0022-538X/92/063803-08$02.00/0
Copyright ©D 1992,American Society for Microbiology
A
Cellular
Function
Is
Required
for
Pseudorabies Virus Envelope
Glycoprotein
Processing and Virus
Egress
M. E. WHEALY,' A. K. ROBBINS,' F. TUFARO,' ANDL. W. ENQUISTl* DuPontMerckPharmaceutical Company, ViralDiseases Research, Experimental Station,
Wilmington, Delaware 19880-0328,' and Department of Microbiology, University of
British Columbia, Vancouver, Canada V6T1232 Received 15January 1992/Accepted 23 March 1992
ThemouseL-cell mutantgro29 is defective foregressof herpes simplex virustype 1 (HSV-1) virionsand is
significantlyreduced inHSV-1glycoproteinexport(B. W. Banfield and F. Tufaro, J. Virol. 64:5716-5729, 1990). In this report,we demonstrate that pseudorabies virus (PRV), a distantly related alphaherpesvirus, shows a distinctivesetof defects after infectionofgro29cells.Specifically,weidentifydefects in therateandextentof viral
glycoproteinexport, infectious particleformation, plaque formation, andvirusegress. Theinitialrateofviral
glycoprotein synthesiswasunaffected ingro29 cells,but theextentofexportfromtheendoplasmicreticulumto
theGolgi apparatuswas impaired and export through theGolgi apparatusbecameessentially blocked late in
infection.Moreover, by using asecretedvariant ofa viralmembraneprotein,wefoundthatexportfrom the
Golgiapparatus outofthecellwasalso defectiveingro29cells. PRVdoesnotform plaquesongro29 monolayers.
A lowlevel ofinfectiousvirus isformedandreleased early afterinfection, but furthervirus egressis blocked.
Taken together, these observationssuggestthat the gro29 phenotype involves eithermultipleproteinsor asingle
proteinused atmultiplesteps in viralglycoproteinexport andvirus egressfrom cells. Moreover,this host cell
protein is required byboth HSV and PRV forefficientpropagation in infected cells.
It is well established that the glycoproteins encoded by herpesviruses use the host cell secretory mechanism for
synthesis, processing, and sorting of the glycoproteins to
appropriate organelles (3, 6, 8, 9, 12, 13, 18, 22). Even
though we know these viral glycoproteins play important
rolesinthe viral life cycle,wedonotunderstand the precise processingandorderedassemblyeventsrequiredfor
forma-tion ofinfectiousvirus. Currently, therole ofthe hostcellas
wellasthevirusintheassembly andtrafficking of virions is
thesubject ofintensiveinvestigation.
Tounderstandthe role of thesecretorypathwayinthe life cycleof thisfamilyofviruses, Tufaroandcolleagues isolated
amurineL-cellmutant,gro29, whichisunableto supportthe propagationofherpes simplexvirustype 1 (HSV-1) (1, 21). Despite normal infection and viralgene expression, HSV-1
virions did not traverse the secretorypathway and instead
weretransportedtocytoplasmic vacuoles, wherethey
accu-mulatedasnoninfectiousparticles.Theinabilitytotransport
HSV-1virionsto the cell surface ofgro29cellswas
accom-panied byareduction in thetransportandprocessingof viral
glycoproteins, although the blockto glycoprotein transport was not as strong as the block to virus egress. It was
proposed that gro29 cells were able to transport and carry
outsomeof theprocessingeventsfor viralglycoproteinsbut
wereunabletoallow thetransportoflargercomplexes,such
as virions.
Pseudorabies virus(PRV)isdistantlyrelatedtoHSV-1 but sharesacommonlifecycleas analphaherpesvirus (3, 25). It
was of some interest to determine first whether the gro29 phenotypewas specificonlyto HSV-1 and second whether PRV was blocked in gro29 cells, to take advantage of the
uniquefeatures of PRVglycoproteinsto testcertain
predic-tions of the modelproposed byTufaro andcolleagues (1,21)
* Correspondingauthor.
andto determine moreprecisely the nature of the defect in
thesemutant cells.
In this report,we show that PRV growth, like HSV-1, is
defective in gro29 cells. The specific modifications and
processing of certain PRV membrane proteins provided insight into which secretory organelle had been traversed duringexportthrough thesecretorypathway.We foundthat in gro29 cells, egress of infectious particles was partially
blockedearlyininfection andwascompletelydefective late
after infection.Moreover,infectedgro29cells hadadefectin exportof viralglycoproteinsfrom theendoplasmicreticulum
(ER) to the Golgi apparatus and did not secrete a
nonan-chored viral membrane protein. Taken together, these
re-sults suggest that gro29 cells have a defect in a cellular
functionrequiredfor ERtoGolgiapparatus exportaswellas
Golgi apparatustocell surface export.Thisdefectbecomes
rate limiting for egress of virus particles as infection
pro-ceeds. It is likelythatherpesvirus particle egressfromcells andglycoproteinmovement throughthesecretory pathway
share acommonmechanism.
MATERUILS AND METHODS
Cells and viruses. ThepropagationofL andgro29cellshas been describedpreviously (21). PK15 cells and the Becker strain of PRV (PRV Be) were prepared as previously
de-scribed (15). PRV1007 encodes a mutant gIll glycoprotein
geneinwhichaTAGstopcodonreplacesatyrosineatcodon 436. Theresultingtruncatedglll protein lacks the
carboxy-terminal transmembrane and anchoring domain. Further characterizationof thisvirushasbeen describedbySolomon
et al. (17).
