs
Hinfl fragments c ofBc
hybridizationtoblotsofc
are shown. As indicated below the restriction mat
tains partof U2, all of IR abbreviations andsymbol
jst :;: Sst-.- Ss6.-- of BamHI fragment H. Since the
BamHI-H-y Sst-I-b fragment doesnothybridizeto the 3-kb
cytoplasmic polyadenylated RNA (see below) but does identify an abundant 1.1-kb nuclear
33 non-polyadenylated RNA, Si nucleaseanalysis
was performed with nuclear non-polyadenylated
4i
3 A g RNAtoseewhether the 1.1-kbprotected
DNA* 41 l w l
sobserved
in Fig.7couldresultfromcontamina-tion of
cytoplasmic
RNA with nuclearnon-polyadenylated RNA. A 1.1-kb protected DNA
resulted (Fig. 7, lane N). S1 nuclease analysis with cytoplasmic polyadenylated RNA which was less contaminated with nuclear RNA
re-Akb vealed only the 1.3-kb protected segment (data
ajT HJ notshown). Protectionof theRstrandof BamHI
Sst b- fragment H by the nuclear non-polyadenylated RNAindicates that this RNA is transcribed from the same strand as the cytoplasmic
polyade-4. 5 nylated RNA.
tHi.
Identification of DNA segments which encodeU ~ the 3-kb RNA. To determine which DNA
seg-ments identified in S1 nuclease analysis encode nalysis of IB4 RNA hybrid- the 3-kb or other possible cytoplasmic RNAs
EBV BamHI fragment H. and which are likely to have been identified non-polyadenylated nuclear because of contamination of the cytoplasmic
rhe blots shown were made RNA with nuclear RNA, small fragments of ;tlfragments a and b and the BamHI fragments V, X, and H were hybridized
7mHI fragment H used for to Northern blots of cytoplasmicpolyadenylated ,ytoplasmic RNA (see Fig. 8)
by the schematic diagram ornuclear RNA (see maps of BamHIfragments
ps,
BamHIfragment H con- V, X, and H in Fig. 5 through 7). DNA from the2, and part of U3. For other Hinflfragment aof BamHIfragmentV
hybrid-Is,seelegendtoFig.4and 5. ized to the 3-kb RNA (Fig. 8); DNA from the
attached to pKH47 homopolymer. RNA-pro-tected DNA segments of 0.56 and 0.9 kb were
identified usingthe Rstrandof HindIII-b. These data confirm that the 0.56-kb protected DNA segmentisentirelytotheright of the HindlIl site within the BamHI-X-HindIII-b fragment and the family of larger protected DNA segments hasacommonright end at or near the right end of BamHI fragment X. The data also indicate that the family of protected DNA segments arisesbecause ofheterogeneity beginning1.1kb from the right end of BamHI fragment X and extending to the left end of BamHI fragment X (Fig. 6).
Cytoplasmic polyadenylated RNA protected 1.3- and 1.1-kb segments of BamHI fragmentH DNA(Fig. 7). Use of the SstI fragments a and b
of BamHI fragment H in hybridizations with
RNA showed that the 1.3-kb protected DNA
maps to theleftoftheSstI site, and the 1.1-kb
protected DNA maps to the right (see map of
BamHIfragmentHin Fig. 7).3 P-labeledHinfl
fragment c (the leftmost Hinfl fragment of
BamHI fragment H) hybridized to the 1.3-kb
protected DNA (data not shown), confirming
thatthis protectedDNA mapsnear theleftend
; Hinf1-G HlndIFo --Vd:d'7,Rt-n,, I- S -_, H1lff-c
; | 0 ¢ 33
* .E
~~~~~~
FIG. 8. Hybridizationoffragmentsof BamHI frag-mentsV, X,and Htothe 3-kb RNAon blots of IB-4
cytoplasmic polyadenylated RNA. The DNA frag-mentsindicated above the laneswerelabeled with32P
and were hybridized to RNA blots. The fragments usedare indicated onthe maps in Fig. 4through 6. Ribosomal RNAmarkers(A)andhybridizationof the DNAprobestothe3-kb RNA(A)areshown. Sizesare
indicated in kb. The bracket indicateshybridizationof the PstI fragment b probe to nuclear RNAs which contaminated thecytoplasmicRNApreparation.
52:>,
i 43 1
2 ) I(
J.VIROL.
