Copyright© 1977 AmericanSociety for Microbiology Printed inVol.23,U.S.A.No. 3
In
Vitro
Synthesis of
a
Unique RNA
Species
by
a
T
Particle
of
Vesicular
Stomatitis Virus
SUZANNE U. EMERSON,* PETER M. DIERKS, AND J. THOMAS PARSONS
Department ofMicrobiology,TheUniversity of VirginiaSchoolofMedicine,Charlottesville, Virginia22901
Received forpublication 7April 1977
A T particle of vesicular stomatitis virus, containing most ofthe L-gene region, has been isolated. In vitro, these T particles synthesize
exclusively
asmalladenine-rich RNA that is
complementary
totheT-particle genome. Par-tial sequenceanalysisofthis small RNAindicatesthat it isanRNA of unique sequence with alengthofapproximately 45nucleotides.Vesicular stomatitis virus (VSV) contains a single strand of minus-sense RNA that codes forfive proteins,N, NS, M, G, andL (28). The linear arrangement of the genes coding for these proteins in wild-type virus has been shown to be 3'-N-NS-M-G-(L)-5' (1, 4). How-ever, when VSVispropagatedat ahigh multi-plicity ofinfection, a new class ofvirusparticles
(T particles) is generated that appear shorter than the normal VSV particles (B particles) (17). Characterization ofa number of T par-ticles of various sizes has shown that theyall contain incomplete genomes and usually are
derived from the region of the viral genome
coding for the Lprotein (25). Since Tparticles
are defective for replication, they require the co-replication of B particles toprovide missing functions. Also, Tparticles apparently replicate more quickly than B particles and interfere with their production. The mechanisms of
T-particle generation and interference with
B-particle replication are not presently
under-stood.
Bparticles contain anRNA-dependent RNA
polymerase that synthesizes mRNA in vitro,
using the B-particle nucleocapsid astemplate (5, 9, 21). This mRNA is capped at the5' end with7mG and ispolyadenylatedatthe3'end (2, 7, 26). Most ofthe mRNA is synthesized from the region ofthe genome coding for the N, NS, M, and G proteins; the region of the genome coding for the L proteindoes notappear tobe
efficiently transcribed in vitro (9, 16). The in
vitro synthesis of RNA byTparticles has also been investigated by anumber oflaboratories (8, 13, 20, 23). Reichmann et al. examined six different T particles and concluded that all T
particles reproducibly synthesized a small quantity of adenine-richRNAthat was
consid-erablyshorter than theRNAsynthesizedbyB
particles (23). The nucleotide and ion require-ments for this RNAsynthesis were similar to
the
requirements
fortranscription
by
Bparti-cles.
However,
Reichmannetal.wereunabletohybridize
theT-particle products
tovirionRNA and thus could not demonstrate that these RNAswerecodedby
theT-particle
template. Incontrast, Mori andHowatsonreportedthat
cer-tain
T-particle
preparations synthesized RNA thatwashydrogen
bondedto T-particlevirionRNA,
indicating
that Tparticlesmayfunctionas
templates
for in vitrosynthesis (20).Reconstitution studies have shown that T
particles
do contain an RNA-dependent RNApolymerase,since soluble enzymefractions
pre-pared
from either BorTparticleswereequally
effective inpromoting synthesisofmRNAfrom
enzyme-deficient B-particle
nucleocapsidtem-plates
(13).However,
only very low levels of RNAsynthesis
were observed when enzyme fractions from either B or T particles wereadded to
T-particle
nucleocapsidtemplates,
suggesting thatT-particle templates were
de-fective for RNA synthesis. These observations have led us to reexamine the question of whether Tparticles actively synthesizeRNAin
vitro that is
template
coded. Inthis paperwereportexperiments demonstratingthataT
par-ticle derived from the L-gene region of VSV doesserve astemplateforRNAsynthesis; how-ever, the T-particle productis notsynthesized from the entire genome, but rather isaunique
RNA species approximately 45 nucleotides in
length.
MATERIALS AND METHODS
Chemicalsand radiochemicals. For the
hybridi-zation studies, pancreatic RNase A was obtained fromSigma ChemicalCo., St. Louis, Mo., andT1 RNase was obtained from Worthington Biochemi-cals Corp., Freehold, N.J. a-32P-labeled triphos-phates (105Ci/mmol) and[32P]orthophosphatewere
purchased from Amersham/Searle, Arlington Heights, Ill. [3H]uridine (27 Ci/mmol),[3H]ATP(17
Ci/mmol),and[3H]UTP(16Ci/mmol)wereobtained 708
on November 10, 2019 by guest
http://jvi.asm.org/
VOL. 23, 1977
fromSchwarz/Mann, Orangeburg, N.Y. Oligo(dT)-cellulose (T2) was from Collaborative Research Inc., Waltham, Mass. Millipore Corp., Bedford, Mass., provided the cellulose acetate filters. Acrylamide andNN-methylenebisacrylamide (Bis) were from Bio-Rad Laboratories, Richmond, Calif., and the agarose wasfrom Marine Colloids, Inc., Springfield, N.J.
