Copyright © 2000, American Society for Microbiology. All Rights Reserved.
RNA Dimerization Defect in a Rous Sarcoma Virus
Matrix Mutant
LESLIE J. PARENT,1,2TINA M. CAIRNS,2JESSICA A. ALBERT,1CAROL B. WILSON,2
JOHN W. WILLS,2ANDREBECCA C. CRAVEN2*
Departments of Medicine1and Microbiology and Immunology,2The Pennsylvania State University College of Medicine,
M. S. Hershey Medical Center, Hershey, Pennsylvania 17033
Received 26 May 1999/Accepted 17 September 1999
The retrovirus matrix (MA) sequence of the Gag polyprotein has been shown to contain functions required for membrane targeting and binding during particle assembly and budding. Additional functions for MA have been proposed based on the existence of MA mutants in Rous sarcoma virus (RSV), murine leukemia virus, human immunodeficiency virus type 1, and human T-cell leukemia virus type 1 that lack infectivity even though they release particles of normal composition. Here we describe an RSV MA mutant with a surprising and previously unreported phenotype. In the mutant known as Myr1E, the small membrane-binding domain of the Src oncoprotein has been added as an N-terminal extension of Gag. While Myr1E is not infectious, full infectivity can be reestablished by a single amino acid substitution in the Src sequence (G2E), which eliminates the addition of myristic acid and the membrane-binding capacity of this foreign sequence. The presence of myristic acid at the N terminus of the Myr1E Gag protein does not explain its replication defect, because other myristylated derivatives of RSV Gag are fully infectious (e.g., Myr2 [C. R. Erdie and J. W. Wills, J. Virol. 64:5204–5208, 1990]). Biochemical analyses of Myr1E particles reveal that they contain wild-type levels of the Gag cleavage products, Env glycoproteins, and reverse transcriptase activity when measured on an exogenous template. Genomic RNA incorporation appears to be mildly reduced compared to the wild-type level. Unex-pectedly, RNA isolated from Myr1E particles is monomeric when analyzed on nondenaturing Northern blots. Importantly, the insertional mutation does not lie within previously identified dimer linkage sites. In spite of the dimerization defect, the genomic RNA from Myr1E particles serves efficiently as a template for reverse transcription as measured by an endogenous reverse transcriptase assay. In marked contrast, after infection of avian cells, the products of reverse transcription are nearly undetectable. These findings might be explained either by the loss of a normal function of MA needed in the formation or stabilization of RNA dimers or by the interference in such events by the mutant MA molecules. It is possible that Myr1E viruses package a single copy of viral RNA.
The retrovirus Gag polyprotein directs the assembly and budding of virus particles from the plasma membrane of in-fected cells. Extensive functional mapping of several Gag pro-teins has led to the identification of three common assembly domains that are required for this activity. The N-terminal membrane-binding (M) domain directs Gag molecules from the cytoplasm to the inner leaflet of the plasma membrane (44, 55, 63) where aggregates of Gag proteins are formed through protein-protein interactions primarily involving the I (interac-tion) domains (4, 12, 22, 57). These interactions lead to the emergence of spherical particles that pinch off the membrane during the final step in the budding process, which is mediated by the L (late) assembly domain (27, 45, 58). Virus maturation occurs as the Gag polyprotein is cleaved into the major virus structural proteins by the viral protease (PR) to yield the matrix (MA), capsid (CA), and nucleocapsid (NC) proteins as well as several small peptides, and in the case of Rous sarcoma virus (RSV), PR itself. Reorganization of the virion architec-ture occurs following cleavage of the Gag protein. MA forms a spherical shell under the lipid envelope. At the center of the virion condenses an electron-dense core, a complex of the
CA and NC proteins, viral RNA, and the replicative enzymes reverse transcriptase (RT) and integrase.
The Gag polyprotein also coordinates the packaging of Pol, Env, and the genomic RNA. The NC region of Gag is the primarytrans-acting factor required for incorporation of the genome into virions (5, 54). Within NC reside two Cys-His boxes and an adjacent basic residue-rich region that interact directly with the viral RNA during encapsidation. Sequences outside of NC have also been implicated to play minor roles in RNA packaging (50, 54). With regard to RSV, sequences in MA are not required for specific genomic RNA incorporation into particles (50) although MA does have weak, nonspecific nucleic acid-binding properties (52). Packaging signals in the RNA itself consist of acis-acting element known aswithin the 5⬘untranslated region. In RSV, RNA encapsidation signals have been identified within a 270-nucleotide (nt) region be-tween the primer binding site and the splice donor ingag(1) and within a 115-nt region spanning the direct repeats at the 3⬘
untranslated end of the genome (2, 51).