.O
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.490.55.250.62.293.2] [image:7.490.260.449.422.571.2]PstI-b fragmentof BamHIfragment V did not,
although ithybridizedwelltonuclearRNA(see Fig. 2) andidentifies asmallamount ofnuclear polyadenylated RNA in the cytoplasmic poly-adenylated RNA preparations. Since the PstI
fragment b sequences are notrepresented in the 3-kb cytoplasmic RNA but are represented in nuclear RNA, thefull-lengthand 1.75-to3.1-kb minor BamHI-Vfragments thatwere protected in theSinuclease analyses of DNA (seeFig.5)
cannotbeexonsof the 3-kb RNA andarelikely
tobepartofcontaminating nuclear RNAs. Simi-larly, the HindIIIfragment bofBamHIfragment
Xhybridizestothe 3 kb RNA,but theHindlIl fragmentadoesnot(Fig. 8). This indicates that the 0.56-kb segment of RNA transcribed from theHindIII fragment b (see Fig. 6) is probably
part of the 3-kb RNA. The longer protected DNAs of BamHI fragment X, in addition to
being heterogeneous, contain the HindIII
frag-ment aand PstI fragment bsequencesof BamHI
fragmentX.Therefore, theycannotbeexonsof the 3-kb RNA since the HindIII-a and PstI-b portionsofBamHIfragmentXdonothybridize
to the 3-kb RNA. Also, as would be expected from theS1 experiments, theleftward SstI
frag-ment aandHinflfragmentcof BamHIfragment
Hand thepartof theHinflfragmenta totheleft of IR2hybridize tothe 3-kb RNA, whereas the rightward SstI fragment b and IR2 donot(Fig.7 and data notshown). EcoRI fragmentJ, which
maps totheleft of IR1 in Ul, doesnothybridize
to any of the IR1-U2 RNAs.
Reconstruction experiments inwhich the hy-bridization of BamHI-X-HindIII fragment b to
the 3-kb RNA wascompared withhybridization
to known amounts of BamHI-X under similar conditions indicate that there is approximately 0.0038ngof the BamHI-Xpartof the3-kbRNA
per 5 ,ug of IB4 cytoplasmic polyadenylated
RNA. This estimate and the polyadenylated
RNA yield per cell indicate that there are
ap-proximately three copies of the 3-kb RNA per
cell.
DISCUSSION
The simplest model (Fig. 1) which would
account for the accumulated data describing
RNA from IR1 and U2 is that the CCAAT-34
nucleotide-TATAA sequence in IR1 is part of
thefunctional promoterfortranscription ofthe
IR1-U2 R strand in latently infected cells. The
smallesttranscript would startin the last
com-pletecopyofIR1, extendthrough0.9kbofIR1,
continue through the BamHI site between
BamHIfragmentsVand Xintothe 0.6 kb of IR1
and 1.4kb ofU2inBamHI fragmentX, and then
continue through at least 1.3 kb of BamHI
fragment H U2. Afterpolyadenylation at a site
about 1.3 kb intoBamHI fragment H U2, this
transcript would be approximately 5.3 kb. Other polyadenylated RNAs would begin inmore left-ward copies of IR1 and would vary in size
upwards from5kb in3-kb increments. The
1.1-kb U3 non-polyadenylated RNA could be a 3'
cleavage product of the putative primary
tran-script since it is also transcribed from the R strand of EBV DNA. However, no RNA
en-coded by IR2 has been detected. The 3-kb cytoplasmic polyadenylated RNA would be spliced from the large IR1-U2 polyadenylated RNAs andconsist (3' to 5')ofa polyadenylate tail (probably less than 0.2 kb), segmentsof1.3
and 0.56 kb from BamHI-H and -X U2 which
may be continuous, and small exons as yet
undetected from the left end of U2 and IR1,
totaling0.9kb (Fig. 1).Alternativelythe1.75-kb RNA-protectedIR1 DNA segmentcouldencode anexon of the 3-kbRNA.