Glycoprotein molecular mass nomenclature. Viral
glyco-proteinsglll, gll, andgp5Ohaveapparent molecularmasses
after infection of L cells different from those after infection
of PK15 cells. These differences are most likely due to differencesinthedegreeofglycosylation. In PK15cells, the
3803
on November 9, 2019 by guest
http://jvi.asm.org/
3804 WHEALY ET AL.
PRVglycoproteins have the following apparent molecular masses: gIII precursor, 74 kDa; gIll mature, 92 kDa; gll precursor, 100 kDa; gll mature, 110 kDa; gll cleavage
products, 68 and 55 kDa; gpSO precursor, 46kDa; and gpSO mature, 55 kDa. In L cells, the PRV glycoproteins have apparent molecular masses as follows: glll precursor, 72 kDa; gIll mature, 82kDa; gll precursor, 92kDa;gII mature, 98kDa;gIlcleavageproducts, 66and 60kDa; gpSO precur-sor, 44kDa; andgpSO mature, 56 kDa. The PRV
glycopro-teins in gro29 cells have thesameapparentmolecularmasses asthose inLcells, except the gllcleavage products,which
are64and 54 kDa.
Fixation and stainingof PRVplaques. PRVplaqueswere
prepared under Methocel byusingPK15, L-cell, and gro29 monolayers. At 3 days postinfection, the Methocel was
removed, and the cells were fixed with 4% formalin for 30 min at room temperature. After 30 min, the formalin was
replaced with 0.5% crystal violet in 22% ethanol for 10 min, and the cellswerewashed withwater.
Growth curves. Single-step growth curves of PRV were
completed as described by Whealy et al. (23) with the following modifications: at 1 h postinfection, the inoculum
wasremoved from thecells, and the cellsweretreatedfor 1 minwith 40mM citric acid-10mM KCl-135 mMNaCl and replaced with Dulbecco modified Eagle medium (DMEM) containing 2% fetal calfserumpluspenicillinand
streptomy-cin.
Antibody reagents. The antisera used in these studies included a mouse monoclonal antibody, 6D8MB4, reactive withPRVgpSO(a kind gift from C.Whetstone, Ames,Iowa), agoatpolyvalent antiserum(labeled282)thatrecognizesthe native and denatured forms of the gIIIglycoprotein(14, 15),
andagoatpolyvalentantiserum(labeled 284)thatrecognizes
the native and denatured forms of thegII glycoprotein (14,
24).
Pulse-chaseexperiments to measurekinetics ofviral glyco-protein export. The kinetic analysis was done as described previously(16), except that infected cellswerepulse-labeled for 5 min rather than for2min. Techniquesfor immunopre-cipitation and autoradiography of viral glycoproteins have been described previously (4, 15).
Measurement ofgll oligomer formation by sedimentation through sucrosegradients. ThetechniquesformeasuringglI
oligomer formation by sedimentation throughsucrose
gradi-entswere asdescribedbyWhealyetal. (24), with modifica-tions as noted in the legend to Fig. 6. Briefly, L cells and gro29 cellswereinfected with10PFUofPRV Bepercell.At
5.5h postinfection, the cellswerestarved forcysteine. At 6 hpostinfection, cells were labeled for 5 min with
[35S]CYS-teine and then chased for 25 and 90 min with coldcysteine. Next, the monolayerswere solubilized in Triton X-100 and fractionated on sucrose gradients. Gradient fractions were immunoprecipitated with anti-gll serum (284 serum) andanalyzed on sodium dodecyl sulfate (SDS)-polyacrylamide gels.
RESULTS
Plaque-forming efficiency of PRV Be on PK15, L, and gro29
cells. Banfield and Tufaro(1) demonstrated that HSV-1 was unable to form plaques on gro29 cells. We measured the plaque-forming efficiency of PRV on swine kidney fibro-blasts (PK15 cells), the parent murine L-cell line, and the gro29mutant. The titer of PRV Be stock prepared on PK15 cellswasdetermined for PK15, L, and gro29 cells. Plaques
werefixed, stained, and counted. A typical dilution series is
PK1 5 CELLS
!
I
S.1
LCELLS
IL. .. .. ..ix X, > v
,. ,1
4/ IX}
;ti.4-:_ .
[image:2.612.336.552.79.416.2]gro29CELLS |
FIG. 1. Plaque-forming efficiencyof PRVonPK15, L,andgro29
cells. Serial 10-fold dilutions of PRV(10-2dilution[topleft ineach dish]through 10-7dilution[bottom rightineachdish])wereplated
on PK15, L and gro29monolayers and allowedtoadsorb for 1 h. Afteradsorption, the inoculumwas replacedwith Methocel. At 3 dayspostinfection,the Methocelwas removed,and the cellswere
fixed with 4% formalin and stained with 0.5% crystal violet, as
described in Materials and Methods.
shown inFig. 1. Theplaque-formingefficiencies of this stock
onthevarious lineswere 1.0 (PK15), 1.3 x 10-1 (L cells),
and>1.7 x 10-6(gro29cells).PRVformsplaquesonLcells about10-fold lessefficientlythan onPK15 cells. Itisclear,
however, that PRV, like HSV, does not form plaques on
gro29 cells.