The genomic RNA of all retroviruses exists within the virion as a noncovalently linked dimer of identical 35S RNA mole-cules. Sequences spanning nt 208 to 274 and 400 to 600 have been implicated in the dimerization. The dimer linkage struc-ture appears to overlap the packaging signal, a region re-quired for in vivo incorporation of viral RNA (5, 6, 21, 31, 42). Dimerization likely occurs very early in the assembly pathway and is independent of PR activation (24, 25, 53). The NC
* Corresponding author. Mailing address: Department of Microbi-ology and ImmunMicrobi-ology, Pennsylvania State University College of Med-icine, 500 University Dr., P.O. Box 850, Hershey, PA 17033. Phone: (717) 531-3528. Fax: (717) 531-6522. E-mail: [email protected].
164
on November 9, 2019 by guest
http://jvi.asm.org/
protein has been shown to promote stability and maturation of dimers, although it is not required for dimerization per se (5). MA has not been previously implicated in genomic RNA dimerization.
Several lines of evidence indicate that MA possesses func-tions during replication in addition to membrane targeting and binding, although what these functions might be remains un-certain. Genetic analyses of RSV, murine leukemia virus and human immunodeficiency virus (HIV) have revealed many MA mutants that have little or no infectivity, even though particle assembly and budding proceed normally (11, 13, 16, 32, 36, 45, 48, 62). Characterizations of numerous HIV type 1 mutants have suggested a role for MA in accommodating the long cytoplasmic tail of its Env proteins (23, 37) as well as a poorly understood postfusion function (32). In contrast to HIV, it is unlikely that RSV MA has a role in glycoprotein packaging, since Env proteins lacking cytoplasmic tails or hav-ing foreign intracytoplasmic domains are packaged efficiently (47, 61). Nonetheless, the identification of numerous assembly-competent, noninfectious MA mutants for several different viruses compelled us to examine our RSV MA mutants more thoroughly.
In this report, we present our unexpected finding that addi-tion of the Src membrane-binding sequence to the N terminus of RSV MA results in particles with altered genomic RNA dimerization. This finding raises the intriguing possibility that the MA sequence influences RNA dimer formation and/or stability.
MATERIALS AND METHODS
Viruses and cells.The infectious RSV genome (pRC.V8) used in these studies was derived from pBH.RCAN.HiSV (pRCAN) (18) and bears the RSV Prague Cgaggene from pATV8. This proviral vector carries a simian virus 40 early promoter-driven hygromycin resistance gene near the 3⬘end of the viral genome, within the boundary of the 3⬘long terminal repeat (15). The JD.Myr2 virus, which produces a myristylated Gag protein due to a E2G substitution, has been described previously (20). Avian cells used for transfection and infection exper-iments were QT6 cells, a chemically transformed quail fibroblast line (41). The maintenance of these cells was previously described (15). The simian virus 40-based pSV.Myr1 and pSV.Myr2 vectors used for expression of the Gag polyprotein in the COS-1 simian cell line have been described elsewhere (59).
Antisera.A polyclonal rabbit serum against RSV (56) was used for immuno-precipitations and immunoblotting. The anti-Env serum used recognizes the subgroup A envelope glycoprotein (gp85) and was kindly provided by Eric Hunter (University of Alabama, Birmingham).
Oligonucleotide-directed mutagenesis of thegaggene.Mutagenesis was per-formed by the method of Kunkel et al. (33), using the previously described recombinant bacteriophage MGAG as the template unless otherwise noted (59). The oligonucleotides used to make the indicatedgagalleles were as follows: for myr1⫺, 5⬘-GGATCAAGCATGGAATCCAGCAAAAGC; formyr1e, 5⬘-CCCG GTGGATCAAGCATGGGATCATCAAAATCTAAACCTAAGGATGAAG CCGTCATAAAGGTGAT; and formyr1e⫺, 5⬘-GGATCAAGCATGGAATCA TCAAAATCTAAAC. M13 DNAs bearing the mutations were identified by dideoxy sequencing. For each mutation, replicative-form DNAs from two inde-pendent clones were digested withSstI (nt 255) andHpaI (nt 2731), and thegag fragments were transferred into pRCAN for expression in avian cells. After digestion of the M13 DNA withSstI andBg1II (nt 1630), eachgagallele was cloned into pSV.GagSX for expression in mammalian cells (45).