Data in support of this model include the
following. (i) There are large nuclear RNAs which differ in length by approximately 3 kb (Fig. 2). IR1 isa3-kb tandem repeat. S1
nucle-ase analysis shows that some RNA molecules includeatleastonefull-lengthcopyof IR1 (Fig. 5). (ii) RNA hybridizes onlytothe R strand of
EBVIR1orU2DNA,indicating thatmost orall of the RNA is transcribed from leftto right as
shown in Fig. 1. (iii) There is a CCAAT-34 nucleotide-TATAAAsequence which ispart of
a promoter for in vitro transcription of the R
strand of IR1 (8; Fig. 4). In latently infected cells, an RNA segment was detected which is complementarytoandnearly equalinlengthto
the partof the IR1Rstrand 3'totheCCAAT-34 nucleotide-TATAAA sequence (Fig. 5). This 1.75-kb segment could be the 5' end of the
precursorRNA. ThecytoplasmicRNA
prepara-tions were contaminated with nuclear RNAs. The discrepancy in size between the 1.9-kb runoff product of in vitro transcription and the 1.75-kb RNA segment detected byS1 nuclease analysis with RNA from latently infected cells could be due to the different types of gels (methylmercury versus formaldehyde) and dif-ferent markers (RNA versus DNA size mark-ers). Alternatively, the 1.75-kb RNA segment
could bepartofapartiallyorcompletelyspliced
RNA. The1.75-kb segmentisunlikely tobean exon of the 3-kb RNA since there are two other
RNA-protected DNA segments totaling 1.9 kb
(seebelow), and althoughtheBamHI-V-HinfI-a
partofthe1.75-kbDNA segmenthybridized to the 3-kb RNA, Hinfl fragmente, therightmost
part of the 1.75-kb segment, did nothybridizeto
the 3-kb RNA (data not shown). This suggests
that not allof the IR1 sequence totheright ofthe
promoter becomes part of the 3-kb RNA. (iv)
The 3-kbcytoplasmic polyadenylated RNAis a
spliced RNA since it is encoded partly by
VOL.46, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
432 VAN SANTEN ET AL.
BamHI-V-Hinfl-a and by BamHI-H-Hinfl-a, whichare 3.5kbapart inthe EBV genome (Fig. 1, 5, and 7), and the intervening BamHI-X-HindIII-a fragment doesnothybridize tothe 3-kb RNA(Fig.1and7 anddatapresented above). The IR1 sequence (8) reveals many potential splicedonorandacceptorsites in thepartof IR1
to theright oftheputativepromoter. (v) AllIR1
and U2 DNA fragments tested, including the BamHI-V-PstI-b fragment (much of which is included in the BamHI-X-HindIII-a fragment), hybridize tothe large nuclearRNAs, aswould be expected if these RNAs are unspliced or
partially splicedprecursors tothe3-kb cytoplas-mic RNA(Fig. 1). (vi)Cytoplasmic polyadeny-latedRNAis complementarytopossibly
contin-uous 0.56- and 1.3-kb segmentsof theR strand of BamHIfragmentXandHU2 DNA.
Further-more, these DNA segments hybridize to the
3-kb cytoplasmic polyadenylated RNA. (vii) The large nuclear RNAs and the 3-kb cytoplasmic RNAdonot extend intoIR2 or U3; thus, they have similar 3' ends.
RNAs having common 3' and variable 5' domainswere detectedfrom IR1 and U2(Fig.5 and 6). Since the cytoplasmic preparations in
which these RNAsweredetectedwere
contam-inated with nuclear RNA, these RNAs could be partially spliced precursors to the 3-kb RNA. Alternatively, they could be mature, less-abun-dant RNAs which are spliced differently from the 3-kb RNA. Partiallyoranomolously spliced globin precursor RNAs have been detected in thenuclei of Friend erythroleukemia cells (13). Thevariablyspliced EBV IR1-U2 polyadenylat-ed RNAs are, in aggregate, as abundant asthe putative fully spliced RNAs. These variably splicedmolecules could inaggregate accountfor the seemingly discrepant finding that there are
fewercopiesof the 3-kb RNAperlatently infect-ed transformed cell than of the RNA specified by the EBV U5 region (van Santen et al., manuscript in preparation), whereas cytoplas-micpolyadenylated RNAhybridizesmore tothe IR1-U2region thanto the U5region.
The modelproposed for generation of the 3-kb
EBV RNA is similar to that proposed for the
formation of polyoma viruslatemessenger RNA
(26, 47). The 5,560-base pair circular polyoma virusDNAencodes giant RNA molecules which are tandemly repeated whole genomic
tran-scripts (1, 26, 47). Multiple copies of a
57-nucleotidesequence which is presentonly once
inpolyomavirusDNAare present at the 5' end
ofpolyomalatemessenger RNAs (26,47). Each copy of the 57-nucleotiderepeat is spliced from
acomplete genometranscript(47).
Sincethe P3HR-1 virusis deleted forU2 and IR2and is unable toinitiategrowth transforma-tion (7, 17, 18, 24, 38, 40), the IR1-U2 RNA is
believed to be important in initiating or
main-taining latent infection or growth transforma-tion. The IR1-U2 RNA would, therefore, be
expected to be a constant feature of latent
growth-transforming infection. However, the U2regionwhich encodes 1.9 kb of the 3' end of
the IR1-U2 RNA varies extensively among
transformation-competent EBV isolates (11, 23).