Production of infectious virus by gro29 cells. We deter-minedthe extentofviral propagation in L cells and gro29
cellsby comparingsingle-step growth curves of intracellular
andreleasedinfectious virions (Fig. 2).Animportant part of theexperimental protocolwas alow-pH citrate washat 1 h
postinfection to remove any virus that had not penetrated the cells. Thus, any infectious virus measured after that
pointmust be newly formed virus.
When Lcellswereinfected with PRV, intracellular infec-tiousviruswasdetected between3 and5hpostinfection and infectious virus begantobe releasedextracellularly between 5 and 7 hpostinfection. Incontrast, when gro29 cells were infectedwith PRV, infectious virus particles were detected
intracellularly between 5 and 7 h postinfection and were releasedtothe media between 7and 9hpostinfection.From
the results in Fig. 2, we conclude that in gro29 cells, the J. VIROL.
I
I
...MEMO-.
molok.
i,
I ...
on November 9, 2019 by guest
http://jvi.asm.org/
L
Cells
I.. 106
010 4
CL 10
102'
.1
10 15 20 25 31
Hours Post-infection
gro29
Cells
107.
106.
105.
E
0I IL
0D 103
102
10
0 5 10 15 20 25 30
Hours Post-infection
FIG. 2. Single-step growth curves of PRV Beon L and gro29
cells. The cellswereinfectedatamultiplicityofinfection(MOI)of
5 andwereincubated at37°C. At1 hpostinfection, the cellswere treatedwithlow-pH citrate,asdescribedinMaterialsand Methods.
At 1,3, 5, 7, 9, 11, and 25 hpostinfection, plateswereharvested, andthevirus titers of the cell and mediumfractionsweredetermined separatelyandplotted.
LCELLS gro29CELLS
0 45 90 120 240 0 45 90 120 240
A 92
-68-=W 0
opmed
46
-
92-
6846
-C
68
-46
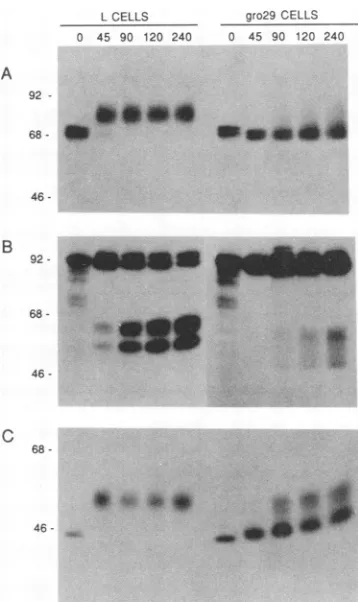
-FIG. 3. Kinetic analysis of PRV glycoprotein synthesis and modification in Land gro29 cells at4 h postinfection. Cellswere
infectedwith PRVatanMOI of 10. The cellswerepulse-labeledat
4 h postinfection and chased for the time (in minutes) indicated above each lane.The cellswerelysed andimmunoprecipitated with
glll-specific antiserum (A), gll-specific antiserum (B), or a
gp5O-specific monoclonal antibody (C). Immunoprecipitates were
re-solvedon a10%polyacrylamide gel and visualized by fluorography. Themolecularmassstandards (inkilodaltons)are indicatedat the
left ofeach panel.
appearance of infectious PRV particles in the media is delayed and thequantityisreduceddramaticallyin
compar-isontovirusproduced bytheparental Lcells. Wenotethat the rate of intracellular accumulation and the release of infectious virus from cells are indistinguishable for L and
PK15 cells(data notshown).
Kinetics of viral glycoprotein export at 4 h postinfection.
Since infectious particle formation and release of extracel-lular virus were aberrant ingro29 cells, we next examined thesynthesisandexportof critical viralglycoproteins. PRV offers a unique set ofglycoproteins for this analysis since their modification andprocessingareindicative of the secre-toryorganelletheyhave traversed during export(3, 11, 16, 17, 22, 24).
We measured the rate and extentof synthesis and
proc-essing byapulse-chase protocolusedpreviously(16). Since
a small burst of infectious particles appeared to be made early,but further infectiousvirusproductionwasblocked in gro29 cells, experimentsweredoneat4and 6 hpostinfection
to examine an early and a later time point during the
infection. The results of the 4-hanalysisareshown inFig.3.
The pulse-chase profile forglycoprotein gIll in Lcells is
shown onthe left side ofFig. 3A. Thepredominant protein
made in the5-minpulse (lane0)was theprecursor
synthe-sized in the rough ER. This form of the glycoprotein was
already fully glycosylated with high-mannose
oligosaccha-rides and was sensitive to endoglycosidase H (data not
shown). By 45 min of chase (lane 45), the precursor was
converted to the diffuse, more slowly migrating species
characteristic of mature gIII. This form is known to be resistant to endoglycosidase H, indicating that the
high-mannose oligosaccharides have been converted to complex
carbohydrates (10). The 5-min pulse sample from infected
gro29 cells was essentially identical to that seen in L cells
(lane 0). This similarity indicated that the primary rate of
synthesisofgIIIwasunaffected ingro29 cells and thatthere
was nodefect in the earlyeventsof PRV infectionorin the
addition of high-mannose oligosaccharide moieties to the
newlysynthesizedpolypeptides. However,while the parent
L cells couldsupportefficientexportof thegIll precursorto
the Golgi cisternae forfurtherprocessing, gro29 cells were
markedly defective. There was little, if any, chase of the precursoruntil 90minpostlabeling,andmostof themassof precursorneverchased tothemature form, suggestingthat
these species had not been exposed to the processing
en-zymesresident in the peripheral Golgicisternae.
a-- cells
-*- media
4wo
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.66.285.76.416.2] [image:3.612.344.523.79.380.2]3806 WHEALY ET AL.