Transfection, labeling, immunoprecipitation, and production of cell lines.
COS-1 cells were transfected by the DEAE-dextran-chloroquine method, met-abolically labeled withL-[35S]methionine or [3H]myristic acid, and
immunopre-cipitated with anti-RSV serum as described previously (56, 60). QT6 cells were transfected by calcium phosphate precipitation and labeled with [35 S]methi-onine, and RSV proteins were immunoprecipitated as described elsewhere (45). Radiolabeled proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), detected by fluorography, and quantitated by laser scanning densitometry.
To create a cell line that stably expresses themyr1egenome, QT6 cells were transfected with pRC.Myr1E, divided into several 100-mm-diameter culture dishes, and selected in primary growth medium (15) containing hygromycin (300
g/ml; Sigma). Hygromycin-resistant colonies were transferred to individual culture dishes after 2 weeks and were screened for the stable production of particles by analysis of the culture supernatants with RT assay and Western blotting (45). Five independent cell lines were established, and the two most
robust particle-producing lines were used in subsequent experiments. Total cel-lular DNA was isolated from each cell line as described below and was sequenced to confirm the presence of the myr1e mutation. The QT6.Myr1E cell lines produce particles that contain normal amounts of Gag-Pol, Env, and RT activity (as described below).
Virus infectivity assays.The ability of mutant and wild-type viruses to stably infect avian cells was measured in two ways. First, QT6 cells transfected with wild-type or mutant genomes were tested for the continued release of RT activity into the medium upon prolonged passage of the cells in culture. Only infectious genomes spread throughout the culture, integrate, and exhibit persistent virus release. For this, aliquots of culture supernatant were tested for RT activity periodically as described previously (15).
In the second assay, persistent viral infections were detected by looking for continued expression of thegaggene (44). Duplicate 60-mm-diameter plates of QT6 cells were transfected with each proviral DNA. One plate was metabolically labeled 12 h later to ascertain the level of expression of Pr76gagand hence the
efficiency of transfection. Cells from the other plate were passaged every 3 to 4 days, and the continued expression of Gag was measured by immunoprecipita-tions of labeled proteins with anti-RSV serum.
Protein and RNA composition of mutant viruses.Virus particles harvested from culture supernatants of QT6 cell lines were concentrated by ultracentrifu-gation, normalized according to RT activity, and subjected to immunoblotting with anti-RSV and anti-Env sera as described previously (15). Exogenous RT activity was determined as described previously (15). Viral RNA content of similarly produced particles was determined by Northern slot blot analysis using a32P-labeled riboprobe consisting of thegagsequence (15).
ERT assays.Endogenous RT (ERT) reactions were performed as previously described (7, 8), with modifications as described below. Equivalent amounts of virus particles (normalized by RT content) were incubated with melittin (100
g/ml, final concentration) at 41°C for 10 min prior to addition of ERT buffer (0.1 M Tris-HCl [pH 8.3], 25 mM NaCl, 3 mM magnesium acetate, 30 mM dithiothreitol, 1 mM dCTP, 1 mM dATP, 1 mM dGTP [final concentrations]). The reaction mixtures were divided equally into two tubes. To one set, nonra-dioactive was added to 1 mM (final concentration), and the reaction mixture was incubated at 41°C for 3 h. The reaction was stopped by addition of an equal volume of buffer containing 1% SDS, 20 mM EDTA, and 100g of yeast tRNA. The reaction products were phenol-chloroform extracted and ethanol precipi-tated. These samples were subjected to PCR analysis for the DNA products of reverse transcription, using the primers and conditions specified below. To the second set of reactions, 10Ci of [32P]TTP was added, and the remainder of the reaction was performed as described above. After ethanol precipitation, the radiolabeled ERT products were denatured with 0.05 M NaOH at 37°C for 30 min and electrophoresed in 1% agarose in Tris-borate-EDTA at 100 V for 3 h, and the gel was dried and subjected to autoradiography.