Alow-abundance 2.0-kb nuclear butnot
cyto-plasmicpolyadenylatedRNA wasalso identified
in this studyto be encoded by the right part of
BamHI fragment H. A more abundant 1.9-kb
polyadenylated early RNA encoded by BamHI fragment H has been previouslydetectedin the
cytoplasm ofproductively infected B95-8 cells
(20). A small amount of this RNA may be
transcribed and polyadenylated but not
trans-ported in latent infection.
ACKNOWLEDGMENTS
Wearegrateful for the gift ofrecombinant EBV DNAs from TimothyDambaugh and Victor Razkowski and for the assist-anceof JuliaDowling in tissue culture. Initial in vitro tran-scription experiments with IR1 DNAweredone byTimothy Dambaugh and Roberto Weinman.
This research was supported by Public Health Service grantsCA 19264 andCA17281 from theNational Institutes of Health and by grant MV 32G from the American Cancer Society. E.K. isafacultyresearchawardee of theAmerican Cancer Society. V.v.S. was supported by aPublic Health Service National Research ServiceAward CA09267from the NationalInstitutes of Health. M.H. is supported by Public HealthService grant AI 07182 from the National Institutes of Health.
LITERATURE CITED
1. Acheson,N.H.,E.Buetti,K.Scherrer,andR.Weil.1971. Transcription of the polyoma virus genome:synthesisand cleavageofgiantlatepolyoma-specificRNA. Proc.NatI. Acad. Sci. U.S.A. 68:2231-2235.
2. Aviv,H.,andP.Leder. 1972.Purification ofbiologically active globin messenger RNA by chromatography on oligothymydylic acid-cellulose. Proc. Natl. Acad. Sci. U.S.A. 69:1408-1412.
3. Bailey, J., and N. Davidson. 1976. Methylmercuryas a reversibledenaturing agentforagarosegel electrophore-sis. Anal. Biochem. 70:75-85.
4. Berger, S.,andC.Birkenmeier. 1979. Inhibition of intrac-table nucleases with ribonucleoside vanadylcomplexes; isolation of RNA fromrestinglymphocytes. Biochemistry 18:5143-5149.
5. Berk, A.J.,F.Lee,T.Harrison, J. Williams,and P. A. Sharp. 1979.Pre-early adenovirus 5 geneproduct regu-lates synthesis of early viral messenger RNAs. Cell 17:935-944.
6.Berk, A.J.,and P.Sharp. 1977.Sizingandmappingof early adenovirus mRNAs by gel electrophoresis ofS1 endonuclease-digested hybrids.Cell 12:721-732. 7. Bornkamm, G. W., J. Hudewentz, U. K. Freese, and
U. Zimber. 1982. Deletion of the nontransforming Ep-stein-Barrvirus strainP3HR-1causesfusionofthelarge internal repeatof theDSL region.J. Virol. 43:952-968. 8. Cheung, A.,andE.Kieff.1982.Longinternal direct repeat
inEpstein-Barrvirus DNA. J.Virol.44:286-294. 9. Dambaugh, T., C. Beisel, M. Hummel, W. King,
S.Fennewald,A.Cheung,M.Heller,N.Raab-Traub,and E. Kieff. 1980. Epstein-Barrvirus DNAVII: molecular cloning and detailedmappingofEBV(B95-8)DNA. Proc. NatI.Acad. Sci. U.S.A.77:2999-3003.
10. Dambaugh,T.,E.Kieff,F. K.Nkrumah,and R.J.Biggar. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
1979. Epstein-Barr virusRNA. IV.Viral RNA in Burkitt tumortissue. Cell 16:313-322.
11. Dambaugh, T. R., and E. Kieff. 1982. Identifiction and nucleotide sequences of two similartandem direct repeats in Epstein-Barrvirus DNA. J. Virol. 44:823-833. 12. Davis, R., D. Botstein, and J. Roth. 1980. Advanced
bacterial genetics. Cold SpringHarbor Laboratory, Cold Spring Harbor, N.Y.
13. Donaldson,D., A. McNab, G. Rovera, and P. Curtis. 1982. Nuclearprecursormolecules of thetwoB-globinmRNAs inFriendErythroleukemiacells. J Biol.Chem. 257:8655-8660.