Further evidence that gro29 was defective in PRV glyco-protein export was obtained from a kinetic analysis of glycoprotein gIl synthesis and processing (Fig. 3B). ThegII glycoprotein is cleaved by a cellular protease located in the trans-Golgi apparatus or trans-Golgi network, and this
proc-essing occurs only after the protein has formed oligomers
and has been exported out of the ER (22, 24). Asobserved forgIll, therewas nodifferencein the 5-min pulse profilefor
gIl in either Lorgro29 cells: the 92-kDa gIl precursor was
synthesized in equal abundance in both. The smaller
gIl-related polypeptides are incomplete translation products,
and their intensities and general profiles are also
indistin-guishable in thetwocell lines. By 45 min of chase (lane 45)
in the Lcells, the mature uncleaved 98-kDaspecies, as well as the 66- and 60-kDa protease cleavage products, was apparent. Significantly, the time and extent ofgIl protease
cleavage were delayed in gro29 cells. Processing was only detected after 90 min of chase, and the apparent molecular massesoftheproductswere 64and55kDa. Whilethere was astrongblockin gIl processing, somemoleculeslabeled at 4 h postinfection can traverse the secretory pathway to the
trans-Golgi ortrans-Golgi network.
One modification that has been difficult to assess in
previous studies using HSV is theextent of 0-linked
glyco-sylation in gro29 cells during infection. Previous
observa-tions of HSV-1 gD suggested, but did not establish, that 0-linked glycosylation was impaired during infection (1).
The PRV gpSO glycoprotein can be used to determine whetherthe gro29 mutantis defective in 0-linked modifica-tions, since this viral glycoprotein has no N-linked sugars
butis modified by 0-linked glycosylation (11, 22). 0-linked glycosylation ispredictedtooccurinitially in thetransitional compartment ofthe ER, and the final complex sugar addi-tionsarecompletedinmedial andtrans-Golgi compartments
(19). Addition of0-linked sugars and the subsequent addi-tion of terminal sialic residues are accompanied by charac-teristic decreasesin electrophoretic mobility. Previously,we have shown that the initial addition of 0-linked sugars resulted in a shift in the apparent molecular mass of gpSO intermediate between the precursor and mature forms (22). Since thisformis detectable only in thepresenceofbrefeldin
A(22),ourinterpretation is that the protein had reached the
transitionalcompartmentof theERbuthad notproceededto the Golgi apparatus.
The kinetics ofgpSO synthesis andprocessing is shown in
Fig. 3C. There was no difference in precursor synthesis in either L or gro29 cells, as indicated by the identical 5-min
pulselanes(lanes 0).Lcells supported thecomplete
conver-sion of thegp5Oprecursor tofully modifiedgp5Oby 45min of chase. However, gro29 cells were delayed considerably in the efficiency and extent of gpSO processing. Since with
gro29 cells we were unable to detect the form of gp5O intermediate between the precursor and mature form
char-acteristic of protein accumulating in the transitional com-partment, we conclude thatmost of thegp5Osynthesized at 4hpostinfectiondid not reach the transitional compartment. Moreover, because a small quantity of gp5O was able to evadethisblock and mature properly, it appeared that once
gp50budded from the ER, it was capable of traversing the
transitional compartment efficiently enroute to the Golgi
organelle,whereit was subsequently modified. It is apparent that a fraction of the newly synthesized gpSO larger than those ofgllandglllwasprocessed, suggesting that the PRV
glycoproteins maybe differentially affectedin gro29 cells. These pulse-chase results indicated that gro29 cells have
anearlyblock inthe secretorypathway. Because precursor
CELLS 0 15 30 45 60' 90 120
MEDIA 15 30 45 60 90 120
LCELLS
92
gro29CELLS am o A:,.
.
92
[image:4.612.332.553.77.266.2]-68
FIG. 4. Kineticanalysis of a secretedformofgIll produced by
PRV1007. L andgro29 cellswere infectedwithPRV1007 at anMOI
of10, pulse-labeledat 6 hpostinfection,and chased forthe time(in
minutes) indicated above each lane. glll-specific antiserum was
used to immunoprecipitate gIll from both the cellandthe medium
fractions. Immunoprecipitates were resolved on a 10%
polyacryl-amide gel and visualized by fluorography. The molecular mass
standards(in kilodaltons) areindicated tothe left ofeachpanel.
glycoproteinsaccumulated and did notreceive typical
post-translational modifications, it was likely that export out of the ER was defective. However, this block was not complete
since some precursor glycoproteins escaped the ER block
and were processed in the Golgi apparatus. As measured by protease processing ofgll, some fractionof the glycoprotein
traveled as far as the trans-Golgi ortrans-Golgi network. It was, therefore, of interest to determine whether gro29 cells were defective in export from the Golgi apparatus to the cell surface. We determined this defect by analyzingexport of a
secreted form of the normally membrane-anchored glll
glycoprotein.