Analysis of products of reverse transcription by PCR.To detect viral DNA products, virus particles were obtained from QT6 cells lines stably expressing viral genomes and used to infect QT6 cells for 18 h. Low-molecular-weight DNA was extracted from duplicate 60-mm-diameter plates of infected cells according to the Hirt protocol (29). DNA was analyzed by using the following pairs of oligonucleotide primers designed to detect the indicated products of reverse transcription: minus-strand strong-stop DNA, primer 5 (R sequence; 5⬘-CTTC ATGCAGGTGCTCGTAGTCG) and primer 3 (U5 sequence; 5⬘-GCCATTTT ACCATTCACCACA); first strand switch, primer 8 (U3 sequence; 5-GGATTG GACGAACCACTGAA) and primer 3; and second-strand switch, primer 5 and primer 7 (5⬘untranslated region); 5⬘-CAACGACTCTCTGAGTTCTC). Prior to the reaction, one of the primers was labeled with [␥-33P]ATP by using T4 polynucleotide kinase. PCR conditions were 97°C for 5 min and then 25 cycles of 94°C, 56°C, and 72°C for 30 s each. The PCR products were separated by electrophoresis in nondenaturing 6 or 8% polyacrylamide gels and detected by autoradiography.
Electrophoretic analysis of RNA dimerization.Virus particles produced from the stable lines or after transient transfection were collected by ultracentrifuga-tion and resuspended on ice in either phosphate-buffered saline (containing 150 mM NaCl) or TNE buffer (10 mM Tris [pH 7.4], 100 mM NaCl, 1 mM EDTA). Virus particles were lysed in 50 mM Tris (pH 7.5)–10 mM EDTA–1% SDS–100 mM NaCl, with 50g of yeast tRNA as a carrier, according to the method of Fu and Rein (25). Equivalent amounts of viral RNA were electrophoresed through a 1% agarose gel in Tris-borate-EDTA (100 V for 3 h or 20 V for 17 h), denatured within the gel, and blotted to a nylon membrane (25). The blots were hybridized by using agagriboprobe (as described above), washed under high-stringency conditions, and subjected to autoradiography.
RESULTS
The initial objective of the experiments described below was to ascertain whether the M domain of a cellular protein could replace all of the functions of the RSV MA protein. We chose the myristylated N-terminal peptide of the Src oncoprotein,
VOL. 74, 2000 RNA DIMERIZATION DEFECT IN AN RSV MA MUTANT 165
on November 9, 2019 by guest
http://jvi.asm.org/
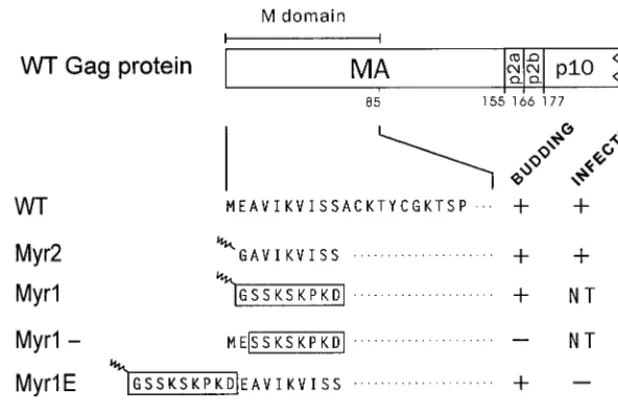
which is a potent signal for targeting and binding of proteins to the inner leaflet of the plasma membrane (10, 46, 49). When the first 10 residues of Gag are replaced with the first 10 residues of Src, the resulting chimera, named Myr1 (Fig. 1), retains the ability to direct particle assembly and budding from mammalian cells (Fig. 2A, lanes 1) (60). Indeed, the entire M domain of RSV Gag can be replaced with this sequence from Src without affecting budding (59). However, because the nu-cleotide substitution in myr1 removes the splice donor site (located at codon 7 ofgag) which is needed for synthesis ofenv
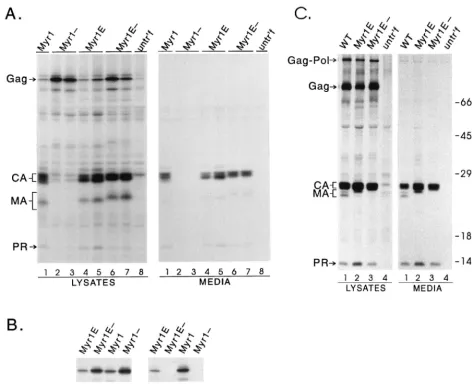
mRNA, this allele could not be used to study infectivity. For that reason we decided to add the coding sequence for the Src membrane-binding domain as a 5⬘extension ofgag, thus pre-serving the splice site and creating a Gag chimera named Myr1E (Fig. 1). The Myr1E protein was found to produce particles at the normal rate when tested in mammalian cells (Fig. 2A, lanes 4 and 5), and like the Myr1 protein, it was labeled with [3H]myristic acid because of the presence of the sequence from Src (Fig. 2B).