14. Gerber, P., J. Whang-Peng, and J. H. Monroe. 1969. Transformation and chromosome changes induced by Epstein-Barr virus in normal human leukocyte cultures. Proc. Natl. Acad. Sci. U.S.A. 63:740-747.
15. Goodman, H.,and R. MacDonald. 1979. Cloning of hor-mone genes from a mixture ofcDNA molecules. Methods Enzymol. 68:75-90.
16. Hayashi, S. 1980. A cloning vehicle suitable for strand separation. Gene 11:109-115.
17. Hayward, S. D., S. G. Lazarowitz, and G. S. Hayward. 1982. Organization of the Epstein-Barr virusDNA mole-cule. II. Fine mapping ofthe boundaries of the internal repeat cluster of B95-8 and identification of additional small tandem repeats adjacent to the HR-1 deletion. J. Virol.43:201-212.
18. Heller, M., T. Dambaugh, and E. Kieff. 1981. Epstein-BarrVirus DNA. IX. Variation among viral DNAs from producer and nonproducer infectedcells. J. Virol. 38:632-648.
19. Heller, M., V. van Santen, and E. Kieff. 1982. Simple repeatsequence in Epstein-Barr virus DNA is transcribed in latentand productive infections. J. Virol. 44:311-320. 20. Hummel, M., and E. Kieff. 1982. Epstein-Barr virus RNA.
VIII. Viral RNA in permissively infected B95-8 cells. J. Virol. 43:262-272.
21. Kawai, Y., M.Nonoyama, andJ.S.Pagano. 1973. Reasso-ciation kinetics for EBV DNA: nonhomologyto mammali-anDNAand homology of viral DNA in various diseases. J. Virol. 12:1006-1012.
22. Kieff, E., T.Dambaugh, M. Heller, W. King, A. Cheung, V. van Santen, M. Hummel, C. Beisel, S. Fennewald, K.Hennessy, and T. Heineman. 1982. Biology and chemis-tryof Epstein-Barr virus. J. Infect. Dis. 146:506-517. 23. King, W., T. Dambaugh, M. Heller, J. Dowling, and
E. Kieff. 1982. Epstein-Barr virus DNA. XII. A variable region of the EBV genome is included in the P3HR-1 deletion. J. Virol. 43:979-986.
24. King, W., A. L. Thomas-Powell, N. Raab-Traub, M. Hawke,and E. Kieff. 1980. Epstein-Barr virus RNA. Viral RNA in a restringently infected, growth-trans-formed cell line. J. Virol. 36:506-518.
25. King, W., V. vanSanten, and E. Kieff. 1981. Epstein-Barr virus RNA. VI. Viral RNA in restringently and abortively infected Raji cells. J. Virol. 38:649-660.
26. Legon, S., A.J. Flavell, A. Cowie, and R. Kamen. 1979. Amplification in the leader sequence of late polyoma virus mRNAs. Cell 16:373-388.
27. Lehrach, H., D. Diamond, J. Wozney, and H. Boedtker. 1977. RNA molecular weightdeterminations by gel elec-trophoresis under denaturing conditions, a critical reex-amination.Biochemistry 16:4743-4751.
28. Lindahl, T., A. Adams, G. Bjursell, G. W. Bornkamm, C.Kaschka-Dierich, and U.Jehn. 1976.Covalently closed circular duplex DNA of Epstein-Barr virus in human lymphoidcell line. J. Mol. Biol. 102:511-530.
29. Manley,J., A. Fire, A. Cano, P. Sharp, and M. Gefter. 1980.DNA-dependent transcription of adenovirus genes in asoluble whole cell extract. Proc. NatI. Acad. Sci. U.S.A.77:3855-3859.
30. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. MethodsEnzymol.65:499-560.
31. McDonnell, M. W., M. N.Simon, and F. W. Studier. 1977.
Analysis of restriction fragments of T7 DNA and determi-nation of molecular weights by electrophoresis in neutral and alkaline gels. J. Mol. Biol. 110:119-146.
32. Miller, G.,J.Robinson, L. Heston, and M. Lipman. 1974. Differences between laboratory strains of Epstein-Barr virus based on immortalization, abortive infection and interference. Proc.Natl.Acad. Sci.U.S.A. 71:4006-4010. 33. Nilsson, K. 1979. The natureoflymphoid cell lines and their relationship to the virus, p. 225-281. In M. Epstein and B. Achong (ed.), The Epstein-Barr virus. Springer-Verlag KG, Berlin.