The glll gene encoded by PRV1007 has a stop codon in
place of tyrosine at codon 436 of the mature protein and
consequently expresses a truncated protein lacking a trans-membrane and anchoring domain. This mutantgIll protein contains the same modifications as the wild-type glllbut is
secreted into the medium and is notfound in virus envelopes (17). Since we can measure the rateand extent of
glycopro-teinprocessing as well as the rate and extent of secretion of the mature protein into the medium, we can deduce the
kinetics of export from the Golgi apparatus to the cell surface and out of the cell.
The results of thekinetic analysis for L and gro29 cells are shown in Fig. 4. In Lcells, the gIII precursor was synthe-sized in the5-min pulse period, and by 45 min of chase, the mature form was clearly detectable in the media. Theresults
with gro29cells were noteworthy. While theglllprecursor was madenormally and a small proportion received mature modifications, no glll-specific protein could be detected in the media, even after 120min. Either the secreted form of
glll was blocked in export from the Golgi apparatus to the media, or it was degraded rapidly after leaving the Golgi apparatus. We conclude from these results that at 4 h postinfection, the movement of newly made viral glycopro-teins through the secretory apparatus effectively has stopped.
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
gro29
MUTATION AND PRV MATURATION 38079ro29 CELLS 0 1530 45 60 904 -2
w
68
-46
c
92
68
-46- 46
FIG. 5. Kinetic analysis of PRV glycoprotein synthesis and
modification in L and gro29 cells at 6 h postinfection. Cells were
infectedwithPRVatan MOI of 10. The cellswere pulse-labeledat
6 h postinfection and chased for the time (in minutes) indicated aboveeachlane. The cellswerelysed and immunoprecipitatedwith
gIll-specific antiserum (A), gIl-specific antiserum (B), or a
gp5O-specific monoclonal antibody (C). Immunoprecipitates were
re-solvedon a10% polyacrylamide-SDSgel and visualized by
fluorog-raphy.The molecular mass standards(in kilodaltons) are indicated
tothe leftofeach panel.
Kineticanalysisof PRV glycoprotein synthesis andexport6 h after infection. In the previoussection, we determined the
kinetics of viral glycoprotein synthesis at 4 h postinfection
and noted at least two characteristic defects in viral glyco-protein export in gro29 cells. To determine whether these
export blocks remained constant throughout infection, we analyzed the pulse-chase kinetics at 6 h postinfection (Fig.
5).
Thekinetics ofgIII andgpSOsynthesis and processingare
shown in Fig. 5A and C, respectively. These data are essentially identical to those seen at the 4-h time point. However, the 6-h pulse-chase profile for gIl was noticeably
differentingro29 cells. Processing of the precursor afterthe
5-min pulse was complete within 15
min
of chase, butprotease processingwascompletelyblocked, even atchase times aslong as 120min. Thus, by comparing Fig. 3 and4,
we concluded that the same defect in the rateand extent of export from the ER to the Golgi apparatus occurred at
both 4 and 6 h postinfection. However, export to the
trans-Golgi or trans-Golgi networkwas defective at 6h, as deduced by the lack of proteolytic processing of the gll glycoprotein at 6 h. An alternative view is that gll export
maynotbedefectivebut ratherthat the cellularproteasethat
processes
gIl
is active at 4 h but not 6 h postinfection in gro29 cells.Oligomer
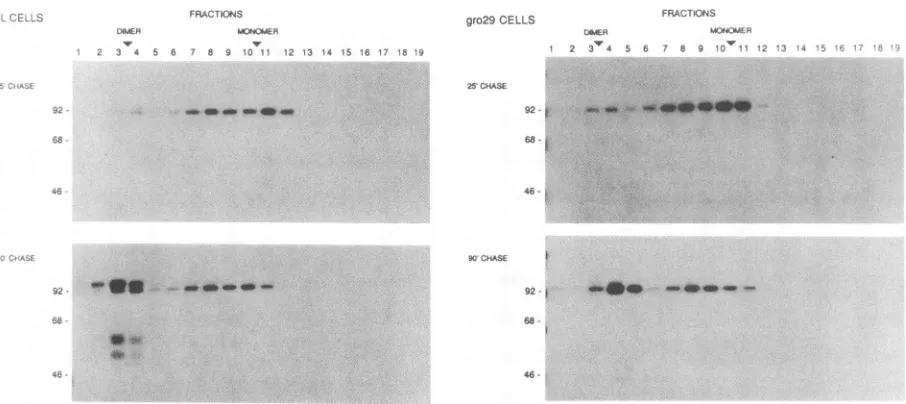
formation is normal in gro29 cells. Pulse-chase results at both 4 and 6 hpostinfection
indicated that gro29cells could
support
normal precursor viral glycoproteinsynthesis
intheER,
but theseprecursors accumulated. Thisfinding suggested
that exit from the ER was defective. Toanalyze
this defectfurther,
wemeasuredthe formation ofgIl
oligomers.
We have demonstratedpreviously
that thegIl
glycoprotein
issynthesized
initially
in theERas a monomerand
subsequently
is converted toanoligomer, presumably adimer
(22, 24).