Placement of the Src peptide as an N-terminal extension potentially preserves the normal M domain of Gag. Evidence in support of this was obtained by eliminating the myristic acid addition sites of Myr1 and Myr1E with a G2E substitution, which inactivates membrane binding (17, 30). This resulted in the creation of Myr1- and Myr1E- (Fig. 1), and as expected, these proteins could not be labeled with [3H]myristic acid (Fig. 2B). The Myr1- protein was unable to direct budding because both M domains were destroyed: the initial insertion of the Src sequence was at the expense of the first 10 amino acids of Gag, which are necessary for activity of the RSV M domain (44); and the Src membrane-binding signal was disabled by the G2E mutation (Fig. 2A, lanes 2 and 3) (3). Therefore, the Myr1 protein was dependent on having a functional Src membrane-binding domain, whereas Myr1E- retained the ability to direct budding even though the inactive form of the Src peptide was
present (Fig. 2A, lanes 6 and 7), indicating that the M domain of Gag remains intact. Thus, Myr1E appears to contain two, independent M domains.
To determine whether the budding activities of the Src ex-tension mutants would be the same in avian cells, themyr1e
andmyr1e⫺alleles ofgagwere cloned into the pRCAN
pro-viral expression vector (18). Levels of particle production were normal upon transfection of these mutants into QT6 cells (Fig. 2C, lanes 2 and 3) and turkey embryo fibroblasts (data not shown). The rate of budding was indistinguishable for the Myr1E mutant compared with wild-type virus, as shown by pulse-chase analysis following transfection (data not shown).
It was not surprising that the presence of the Src sequence was compatible with budding, since a variety of other peptide sequences have been added as extensions from RSV Gag with-out affecting budding or infectivity (39; T. Nelle and J. W. Wills, unpublished data). Therefore, we fully expected recom-binant RSV genomes carrying themyr1eormyr1e⫺ allele of
gag to be infectious in avian cells. However, this was not the case.
Infectivity of Myr1E.To analyze infectivity, RCAN vectors that express the myristylated and nonmyristylated forms of Myr1E were transfected into QT6 cells. Proviral plasmid DNAs containing myr2 (the E2G substitution that creates a myristylated form of Gag) or wild-typegag sequences were included as infectious controls. One of two sets of duplicate transfected plates was labeled with [35S]methionine at 18 h posttransfection to see whether the transfections were success-ful. In each case, Gag proteins were readily detected within cells after a 1-h labeling period (Fig. 3A). To monitor the persistence of infection, cells from the second set of plates were passaged every 3 to 5 days to allow infectious viruses to spread throughout the cultures, and periodically the cells were tested for continued production of Gag proteins by radiolabel-ing and immunoprecipitation. Although abundant Myr1E par-FIG. 1. Schematic representation of wild-type and mutant MA proteins. The N-terminal region of the RSV Gag protein is shown at the top with the MA, p2, and p10 cleavage products indicated; the numbers below refer to amino acid residues. The M domain, as indicated, consists of the first 85 residues of MA. Expanded below are the N-terminal amino acid sequences of wild-type (WT) and mutant proteins. Residues identical to WT are indicated by dots. Boxes are drawn around residues derived from the Src membrane-binding domain, with the squiggle representing myristic acid modification. Myr2 has an E2G substitution, resulting in myristylation and removal of initiator methionine. In Myr1, the first 10gagcodons were replaced with the sequence encoding the myristylated Src N terminus. Myr1⫺has the wild-type glutamate substituted at position 2 of Myr1, resulting in the prevention of myristylation and the retention of methionine. Myr1E has nine codons specifying the Src M domain inserted after thegagAUG so that the Src sequence is extended from the Gag N terminus. In Myr1E⫺, myristylation is abolished by a G2E substitution. The ability of each virus to bud and to infect avian cells is indicated by a plus sign (equivalent to wild-type level), minus sign (ⱕ1% of wild-type level), or NT (not tested).