Virus. VSV, Indiana serotype, was originally from the U.S. Agricultural Research Center, Belts-ville, Md. A cloneobtained from Robert R. Wagner and passaged in mouse L cells was recloned and adapted to growth in BHK-21 cells, whereupon it
generatedthe Tparticle used in these experiments. Forthepreparation of 3H-labeled VSV RNA, virus was grown in BHK-21 monolayers at 370C in the presence of 90% BHK-21 medium(containing gluta-mine)and 10%tryptosephosphate broth with 5 ,uCi of [3H]uridine per ml. Labeled virions were har-vested 16 to 18 h after infection by differential, rate
zonal,andequilibriumcentrifugation as previously described, except that three rate zonal centrifuga-tions were included (15). A virus clone from the samestock,which did not generate T particles, was used to infect cells for preparation of viral mRNA.
RNApolymerase assays. RNA polymerase assays were carriedout as previouslydescribed (14). One
and one-halfvolumesofpurifiedB or T virions in10 mM Tris-hydrochloride (pH 7.4) containing 15% glycerol were mixed with 1 volume of1 x high-salt solubilizer and2.5volumes of reaction mix toyield a final concentration of 30 mMTris-hydrochloride(pH 8.0),4mMmagnesium acetate, 0.65 mM
dithiothrei-tol, 0.8 mMconcentrations of the three unlabeled
triphosphates, and 0.5 mMconcentrations of the a-32P-labeled triphosphates (500
,tCi/tmol),
7.5 tM[3H]UTP (625 uCi/,umol), or 0.6 mM [3H]ATP (83
,uCi/,tmol). Sampleswereincubated at 31°C for the
indicatedtimes, andthelabeledRNA waspurified
by sodium dodecyl sulfate (SDS)-phenolextraction or trichloroacetic acid precipitated and then
col-lectedoncellulose acetate filters and counted. RNApurification.LabeledRNA waspurifiedby extraction with phenol-chloroform-isoamyl alcohol
(1:1:0.04).The RNA was thenprecipitatedfrom the aqueousphase by addition of2volumes ofethanol.
(i)Invitro RNA. If the labeledproductofan in vitro reaction was to beused for hybridization
ex-periments, the nucleocapsid was first removed by centrifugationfor 90 minat38,000 rpmin anSW50.1
rotorbeforephenol extraction. Freenucleotides and small molecules were removed by Sephadex G-50
(Sigma)chromatography.
(ii)Virion RNA.3H-labeledvirionRNA was puri-fiedfrompreparations ofBandTparticlesobtained
by one rate zonalcentrifugation step. The42Sand 30S virion RNAs were separated by centrifugation
at16,500 rpm for16hin anSW27 rotor,usinga10to 30%sucrosegradientcontaining TESbuffer (50 mM
Tris-hydrochloride [pH 7.6]-2 mM EDTA-0.2%
SDS). RNA sedimenting as 42S and 30S, respec-tively,waspooledandreprecipitatedwithethanol. ThevirionRNAwaspartiallyfragmentedby incu-bation at 50°C for 7 min in 50 mM Na2CO3 and ethanolprecipitatedbeforehybridization.
(iii) mRNA. 32P-labeled viral mRNA was har-vested from infected L cells incubated for 5 h in phosphate-free medium 199 containing 80 ,uCi of
[32P]orthophosphate per ml. The monolayers were washed twice with Earle basic salts solution and lysed with basic salt solutioncontaining 1% Triton X-100, 1 mg ofheparin per ml, 90
jig
of cyclohexi-mide per ml, and 250 ,ug of spermidine per ml. Contaminating nuclei were removed by low-speed centrifugation, and the supernatant was layered over1mlof 1 M sucrose in 50mM Tris-hydrochloride(pH 7.4)-25mM KCl-5 mMMgCl2 andcentrifuged intheSW50.1 rotor at 41,000 rpm for 2.5 h. RNA was extracted from the resultant polysome pellet and ethanol precipitated, and the poly(A)-containing
material was selected by chromatography on oligo(dT)-cellulose columns. Thepoly(A)-containing
RNA wasresolved on 10 to 30% sucrose gradients containing TES buffer by centrifugation at 20,000 rpm for 16.5 h in theSW27 rotor. RNA waslocated by Cerenkov counting and the labeled RNA was recovered from the appropriate fractions by ethanol precipitation.