Thedimerform,
butnotthe monomeric form,is
transported
to theGolgi
apparatus where it receives further modifications andproteolytic
processing. Therefore,one
explanation
for the defect ingIl
export from the ER ingro29
cells may be thatoligomer
formation is defective. To determine whethergIl
iscapable
of forming oligomers ingro29
cells,
we carried out apulse-chase
analysis at 6 hpostinfection
followedby
sucrosegradient
fractionation asdescribedin Materials and Methods(Fig. 6). In both L cells and the
gro29
cells,
thegIl
precursor sedimentedpredomi-nantly
asa monomer at25min
ofchase, and by90min
aftersynthesis,
most of thesemonomers were converted tooligo-mers. We conclude that
gIl
oligomerized
normally in themutant cells and that the lack of
oligomer
formation cannot accountfor the ERexport
defect ingro29 cells. In addition, these results enable us to confirm and extend the previous observations of the defective protease processing ofgII
ingro29
cells. In the L-cellgradient
profile,
protease cleavageproducts
wereclearly
visible in the dimer region of thegradient,
whereas no suchcleavage
productswere detected in the dimerregion
in the gro29 profile.DISCUSSION
The
gro29
cell linewasselected forits inabilitytosupportpropagation
ofHSV-1(21).
In the present experiments, weshowthat this
inability
tosupportpropagationistruealsoforPRV,
a distant relative of HSV-1. We were able to extendand define the
gro29
defect in PRV propagation bystudyingthe
synthesis
and modifications of three PRV glycoproteinsas well as
by conducting
single-step
virus growthexperi-ments. It is
apparent
thatgro29
cells are able to supportnormal
synthesis
of viralglycoprotein
precursors but thatthese precursors accumulate in the ER. This early export
block is not
complete,
and someprecursors enter the Golgiapparatus,
as demonstratedby
addition of complex sugars andprotease
processing
ofthegIl
glycoprotein.
It isnote-worthy
thatastheinfectionproceeds
ingro29 cells, proteaseprocessing
of thegII
glycoprotein
becomes less efficient. Sincethe cellularprotease
isthought
toreside inalateGolgicompartment,
perhaps
the trans-Golgi network, gro29 cells may also be defective in entrance to late Golgicompart-ments. A schematic
representation
of these blocks at 4(early)
and 6(late)
hpostinfection
is shown in Fig. 7.Export
ingro29
cells is defective at multiple steps,sug-gesting
that the defect is due either toasingle mutation in a functionrequired
atmultiple
steps in the export pathway orto
multiple
mutationsaffecting
avarietyoffunctions. Thereisa
precedent
forthe formerhypothesis.
In yeastcells, thesecl8
mutation defines afunction requiredat multiplestepsin the
export
pathway
(7).
Similarly,
the N-ethylmaleimide-sensitive fusionprotein
in mammalian cells is required in fusionofER-derived andGolgi-derived
transportvesicles(2,5).
Ifthishypothesis
iscorrect, the gro29 mutation would affect either a common step in transport vesicle formationL CELLS
0 1 5 30 45 60 90 120
A
92
-
6846
-B
92- a __
-VOL.66, 1992
1w
404*
4W.- 4040.
:z,
1-
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.93.497.362.629.2]3808 WHEALY ET AL.
FRAU iCJw4S
)iMER MONOMER
34 A.6 76 610 l.2l 3s3 1,4 1i 16 1.7 19
_ _- _
-gro29 CELLS
92
68 X
46
-K0 OAS*SE
92-1 "w ft a___4i
_-** .:
FIG. 6. PRV gIl dimer formation in L and gro29 cells. Cells were infected at an MOI of 10. At 6 h postinfection, the cells were pulse-labeled and chased for 25 or90min as indicated. The cellswere solubilized in 1% Triton X-100 and fractionatedbysedimentation
througha5to15%sucrosegradient containing0.1% Triton X-100. The fractionswereimmunoprecipitatedwithagIl-specificantiserum.The immunoprecipitates were resolved on a 10% polyacrylamide-SDS gel andvisualized by fluorography. The positions ofmolecular mass standards (in kilodaltons)areindicatedtotheleft of thepanel.The relativepositionsof theexpectedmonomersand dimers ofgllareindicated
atthe topof thefigure.
from ER and Golgi compartments or a common step in
transport vesicle fusion to intermediate compartment or
trans-Golgi network compartments. Moreover, since such functions wouldmostlikelybeessential, thegro29mutation
mustbe leaky. Indeed, Tufaro has demonstrated that unin-fectedgro29 cells contain at least 35% of normal secretory activityfor endogenous proteins (20). This level ofactivity clearly is not sufficient to handle the increased demand for
sorting and export of viral glycoproteins as the infection
proceeds.
Astrikingphenotype ofgro29 cells is the block in HSV-1 and PRV plaque formation and virus egress. Single-step
growth curve studies of PRV in gro29 cells indicate that
infectiousparticlesformatearlytimesafter infection andare
slowly releasedfromcells, but thisprocessisblockedasthe
infection proceeds. These observations are consistent with
the hypothesisthatacellular function becomes ratelimiting
forviral egressduring herpesvirus infection. It maybe the case that the decrease in host cell protein synthesis that accompanies infectionbyboth PRV andHSV results inthe
disappearance ofan already labile host protein involved in
glycoprotein transport, processing, or virus egress. This
resultcouldexplainthenearly complete blocktosecretion of glycoproteins and viralparticlesasthe demand foracritical protein(s)increases during infection.