on November 9, 2019 by guest
http://jvi.asm.org/
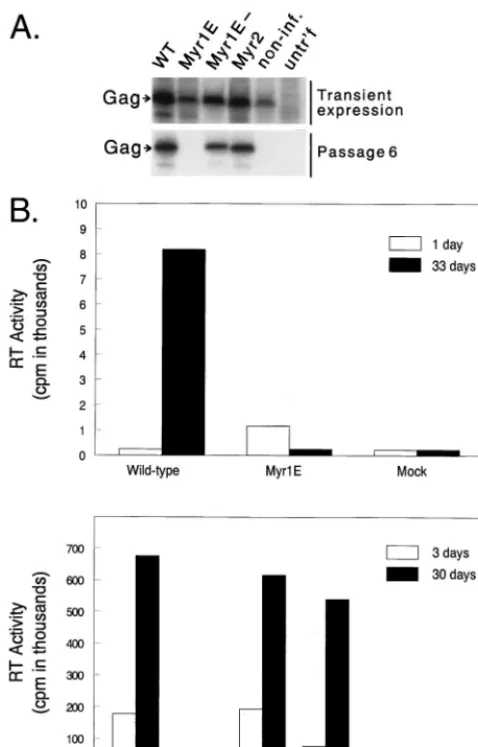
[image:3.612.148.457.73.279.2]ticles had been released immediately after transfection, no evidence of infection was found after six passages (approxi-mately 30 days), and the Myr1E virus had disappeared from the culture (Fig. 3A). Numerous transfection experiments of this type have been repeated by different investigators in our laboratory, using multiple clones of themyr1eallele expressed in the Prague A and RCAN genomes in both QT6 cells and turkey embryo fibroblasts, and persistent infection has never been observed. On the other hand, the Myr1E⫺ and Myr2 mutants spread in QT6 cultures at the same rate as the wild-type virus and readily establish infection (Fig. 3A). The precise magnitude of the defect in Myr1E virus has not been deter-mined; however, our previous experience with these assays indicates that the defect must be on the order of 10⫺5 or greater relative to the wild type.
Additional evidence for an infectivity defect in Myr1E was obtained by measuring the release of RT activity in the me-dium of transfected cells (Fig. 2C). While there was an initial
burst of particle production immediately after transfection of cells with pRCAN.myr1e, at later time points the RT activity in the culture medium decreased to background levels. This re-sult confirms the lack of detectable infectivity for the Myr1E virus. However, in cells transfected with infectious genomes includingmyr1e⫺, myr2, and wild type, RT activity increased rapidly as the virus spread throughout the culture.
[image:4.612.66.538.74.457.2]The striking lack of infectivity observed for Myr1E is not simply due to the presence of myristate. As shown here and previously (20), the E2G substitution in Myr2 (Fig. 1) converts the normally acetylated RSV Gag protein into one that is myristylated, with no effect on infectivity (Fig. 3). Furthermore, the addition of foreign peptides to the N terminus is, in itself, not lethal to the virus, as shown by the infectivity of the Myr1E⫺virus (Fig. 3). As well, other peptides have been fused onto Gag without destroying virus infectivity (39). Together, these data suggest that the impaired infectivity of Myr1E may FIG. 2. Effects of MA alterations on particle assembly and myristylation. (A) Analysis of budding in COS-1 cells. Transfected COS-1 cells were radiolabeled with [35S]methionine for 2.5 h, lysates and media were separated, and viral proteins were detected by immunoprecipitation with anti-RSV serum. Positions of the bands corresponding to Gag precursor, CA, MA, and PR proteins are indicated at the left. untr’f, untransfected. (B) Myristate labeling. Duplicate plates of COS-1 cells were transfected, labeled for 70 min with [35S]methionine or [3H]myristic acid, and immunoprecipitated as for panel A. (C) Budding in avian cells. QT6 cells were transfected with wild-type (WT) and mutant proviral plasmids and analyzed as for panel A. The position of the Gag-Pol protein is indicated at the left, and positions of molecular weight standards are shown at the right.
VOL. 74, 2000 RNA DIMERIZATION DEFECT IN AN RSV MA MUTANT 167
on November 9, 2019 by guest
http://jvi.asm.org/
be due to the addition of the functional Src membrane-binding domain at the N terminus of Gag.
Biochemical analysis of Myr1E virus particles. To deter-mine whether the replication defect of Myr1E was due to a failure to package required viral components, virus particles were examined for incorporation of Env glycoproteins, genomic RNA, and Gag and Gag-Pol proteins. With regard to Env, it was possible that extension of the Src sequence from the N terminus of Gag might interfer with its synthesis, trans-port, or incorporation into the viral envelope because the splice donor for env mRNA resides within the gag gene in RSV. Therefore, the first few amino acids of Gag are initially attached to the nascent Env protein. If the plasma membrane-binding signal of Src was dominant over the Env signal peptide, then targeting of Env to the endoplasmic reticulum would not
occur and little or no glycoprotein would be produced and packaged in the Myr1E virions. This is not the case, however, because Myr1E particles produced by transfection of QT6 cells contained abundant Env (gp85) as do wild-type and Myr1E⫺
viruses (Fig. 4A).