Acrylamide gels. The 2% acrylamide gels con-tained 2% acrylamide, 0.1%Bis, and0.9% agarose, whereas the 20% gels contained 20% acrylamide,
0.075% Bis, and 8 M urea (11). Electrophoresis buffer consisted of 35 mM Tris base, 30 mM
NaH2PO4, 1mMdisodium EDTA, and 0.2% SDS, pH 7.6.Electrophoresis was at5mA/cylindricalgel un-til thebromophenol blue markerwas 1 to 2 cmfrom the bottom. Gels werefractionatedwithaMicklegel
slicer, incubated at 50°C in0.5 ml of Nuclear-Chi-cagosolubilizer diluted with water (9:1), mixed with 10 ml of toleune-based scintillation fluid, and counted on a Beckman scintillationcounter.
Oligonucleotide fingerprinting. RNA products synthesized invitrowith[3H]ATP, [a-32P]GTP,
[a-32P]CTP,or[a-32P]UTPwerepurifiedfrom polymer-ase assays, and unincorporated ribonucleoside tri-phosphates were removed by chromatography on Sephadex G-50. Digestion oflabeled product with bacterial alkaline phosphatase (Worthington) be-fore sequence analysis followed published proce-dures (19). Bacterial alkaline phosphatase was re-moved from thereactionmixby additionofSDSto 0.5% and extraction with water-saturated phenol.
RNAwasdesalted before digestion with RNase T1 bychromatographyonSephadexG-10.
ForRNase T1digestion, 20 jigofyeast RNAwas addedtoeachsample, and the samplewasethanol
precipitated. PrecipitatedRNAwasdissolvedin 2,ul
ofabuffer containing 10mMTris (pH 7.6), 0.5mM
EDTA,and8UofRNase T1(Calbiochem)and incu-bated at 37°C for 30 min. 32P-labeled RNase T-resistant oligonucleotideswere resolved by a two-dimensionalfingerprinting system utilizing electro-phoresis on strips of cellulose acetate (Schleicher
andSchuell)atpH3.5 inthefirstdimension(24)and homochromatography on
polyethyleneimine-cellu-lose thin layers (Polygram Cel-300 PEI,
Machery-Nagel) in the second dimension. Pretreatment of
polyethyleneiminesheetsandhomochromatography
were essentially as described by Volckaert et al. (27). The homomixture used fordevelopment was
on November 10, 2019 by guest
http://jvi.asm.org/
madeby hydrolysis of a30%RNA solution for5 min at370Cin 0.75NKOH, followedby rapid neutraliza-tion withconcentrated HCI and dilutiontoyielda solutionat a finalconcentrationof5%RNAand7M urea, pH 7.5. Homochromatographywasperformed at 60TC. Oligonucleotides were located by autora-diography, collected, and then eluted with
triethyl-ammonium-carbonate, pH 10 (27). Compositions of the eluted materialweredeterminedbysubsequent digestion with RNaseA(3).Nearest-neighbor anal-ysis ofa-32P-nucleoside triphosphate-labeled oligo-nucleotides was performed bydigestion with RNases T1, T2, and A(10).
RESULTS
PurificationofLT virions. TheVSVIfldvirus used in our laboratory, when propagated in BHK cells infected at a high multiplicity of infection, produced T particles that sedimented on sucrose velocity gradients as a single band clearly resolved from the B-particle band. RNA isolated from pure preparations of B and T particles migratedonpolyacrylamide gels with estimated positions equivalent to RNA with sedimentation values of 42S and 30S, respec-tively. Since thisT-particleRNA islonger than the RNAfoundinmost of the other T particles, we have called it LT for "long T particle." The LT and B particles could be purified by three sucrose velocity gradient centrifugations. The purity of thesepreparations was routinely mon-itored by extracting [3H]uridine-labeled RNA from either the BorLT virions and
determin-ingthepercentage of labelmigratingas42S B-or30S LT-virion RNA on polyacrylamide gels. Apolyacrylamidegel profiledemonstratingthe purity of a typical LT-particle preparation is shown in Fig. 1. The purification procedure
removedvirtually all of the B particles from the LT population, although B-particle prepara-tionsnormallycontain 10 to20%contaminating
LTparticles.
LT-virion RNA hybridizes to 28S viral
mRNA.Todetermine thesequencecomposition
of the LTRNA, LT-virion RNA was annealed
with purified viral mRNA. 32P-labeled viral mRNA purified from cells infected with a
T-particle-free stock of VSVwas fractionated by
sucrose velocity centrifugation. Fractions
con-taining labeled 28S mRNA (which codes forthe Lprotein) and 13-18SmRNA's (which code for the remaining four viral proteins) were
col-lected, and thepurity of the mRNA
prepara-tions was verified bypolyacrylamide gel elec-trophoresis (Fig. 2). When the 3H-labeled LT-virion RNA was annealed with increasing amountsof28S or 13-18SmRNA's, onlythose
samples containing 28S mRNA showed
in-creased RNase resistance (Fig. 3). With the
rn28S 18S
x_4
E
2
0 10 20 30 40 50 60
- Gel Fraction +
FIG. 1. Purity of the LT-virion preparation. Virus particles labeled with [3H]uridine were purified by onedensity and threeratezonalgradient centrifuga-tions. The RNAwasextractedfrom the LT prepara-tion, and itspurity was determined by electrophore-sison 2%acrylamidegels. Arrows mark thepositions of B- and LT-virion RNAs and 28S and 18S ribo-somalRNA markers.