To explain both the glycoprotein export defect and the virusegressdefect,weproposethat thetwoprocessesshare
acommon mechanism. Thus, if gro29 is defective in
trans-portvesicleformationorfusion, itwould followthat
herpes-virusegressalsousessimilarmechanismsasparticles travel
from thenucleustothe cellsurface.Whealyetal. (22) have
proposed a model for the assembly and egress of PRV invoking primary envelopment at the inner nuclear mem-brane, de-envelopment asthe capsid leaves the ER, and a secondary envelopment at the transface of the Golgi
appa-ratus, similartothe envelopment pathway used byvaccinia
virus. In this model, the gro29 defect could affect
de-envelopment at the ER, secondary envelopment at the
trans-Golgi, orfusion of theGolgitransportvesicle with the
plasma membrane(Fig. 7).
An alternative possibility is that virus infection may
change the character of thesecretoryorganellessothatthey canfunctionduringviralmaturation,possibly bythe
synthe-sis ofnewER-orGolgi-resident proteins.Thisvirus-induced
change may not occur ingro29 cells because of its already
inefficientsecretory capacity, resultingin the observed gly-coproteinexportand virusegressphenotypes.Further work
isnecessaryto distinguish thesepossibilities.
We have also noted apparent differences in the secretion ofproteinsinHSV-infectedversusPRV-infectedcells. It has been demonstrated previously that by 18 h postinfection,
secretion of human growth hormone, a nonglycosylated
protein, was not seriously impeded in HSV-infected gro29 cells, despite the apparent lack of virion egress (20). This lack of impedance was explained by assuming that the
requirements for intracellular movement of virions and
nonglycosylated proteins were different. In the present study,we observed that a soluble form ofaviral
glycopro-teinwasblocked insecretion. Thisfindingsuggeststhat viral
glycoproteins are affected to a larger extent than are
non-glycosylatedcellularproteinslate inalphaherpesvirus infec-tion. Perhaps after lytic infection, improperly glycosylated
viral glycoproteins are produced that are less soluble and
therefore accumulate aberrantly in the gro29 secretory
or-ganelles. In contrast, proteinslike human growth hormone thatdonotrelyonglycosylation for their efficienttransport
would be solubleandtherebyremain mobileastheytraverse
theinfected-cellsecretoryorganelles. It is alsopossiblethat
PRVcauses a more rapid andextensive shutdown of
trans-portcapacity than does HSV in infected gro29 cells. Addi-tional studiesare neededto resolve these issues.
The gro29 block of both PRV and HSV-1 is a striking
observation. The mutation(s)present ingro29cells affectsa
cellular function required for efficient production of virus
FRA_ iONS
[AMER MiA(MER
2 3 5 6 7 8 9 10 ,1 2 3 _
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.92.545.76.278.2][;.N.';:.hiS7 1
FIG. 7. A schematicrepresentation of PRVglycoprotein export andvirus egress in gro29 cells. The general model has been described
previously (22).The toppanelshowseventsearlyafterinfection,and the bottompanel shows events late after infection of gro29 cells. Blocks in theproposed pathwaywerededucedbydeterminingthe extent of PRVglycoproteinmodifications,as described in the text. At both early and latetimes, there isapartialblock of PRVglycoproteinexportfrom the ER to theGolgi apparatus (indicated by the dotted X). Early in
infection,there is a secondblocktoexportfromlate Golgi compartments(possible the trans-Golgi network) to the cell surface (indicated by the solidX).Inaddition,late afterinfection,thereisathird block from lateGolgicompartments to the trans-Golgi network (indicated by the solidX).Virus egress is blockedonlylate afterinfection,and thelocation of this block is unknown (indicated by question mark). The cisternae of theGolgiapparatusarelabeledC, M,and Tfor thecis,medial, and trans compartments, respectively. The trans-Golgi network is indicated
by the vesicleleavingthetrans-Golgi apparatus andmovingtothecell surface. Viralglycoproteins are designated as follows: " indicates
proteinswithonlyERmodifications, and t indicatesproteinswithGolgiapparatusmodifications.
progeny of these tworather diverse alphaherpesvirusesyet has only a marginal effect on uninfected cell growth in culture. Theseobservations suggestthatgro29definesa new
class ofcellulartargets forbroad-spectrumantiviral agents. REFERENCES
1. Banfield, B. W., and F. Tufaro. 1990. Herpes simplex virus
particles are unable to traverse the secretory pathway in the mouseL-cellmutantgro29. J. Virol.64:5716-5729.
2. Beckers,C.J. M.,M. R.Block,B.S.Glick, J.E.Rothman,and W. E. Balch.1989.Vesicular transport between theendoplasmic
reticulum and theGolgistackrequiresthe NEM-sensitive fusion
protein. Nature(London)339:397-398.
3. Ben-Porat, T., andA. S. Kaplan. 1985. Molecular biology of
pseudorabies virus, p. 105-173. In B. Roizman (ed.), The
herpesviruses.PlenumPublishing Corp.,New York.
4. Chamberlain, J.P.1979.Fluorographicdetection of
radioactiv-ityinpolyacrylamide gelswith thewatersoluble fluor sodium
salicylate.Anal. Biochem. 98:132-135.
5. Diaz, R., L.S. Mayorga,P. J. Weidman,J. E.Rothman, and P. D. Stahl. 1989. Vesicle fusion following receptor-mediated
endocytosisrequiresaprotein active in Golgi transport. Nature (London) 339:398-400.