To characterize their Gag products, Myr1E and Myr1E⫺
[image:5.612.54.293.72.445.2]virions produced after transfection were analyzed by immuno-blotting (Fig. 4B). Both mutants packaged normal amounts of the CA protein. For Myr1E, the amount of MA detected was comparable to the wild-type amount, although the MA band runs as a single band with slower mobility (also see below). The Myr1E⫺MA band appears as a more slowly migrating doublet and has lower intensity than the wild type owing to conforma-tional changes in the epitope recognized by the polyclonal anti-RSV serum. The NC and PR bands run together in our gel system; however, the intensity of the combined NC-PR band is the same in all lanes (data not shown). We also found that for Myr1E and Myr1E⫺, the ratio of CA protein to RT activity (measured by using an exogenous template) was equal to that FIG. 3. Persistence of virus production in long-term cultures. (A) Duplicate
plates of QT6 cells were transfected with proviral constructs expressing the indicatedgagalleles. At 18 h posttransfection, the Gag precursor in cell lysates was detected after a 1-h labeling period by immunoprecipitation with RSV antiserum (transient expression). Persistent expression of Gag was measured similarly after passage 6. non-inf., noninfectious CA mutant L171I, included as a negative control; untr’f, untransfected. (B) In two separate experiments, infec-tions of QT6 cells were initiated by transfection as for panel A; then the accu-mulation of RT activity in the culture medium was monitored over a period of 30 or 33 days posttransfection. The large difference between the two experiments in the scale on the RT activity axis was due to differences in specific activity of the isotope and in the particle concentration factor.
FIG. 4. Biochemical properties of virus particles. (A) Envelope incorpora-tion. Equivalent amounts of virions from culture supernatants were collected by ultracentrifugation from QT6 cells transfected with mutant or wild-type (WT) proviral plasmids. Levels of Env (SU) expression were detected by immunoblot-ting with anti-Env serum. untr’f, untransfected. (B) Gag processing. Virus par-ticles from panel A were analyzed by immunoblotting with anti-RSV serum to detect processed Gag proteins CA and MA. (C) RNA genome incorporation. Virus particles were normalized for RT content, RNA was purified, serial four-fold dilutions were performed, and the RNA was applied to a slot blot apparatus, bound to nitrocellulose, and detected with a radiolabledgagriboprobe.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.360.503.72.416.2]of the wild-type virus, indicating that normal amounts of this enzyme, and hence Gag-Pol, are packaged (data not shown).
When analyzed by SDS-PAGE, the wild-type MA (p19) protein appears as a doublet, which reflects modification of a subpopulation of the molecules by serine phosphorylation (34). Myr1E and Myr1E⫺showed different patterns with regard to MA. The Myr1E protein migrates more slowly than wild-type MA, consistent with its larger size, increased positive charge, and the presence of myristate (Fig. 4B). Its appearance as a single band rather than a doublet suggested that the Src mod-ification might alter phosphorylation. This was not the case, however, because analysis of Myr1E-infected cells grown in [32P]orthophosphate did not reveal any reduction in phosphor-ylation of the Myr1E MA protein (data not shown). Moreover, replacement of the serine at position 106 (the only site of serine phosphorylation in MA) with alanine does not alter the infectivity of wild-type (43), Myr1E, or Myr1E⫺virus (data not shown). Hence, this alteration of MA does not hold the key to explaining why Myr1E is not infectious.
Although it has been demonstrated that sequences through-out RSV MA are not essential for viral RNA packaging (50), it was important to determine whether the Src extension had interfered with RNA packaging in Myr1E particles. RNA was extracted from virus particles produced in avian cells that were normalized for exogenous RT activity, and serial dilutions were compared (Fig. 4C). In these analyses, the levels of RNA isolated from Myr1E were consistently approximately twofold lower than wild-type levels. Thus, there is a mild decrease in levels of Myr1E genomic RNA, which alone cannot explain the severe infectivity defect. To more fully characterize the nature of the replication block, the ability of Myr1E virus particles to undergo DNA synthesis was assessed.