N
I0
E
(._
0
N
xo
0 10
20
30 40
50
60
-
Gel
Fraction
+FIG. 2. Acrylamide gel profiles ofpurified 28S and13-18S viral mRNAs. 32P-labeled mRNA was isolated from polysomes andpurified by oligo(dT)-cellulosechromatography andsucrosegradient sedi-mentation. Thepeak fractions from thesucrose gra-dients were analyzedon 2%acrylamidegels.(A) 28S mRNAfraction; (B)13-18S mRNAfraction.
addition of thehighest level of 28S mRNA, 82% ofthe 3H-labeled LT-virion RNA wasfound in RNase-resistant hybrids, whereas 53% of the added 28S mRNA was also hybridized. 3H-la-beled LT-virion RNA did not anneal signifi-cantly with the 13-18S mRNA fractions, whereas 3H-labeled B-virion RNAbecame37%
RNase resistant afterannealing tothe 13-18S mRNA (data not shown). These results show thatmost, if notall, of theLTRNAsequences
are homologous tothe regionofthe B-particle
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.503.290.448.55.209.2] [image:3.503.271.462.291.466.2]VSV IN VITRO RNA SYNTHESIS 711
'a
.G)
.r_
z
C
0
->d
0 2 4 6 8 10
cpmxI0-3of(32P)mRNA added
FIG. 3. Sequence homology of LT-virion RNA withviral mRNA's.Differentamountsof32P-labeled mRNA were mixed with [3H]uridine-labeled LT-virionRNA in 0.4 MNaCl-10 mM Tris-hydrochlo-ride (pH7.4)inatotal volumeof80pl.Sampleswere
boiled for30sand thenannealedat68°C for2.5 h. Each sample was diluted to 2.1 ml with 0.15 M NaCl-10 mM Tris-hydrochloride (pH 7.4) and di-vided intofour 0.5-ml fractions. Nine units each of RNaseA and RNase T1 were added to half ofthe tubes,andallsampleswereincubatedat37°Cfor 30 minand thenprecipitated withtrichloroaceticacid, collected on cellulose acetate filters, and counted. Each tube contained2,500 cpm of 3H-labeled
LT-virion RNA. The percentage ofhybridization was
calculatedasin Table1,footnotec. LT-virionRNA
annealed with13-18SmRNA (O)or28SmRNA(0).
genome coding for the 28S mRNA, which
meansthat theyrepresentall orpartof the
L-proteingene.
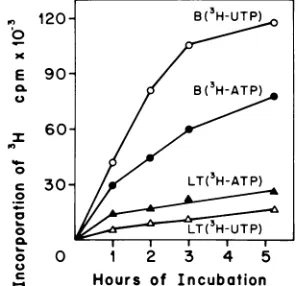
LTparticlessynthesizeRNA invitro. B and LT particles werepurified from asingle
infec-tionand testedinastandardinvitro polymer-ase assay. LT particles incorporate either
[3H]ATP or [3H]UTP into trichloroacetic
acid-precipitable material forup to 5 h (Fig. 4). A
comparison of the extent of incorporation of
[3H]UTPbysimilaramounts of B and LT
parti-cles showed that LT particlessynthesized less than 10%asmuchRNAasBparticles (datanot
shown). The LTproductdidnotcontain
signifi-cant regions of poly(A) since the
[3H]ATP-la-beled LT product was completely acid soluble
afterdigestion withamixtureof RNase Aand
RNase Ti (Table 1) and did not bind to
oligo(dT) columns in 0.5 M NaCl (data not
shown). Characterization of labeled in vitro
products on 2% polyacrylamide gels
demon-stratedthatmostof the RNAsynthesized byB
particles wasofmRNA size, whereas all of the
RNA synthesized by LT particles was much
smaller and migrated ahead of the
bromo-phenolbluedyemarker(Fig. 5A andB).
Analy-sis of the in vitro products on 20%
polyacryl-amide gels showed that the majority of the
labeled B product failedtoenter the gel (Fig. 5C).Incontrast,virtually all of the LT product
entered the gel and migratedas a sharp band
ahead ofa4S marker (Fig. 5D).
LT product hybridizes to LT-virion RNA. To demonstrate that LT-virion RNA was the
template for the synthesis of the small RNA species, LT product labeled with [ -32P]CTP or
[3H]UTPwasannealedwitheither 42S
B-parti-cleor30S LT-particlevirionRNAthat had been
partially fragmented with dilute alkali.