6. Gong, M., and E. Kief. 1990. Intracellular trafficking of two
majorEpstein-Barrvirusglycoproteins,gp350/220 andgpllO.J. Virol. 64:1507-1516.
7. Graham,R.R.,andS.D. Emr.1991.Compartmental organiza-tion ofGolgi-specific protein modification and vacuolar protein
sorting events defined in a yeastsecl8 (NSF) mutant. J. Cell Biol. 114:207-218.
8. Johnson, D. C., and P. G. Spear. 1982. Monensin inhibits the
processing ofherpes simplex virus glycoproteins, their trans-porttothe cellsurface,and the egress of virions from infected cells.J.Virol. 43:1102-1112.
9. Jones,F., and C. Grose. 1988. Role ofcytoplasmicvacuoles in varicella-zoster virusglycoprotein traffickingand virion
envel-opment. J. Virol. 62:2701-2711.
10. Kornfeld, R.,and S. Kornfeld. 1985. Assemblyof
asparagine-linkedoligosaccharides. Annu. Rev. Biochem. 54:631-664. 11. Petrovskis, E. A., J. G. Timmins, M. A. Armentrout, C. C.
Marchiolli, R. J. Yancey, Jr., and L. E. Post. 1986. DNA
sequence of the gene forpseudorabiesvirusgp5O, a
glycopro-tein withoutN-linkedglycosylation. J.Virol. 59:216-223.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.162.446.82.422.2]3810 WHEALY ET AL.
12. Pizer, L. I., G. H. Cohen, and R. J. Eisenberg. 1980. Effect of
tunicamycin on herpes simplexvirus glycoproteins and infec-tiousvirus production. J. Virol. 34:142-153.
13. Poliquin, L., G. Levine, and G. C. Shore. 1985. Involvement of theGolgi apparatus andarestructured nuclearenvelopeduring biogenesis and transport of herpessimplexvirusglycoproteins.
J. Histochem.Cytochem. 33:875-883.
14. Robbins, A. K., J. P. Ryan, M. E. Whealy, and L. W.Enquist. 1989. The geneencoding theglll envelopeproteinof pseudora-bies virus vaccine strain Bartha containsa mutation affecting
protein localization. J. Virol. 63:250-258.
15. Robbins, A. K., R. J.Watson, M. E.Whealy, W. W. Hays, and L. W. Enquist. 1986.Characterization ofa pseudorabies virus
glycoproteingenewith homology toherpessimplex virus type 1 and type 2glycoproteinC. J. Virol. 68:339-347.
16. Ryan, J. P., M. E.Whealy, A. K. Robbins, and L. W. Enquist. 1987.Analysis ofpseudorabiesvirusglycoprotein gIII localiza-tion and modification by using novel infectious viral mutants
carryinguniqueEcoRI sites. J. Virol. 61:2962-2972.
17. Solomon, K. A., A. K. Robbins, and L. W. Enquist. 1991. Mutations in the C-terminal hydrophobic domain of pseudora-bies virus glll affect both membrane anchoring and protein export.J. Virol.65:5952-5960.
18. Spear, P. G. 1984. Glycoproteins specified by herpes simplex viruses, p. 315-356. In B. Roizman (ed.), The herpesviruses,
vol. 3. Plenum Publishing Corp., New York.
19. Tooze, S. A., J. Tooze, and G. Warren. 1988. Siteof addition of N-acetyl-galactosamine to theEl glycoprotein of mouse hepa-titis virus-A59. J.CellBiol. 106:1475-1487.
20. Tufaro, F.Unpublishedobservations.
21. Tufaro, F., M. D.Snider, and S. L. McKnight.1987. Identifica-tion and characterizaIdentifica-tion of a mouse cell mutant defective in the intracellular transport ofglycoproteins. J. Cell Biol. 105:647-657.
22. Whealy, M. E., J. P.Card, R. P. Meade, A. K. Robbins, and L.W.Enquist. 1991. Effect of brefeldin A onalphaherpesvirus
membrane protein glycosylation and virus egress. J. Virol. 65:1066-1081.
23. Whealy, M. E., A. K. Robbins, and L. W. Enquist. 1988. The
pseudorabies virus glycoprotein glll is required for efficient
virus growth in tissue culture. J. Virol. 62:2512-2515.
24. Whealy, M. E., A. K. Robbins, and L. W. Enquist. 1990. The export pathway of the pseudorabies virus gB homolog gll
involves oligomerformation in the endoplasmic reticulum and protease processing in the Golgi apparatus. J. Virol.
64:1946-1955.
25. Wittmann,G., and H.-J. Rziha. 1989. Aujesky's disease (pseu-dorabies) in pigs, p. 230-233. In G. Wittmann (ed.), Herpesvirus
diseases ofcattle,horses andpigs. Kluwer Academic Publish-ers, Boston.
J. VIROL.
![FIG. 1.Afterfixedoncells.describeddaysdish] Plaque-forming efficiency of PRV on PK15, L, and gro29 Serial 10-fold dilutions of PRV (10-2 dilution [top left in each through 10-7 dilution [bottom right in each dish]) were plated PK15, L and gro29 monolayers](https://thumb-us.123doks.com/thumbv2/123dok_us/1308233.84077/2.612.336.552.79.416/afterfixedoncells-describeddaysdish-plaque-efficiency-dilutions-dilution-dilution-monolayers.webp)