34. Nonoyama, M., and J. S. Pagano. 1972. Separation of Epstein-Barr virus DNA from large chromosomal DNA in non-virus producing cells. Nature (London) New Biol. 238:169-171.
35. Orellana, T., and E. Kieff. 1977. Epstein-Barr virus-specific RNA. II. Analysis of polyadenylated viral RNA in restringent, abortive, and productive infections. J. Virol. 22:321-330.
36. Pope,J.H., M. K. Horne, and W. Scott. 1968. Transfor-mation of fetal human leukocytes in vitro in filtrates of a human leukemic cell line containing herpes-like virus.Int. J. Cancer 3:857-766.
37. Powell, A. L. T., W. King, and E. Kieff. 1979. Epstein-Barr virus-specific RNA.III.Mapping of the DNA encod-ing viral RNA in restrencod-ingent infection. J. Virol. 29:261-274.
38. Pritchett, R. F., S. D. Hayward, and E. Kieff. 1975. DNA of Epstein-Barr virus.I.Comparative studies of the DNA of Epstein-Barr virus from HR-1 and B95-8 cells: size, structure, and relatedness. J. Virol. 15:556-569. 39. Pritchett, R. F., M. Pedersen, and E. Kieff. 1976.
Com-plexity of EBV homologous DNA in continuous lympho-blastoid cell lines. Virology 74:227-231.
40. Raab-Traub, N., R. Pritchett, and E. Kieff. 1978. DNA of Epstein-Barr virus. III. Identification ofrestriction en-zyme fragments that contain DNA sequences which differ amongstrainsof Epstein-Barr virus.J.Virol. 27:388-398. 41. Ragona, G., I. Ernberg, and G. Klein. 1980. Induction and biological characterization of the Epstein-Barr virus (EBV) carried by the Jijoye lymphoma line. Virology 101:553-557.
42. Reedman,B. M., andG. Klein. 1973. Cellular localization of an Epstein-Barr virus (EBV)-associated complement-fixing antigen in producer and non-producer lymphoblas-toid cell lines.Int.J. Cancer 11:499-520.
43. Rigby, P. W.J.,M. Dieckmann, C. Rhodes, and P. Berg. 1977. Labeling deoxyribonucleic acid to high specific activityin vitro by nick translation with DNA polymerase. J.Mol. Biol. 113:237-251.
44. Southern, E. M. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J.Mol. Biol. 98:502-517.
45. Svedmyr, E.,and M.Jondal.1975.Cytotoxic effectorcells specificfor B cell lines transformed by Epstein-Barr virus are present in patients with infectious mononucleosis. Proc. NatI.Acad. Sci. U.S.A. 72:1622-1666.
46. Thomas,P. A.1980.Hybridization of denatured RNA and small DNAfragmentstransferred to nitrocellulose. Proc. Natl. Acad. Sci.U.S.A.77:5201-5205.
47. Treisman, R. 1980. Characterization of polyoma late mRNAleader sequencesby molecular cloning and DNA sequence analysis.Nucleic Acids Res. 8:4867-4888. 48. vanSanten,V., A.Cheung, and E. Kieff. 1981.
Epstein-Barr virus RNA. VII. Size and direction of transcription ofvirusspecified cytoplasmic RNA in a cell line trans-formed by EBV. Proc.NatI.Acad. Sci. U.S.A.
78:1930-1934.
49. Wellauer, P., and I. Dawid. 1973. Secondary structure maps ofRNA:processing of HeLa ribosomal RNA. Proc. Natl.Acad. Sci.U.S.A.70:2827-2831.
50. zur Hausen, H., and H.Schulte-Holthausen. 1970. Pres-ence of EB virusnucleicacid homology in a "virus free" line ofBurkitt's tumor cells. Nature (London) 227:245-248.
VOL.46, 1983
on November 10, 2019 by guest
http://jvi.asm.org/

![FIG. 2.probesof[PA(-)]ribosomalRNADNA BamHI Sizes of IB4 nuclear RNAs transcribed from EBV BamHI fragments V, X, and H](https://thumb-us.123doks.com/thumbv2/123dok_us/1446759.97143/4.490.287.409.429.527/probesof-ribosomalrnadna-bamhi-sizes-nuclear-transcribed-bamhi-fragments.webp)
![FIG.:2ized 7. SI nuclease a] to the R strand ofnalysis](https://thumb-us.123doks.com/thumbv2/123dok_us/1446759.97143/7.490.260.449.422.571/fig-ized-si-nuclease-r-strand-ofnalysis.webp)