Sev-enty-threepercentof theLTproducthybridized
in 120- BH-UTP
E
90-06/ B(3H-ATP)
60-0o3
C 30- LT( H-ATP)
0
o T(H-UTP)
o 0 1 2 3 4 5
Hours of Incubation
FIG. 4. Kinetics ofin vitroRNAsynthesis by
puri-fiedB and LT virions. [3H]uridine-labeled virions
werepurified and assayed for polymeraseactivityas
described in the text. Duplicate samples were
as-sayed, and the background at zero time (5,500 to
7,500 cpm) was subtracted. B virions (circles) and
LTvirions(triangles) wereincubatedwith[3H]UTP
(open symbols)or f3H]ATP(closedsymbols).
TABLE 1. RNasesensitivityofin vitropolymerase products
Trichloroacetic acid- RNase
Sl~~~~~nsoluble cpmb
Sample
insoluble_____
resist--RNase +RNaseant'
(%)LT product 4,772 47 0.98
([3H]ATP)
LTproduct 1,560 46 2.95
([3H]UTP)
Bproduct ([3H]ATP) 14,870 1,941 13.05 Bproduct([3H]UTP) 10,585 49 0.46
aIn vitroproductsfromthe5-h, 15-min reactionshown
inFig. 4werecentrifugedtoremovetemplate, phenol
ex-tracted, andchromatographedonSephadexcolumnsto
re-movelow-molecular-weightcomponents.
bRNAwasannealedfor 2hat72°Cin 1001.lof0.4 M
NaCl-10mMTris-hydrochloride (pH 7.4) andthentreated withRNaseAand RNase T1 as describedinthelegendto
Fig. 3.
eCalculatedas(trichloroacetic acid-insoluble cpmafter RNase/trichloroacetic acid-insolublecpmbeforeRNase) x
100.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.503.89.206.65.220.2] [image:4.503.276.425.221.364.2]712 EMERSON, DIERKS, AND PARSONS
cq
x
E
CI
I
10 20 30 40 50 60 70
- Gel Fraction +
0 10 20 30 40 50 60 70
- Gel Fraction +
FIG. 5. Acrylamide gel profiles of RNAs synthesized in vitro by purified B and LT virions. [3H]UTP-labeled productsfroma3-h in vitropolymerase reaction were separated from template by centrifugation,
phenol extracted, and separated from unincorporatednucleotides and small molecules by Sephadex chroma-tographybefore analysisonacrylamide gels.Arrows denote the position of marker RNAs. (A) 2% gel, B-virion
product; (B) 2% gel, LT-virion product; (C) 20% gel, B-virion product; (D) 20% gel, LT-virion product.
to LT-virion RNA atapproximately a3:1
(vir-ion:product) molar ratio of input RNAs (Fig. 6). Annealing of labeled LTproductwith B-parti-cle RNA yielded little, ifany, hybridization,
although bothB- and LT-virion RNAs hadthe
same specific activity and similar amounts of
these RNAswere addedtothe annealing
mix-tures. Lack ofannealingtoB-virion RNAmay
reflect technical difficultiesinthehybridization procedure or could indicate that generation of
the LT particle resulted in the acquisition of
new or modified sequences. We are currently
tryingtodifferentiate between thesetwo possi-bilities. Although we have not been able to
demonstratethepresenceofsequences
comple-mentary to LT product in B-particle virion RNA, it is clear from ourdata that LT-virion
RNA is the template for the synthesis ofLT
product.
Oligonucleotide fingerprint of LT product. Todirectly estimate the complexity of the small LTRNA synthesized by LT particles, LT
prod-uct was synthesized in vitro with labeled
[a-32P]GTP and subjected toRNase T1 digestion. The resulting T1 oligonucleotides, containing
atleastone labeledP residueper
oligonucleo-tide,were separated byelectrophoresison
cellu-lose acetatefollowed byhomochromatography.
Figure 7 showsanoligonucleotide fingerprint
of LT product synthesized using [a-32P]GTP anddigested with RNaseT1. Six major oligonu-cleotides canbeidentified, suggesting that the
LTproduct isarelatively homogeneousspecies
._4,
N
._
0
I--J
0
15
CPm X10 of(3H) RNA added FIG. 6. Hybridization ofLT in vitroproduct to
LT-virion RNA. [32P]CTP-labeledLTproduct was
mixed with increasing amounts of [3H]uridine-la-beledpurifiedB-orLT-virion RNA inavolumeof55
gl of0.3 M NaCI-0.3 M sodium citrate,pH 7.0.
Sampleswereboiledfor30sand incubatedat70'C for17.5 h. Eachsamplewas dilutedto2.1 mlwith 0.3 MNaCl-10 mM Tris-hydrochloride (pH 7.4), and 0.5-mlfractionswereanalyzedasinFig.3. Each 0.5-ml fraction contained 1,600 cpm of32P-labeled
product. The 3H-labeled virion RNAs contained
ap-proximately 13,500 cpm/lg.LTproduct hybridized
toB-virionRNA(0)orLT-virion RNA (0).
with low complexity. Oligonucleotide finger-print analysis of[a-32P]GTP-labeled LT product digested first with bacterial alkaline
phospha-taseandsubsequently with RNase T1 revealed
re
0
x
E 0.
Z-,
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.503.114.405.63.262.2] [image:5.503.288.433.324.472.2]A
4
03
o2
1
e
5
6
4
3
6'
_b
_
2
c
02
5
FIG. 7. Two-dimensionaloligonucleotide fingerprintsof[Ca32P]GTP-labeledRNAsynthesizedin vitro by LTparticles. 32P-labeledproductfrom a singlepreparation was divided into two equal portions. One portion
(A) was digested with RNase Ti andfingerprinted as described in Materials and Methods. The second portion(B)wasfirsttreated with bacterial alkalinephosphataseand thenRNaseTiasdescribedinMaterials andMethods. The direction of electrophoresis in the first dimension on strips of cellulose acetate in 5 M urea, pH3.5, is from left to right.Homochromatography in thesecond dimension onpolyethyleneimine-cellulose
thinlayersatpH 7.5 isfrom bottomto top.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.503.106.398.76.585.2]theconcomitantloss ofoligonucleotideT-6and theappearanceofa newoligonucleotide,
desig-nated T-6' (Fig. 7B). The sequence of
oligonu-cleotide T-6 was determined to be (p)ppAp-CpGp, and that of T-6' was determined to be ApCpGp. These data suggestthat oligonucleo-tide T-6 represents the 5' terminus of the LT product.
Thenucleotide composition,length, and rela-tiveamountsof each of themajorTi oligonucle-otides were determined by using LT product labeled with either [3H]ATP, Ia-32P]CTP, a-32P]UTP, or [a-32P]GTP (Table 2). The relative
yield of the largerTioligonucleotides (T-1, T-5)
was found to vary in different experiments. This observation may reflect the nonspecific degradation of these oligonucleotides duringin
vitrosynthesis and/or their lower efficiencyor
transfer from cellulose acetate during
finger-printanalysis. From thesequence composition
of the Tioligonucleotides (basedonanalysis of RNase Adigestionproducts and nearest-neigh-bor data) and their apparent length, we
con-clude that the LT product synthesized by the LTparticlesrepresents auniquenucleotide se-quence of about45nucleotides.
TABLE 2. Sequence analysis ofRNAseTl-resistant oligonucleotides
Relative molar
Oligo- yields
nucleo-
Com-tide plexityb Un- Phos- Compositiond
no.a treated phatase
treated
T-1 13 0.64 0.51 [(A4C,) (AC) (AC2)] AG(A)
T-2 3 1.00 1.00 AG(A) T-3 2 0.82 0.76 AG(G)
T-4 1 0.72 0.71 G(G,U)e
T-5 20-21 0.38 0.20 [(A,-, U) (AC2) (AU) (AC)]A2G(A)
T-6 3 0.41 ppACG(AY
T-6' 3 0.79 ACG(A)
aSee Fig.7.
"Complexity was calculated from the composition and
does notinclude theresidue thatnearest-neighbor analysis labels the guanosine residue.
'A2G= 1.0,using[a-32P]GTPas source oflabel.
dCompositions were deduced from analysis of products obtainedby digestionwithRNase A and with RNases T1, T2, and A (see Materials and Methods). The last nucleotide
inparentheses was deduced to be the nearest neighbor to the3-terminalguanosine residue. A detailed discussion of
thesequenceanalysis will be presented elsewhere (manu-scriptinpreparation).
INearest neighbor labeled with both [a-32P]GMP and
[a-32P]UMP.
'Oligonucleotide 6, labeled with [3H]ATP and [a-32P]CTP, released a product containing both 32p and 3H labels and migrated on 3MM paper (pH 3.5) as ppAp after digestion with RNases T1, T2, and A.
DISCUSSION
Because T-particle preparations incorporate
verysmallamountsof nucleosidetriphosphates into RNA compared with B-particle
prepara-tionsandsynthesizeamuch smaller RNA spe-cies than B particles do, there has been some
disagreement as to whether T-particle
nucleo-capsids are actually functioning as templates
for RNAsynthesisinvitro(8, 13, 20, 23).Inthis paper,wedemonstrate that an LT particle from theL-gene region ofVSVI1d does synthesize a small RNAin vitro. This RNA product iscoded by the LT-virion RNA and is homogeneous in size and nucleotidecomposition.
The LT product issynthesized in its entirety de novo and does not represent elongation of preexistingmolecules. Since the 5'-phosphoryl-ated oligonucleotide ([p]ppApCpGp) canbe la-beled with either [3H]ATP, [ a-32P]CTP, or
[a-32P]GTP,initiationof LTproductmustoccur in vitro. The LTproductis homogeneous when
analyzed by polyacrylamide gel
electrophore-sis, suggestingthat termination isoccurringat aspecific site or region. Termination resultsin
release of the product from the template, for after high-speed centrifugation the LT product
is recovered in the supernatant fraction,
whereas thetemplate is in the pellet fraction. Therefore, whereas both LT and B particles initiateRNAsynthesisinvitro,Bparticles
syn-thesize mainly large products complementary
to -50% of the genome and LT-particle RNA synthesis is restrictedto a region only45 nu-cleotides inlength.
Thefunction orbiological significance of the
LTproduct is presently undefined, but a
com-parisonwith the "leader" RNAreported by Co-lonno and Baneijee suggests that these two RNAs may be analogous (11, 12). "Leader"
RNA isasmall RNAspecies (less than4S)that
is synthesized by B virions in vitro. Colonno
andBanerjeehavereportedthat B virions
con-tainonlyone copyofthesequence
complemen-tary to"leader," andthis sequenceconstitutes the3'endof thegenome. "Leader" RNA and LT
product both have the same 5' sequence
(p)ppApCpGp and contain an unusually high percentageofadenine residues (approximately 50%)that do not constitute poly(A). However,
preliminary sequence comparisons indicate
that the primary sequences of "leader" and
LT product are not identical (A. K. Banerjee, personal communication). Thismayreflect
dif-ferencesinthevirusstocks usedorthe
synthe-sisofLTproduct fromadifferentregion of the
viralgenome. Atthistime wehave beenunable
topurify enough "leader" fromourpreparations
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.503.61.256.373.521.2]of B-virion transcription products to allow a direct comparison.
Are the "leader" and LT-product RNAs
pro-duced by the same mechanism? Colonno and
Banerjee
have postulated that "leader" RNArepresents a by-product of mRNA processing (11). That is, they suggest that during tran-scription, the RNA polymerase initiates at the ultimatebaseof the3' end ofthe Bgenome and synthesizes "leader" as the 5' terminus of a longer RNA, which is subsequently cleaved by
anuclease to generate"leader" and the individ-ual mRNA's. During replication, processing would not occur and genome-length RNA would be synthesized. However, to date no one has reported the detection of either such a nuclease
or a precursor molecule. On the other hand, sincethe 45-nucleotide-long LT product appears homogeneous and no large quantities of other products have been detected, we mustconsider thepossibility thatitsdiscretesizereflects
ter-mination of synthesis rather than cleavage fromalargermolecule.
It may be that LT product synthesis is a
result of replication starts rather than tran-scription. Invivo replication of VSV nucleic acid requires concomitant proteinsynthesis, for pro-duction ofgenome-length RNA quickly stops
when cycloheximide is added to infected cells (18, 22). Since acharacteristic feature of VSV T particles is theirabilityto replicateefficiently in cells when co-infecting B particles are pres-ent, we suggest that the in vitro synthesis of the LT product, as well as the "leader" RNA, represents replication that is aborted because
one or more factors normallyprovided invivo
by newlysynthesizedproteins areabsent. Fur-ther experimentsshould resolve these alterna-tives, and the information obtained should
greatly increase our understanding of both
VSVreplication andT-particle generation.
ACKNOWLEDGMENTS
Thisresearch wassupported byPublicHealth Service grants AI-11722from the National Institute ofAllergyand Infectious Diseases (S.U.E.) and CA 16553 from the Na-tionalCancer Institute(J.T.P.)andbygrant VC-186 from the American CancerSociety(J.T.P.).S.U.E. isthe recipi-entofPublic Health ServiceResearchCareerDevelopment Award AI-00013 from theNationalInstitute ofAllergy and Infectious Diseases.
LITERATURE CITED
1. Abraham, G., andA. K. Banerjee. 1976. Sequential transcription of the genes of vesicular stomatitis
vi-rus. Proc. Natl.Acad. Sci. U.S.A. 73:1504-1508.
2. Abraham,G.,D.P.Rhodes,and A. K.Banerjee.1975.
The 5'-terminal structure of themethylated mRNA synthesized in vitro by vesicular stomatitis virus. Cell5:51-58.
3. Adams, J. M., P.G. N. Jeppesen, F. Sanger, and B. G. Barrell.1969. Nucleotide sequence from the coat pro-tein cistron of bacteriophage R17. Nature (London) 223:1009-1014.
4. Ball,L. A., and C. N. White. 1976. Order of transcrip-tion ofgenes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U.S.A.73:442-446.
5. Baltimore, D., A. S.Huang, and M. Stampfer. 1970. Ribonucleicacid synthesis ofvesicular stomatitis vi-rus. II. An RNApolymerase in the virion. Proc. Natl. Acad. Sci. U.S.A. 66:572-576.
6. Banerjee, A. K., G.Abraham, and R. J. Colonno. 1977. Vesicular stomatitisvirus: mode of transcription. J. Gen. Virol. 34:1-8.
7. Banerjee, A. K., and D. P. Rhodes. 1973. In vitro syn-thesis of RNA that contains polyadenylate by virion associated RNApolymerase ofvesicular stomatitis virus.Proc. Natl. Acad. Sci. U.S.A. 70:3566-3570. 8. Bishop, D. H. L., andP. Roy. 1971. Kinetics of RNA
synthesisby vesicular stomatitis virusparticles. J. Mol. Biol. 57:513-527.
9. Both, G.W., S. A. Moyer, and A. K. Banerjee. 1975. Translation and identification of the mRNA species synthesized in vitro by the virion-associated RNA polymerase ofvesicularstomatitisvirus.Proc. Natl. Acad. Sci. U.S.A. 72:274-278.
10. Cashion, L. M., R. H. Joho, M. A. Planitz, M. A. Billeter, and C. Weissmann. 1976. Initiation sites of Roussarcoma virusRNA-directed DNAsynthesis in vitro. Nature(London) 262:186-190.
11. Colonno, R. J., and A. K. Banerjee. 1976. A unique RNAspeciesinvolved in initiation of vesicular stoma-titis virusRNA transcription in vitro. Cell 8:197-204. 12. Colonno, R. J., and A. K.Banerjee.1977. Mappingand initiation studies on the leader RNA of vesicular stomatitisvirus.Virology 77:260-268.
13. Emerson,S. U., and R. R. Wagner. 1972. Dissociation and reconstitution of the transcriptase andtemplate activitiesof vesicular stomatitis B and T virions. J. Virol. 10:297-309.
14. Emerson, S. U., and R. R. Wagner. 1973. Lprotein requirement for in vitro RNAsynthesisby vesicular stomatitis virus. J. Virol. 12:1325-1335.
15. Emerson,S. U., and Y-H. Yu. 1975. Both NS and L proteins arerequired for in vitro RNAsynthesisby vesicular stomatitisvirus. J. Virol. 15:1348-1356. 16. Galet, H., D. Hallet, and L. Prevec. 1976. Analysis of in
vitrotranscriptionproducts of intracellularvesicular stomatitisvirusRNApolymerase. J. Virol. 19:467-474.
17. Huang, A. S. 1973. Defectiveinterferingviruses. Annu. Rev.Microbiol. 27:101-117.
18. Huang, A. S., and E. K. Manders. 1972. Ribonucleic acidsynthesis of vesicularstomatitis virus. IV. Tran-scriptionbystandard virus in the presence of defec-tiveinterfering particles.J.Virol. 9:909-916. 19. Min Jou, W., and W. Fiers. 1969. Studieson
bacterio-phage MS2. J. Mol. Biol. 40:187-201.
20. Mori,H.,and A. F. Howatson. 1973. In vitro transcrip-taseactivity of vesicular stomatitis virus B and T particles: analysis ofproduct. Intervirology 1:168-175.
21. Moyer, S. A., and A. K. Banerjee. 1975. Messenger RNAspeciessynthesizedin vitrobythe virion-associ-ated RNApolymeraseof vesicular stomatitis yirus. Cell 4:37-43.
22. Perlman, S.M.,andA.S.Huang.1973.RNAsynthesis of vesicular stomatitisvirus.V.Interactions between transcription andreplication.J.Virol. 12:1395-1400.
23. Reichmann,M.E.,L. P.Villarreal,D.Kohne,J. Les-naw,and J. J. Holland. 1974. RNApolymerase activ-ity andpoly(A) synthesizing activityindefective T
on November 10, 2019 by guest
http://jvi.asm.org/
particles of vesicular stomatitis virus. Virology 58:240-249.
24. Sanger, F., G. G. Brownlee, and B. G. Barrell. 1965. A two-dimensionalfractionation procedure for radioac-tivenucleotides. J. Mol. Biol. 13:373-398.
25. Schnitzlein, W. M., and M. E. Reichmann.1976. The size and the cistronic origin of defective vesicular stomatitis virusparticle RNAs in relationto homo-typic and heterotypic interference. J. Mol. Biol. 101:307-325.
26. Villarreal, L. P., and J. J.Holland. 1973. Synthesis of poly(A) in vitro by purified virions of vesicular
stoma-titisvirus.Nature (London) New Biol. 246:17-19. 27. Volckaert, G., W.Min Jou, and W. Fiers. 1976.
Analy-sis of3P labelledbacteriophage MS2 RNA bya
mini-fingerprinting procedure. Anal. Biochem. 72:433446. 28. Wagner,R.R. 1975. Reproduction of rhabdoviruses,p.
1-93.In H.Fraenkel-Conrat and R. R. Wagner(ed.), Comprehensivevirology, vol. 4. Plenum Press, New York.
on November 10, 2019 by guest
http://jvi.asm.org/


![FIG. 7.portionpHLTandthin(A) Two-dimensional oligonucleotide fingerprints of[Ca32P]GTP-labeled RNA synthesized in vitro by particles](https://thumb-us.123doks.com/thumbv2/123dok_us/1541239.106673/6.503.106.398.76.585/portionphltandthin-dimensional-oligonucleotide-fingerprints-labeled-synthesized-vitro-particles.webp)