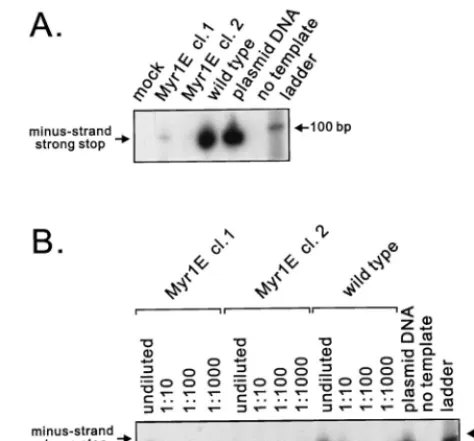
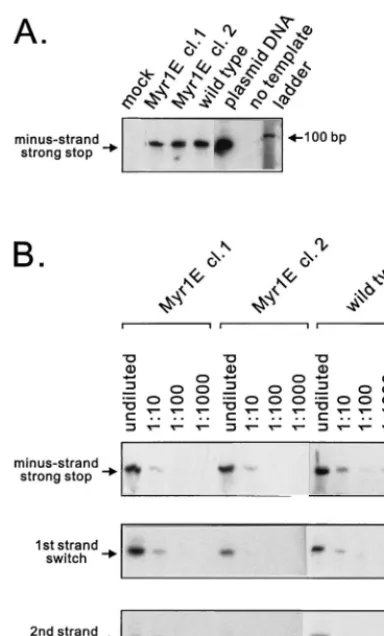
Postinfection levels of reverse transcription. Wild-type or Myr1E virus particles produced by cell lines stably expressing each proviral genome were used to infect monolayers of QT6 cells. Eighteen hours later, low-molecular-weight DNA was isolated from infected cells (29) and used as a template for PCR amplification with primers designed to detect the prod-ucts of reverse transcription (Materials and Methods). As shown in Fig. 5A, the level of minus-strand, strong-stop DNA was markedly reduced in Myr1E-infected cells. To determine the magnitude of the defect, the low-molecular-weight DNA was diluted serially from 10⫺1to 10⫺3and subjected to PCR analyses. The amount of strong-stop DNA isolated from Myr1E-infected cells was reproducibly 100- to 1,000-fold re-duced compared with wild-type infection (Fig. 5B). DNA prod-ucts corresponding to later steps of reverse transcription (first-and second-str(first-and switches) were undetectable after Myr1E infection (data not shown). These findings led us to investigate whether there was an intrinsic defect in reverse transcription attributable to the mutant virus particles.
ERT activity in Myr1E virus particles.To address the pos-sibility that the Myr1E mutation had disrupted a structure within the virion needed for reverse transcription, ERT assays were performed. Virus particles from two independent cells lines stably expressing the myr1egenome or cells chronically infected with wild-type virus were permeabilized with melittin and incubated with deoxyribonucleotides. Viral DNA was iso-lated from the particles and analyzed by PCR using primers to detect the earliest products in the reverse transcription reac-tion, the minus-strand strong-stop DNA. As shown in Fig. 6A, approximately equivalent levels of strong-stop DNA were syn-thesized in virus particles from both clones of Myr1E and the wild-type virus. To allow more quantitative comparisons among samples, limiting serial dilutions of the viral DNAs from the ERT reactions were subjected to PCR with primers
that amplify minus-strand strong-stop DNA and products of the first- and second-strand transfer reactions. In each step, similar amounts of viral DNA were detected for the mutants and wild type (Fig. 6B). When the same population of virus particles was analyzed for ERT activity using radioactively labeled nucleotides, a comparable result was obtained (data not shown).
The ERT findings demonstrate that there is no intrinsic defect in RT activity, incorporation and positioning of the primer tRNA, or integrity of the genomic RNA that alters in vitro viral DNA synthesis for the Myr1E mutant. That is to say, under in vitro conditions within the environment of the virus particle, the genomic RNA serves as an efficient template for reverse transcription. This finding was surprising given the paucity of proviral DNA synthesis that occurs after infection with Myr1E particles. The discrepancy between the normal ERT activity and the apparent block in initiation of reverse transcription after infection led us to further investigate the stability of the secondary structure of the genomic RNA.
[image:6.612.313.550.74.294.2]Analysis of genomic RNA secondary structure. To deter-mine whether the Myr1E genome was normal with respect to the formation of dimers and the stability of the dimer complex, RNA was isolated from equivalent amounts of particles pro-duced by cells transiently transfected with mutant or wild-type proviral DNA (25). The RNA was electrophoresed under non-denaturing conditions prior to Northern analysis using a radio-actively labeled gag riboprobe. Under these electrophoretic conditions, the majority of the wild-type genomic RNA mi-grates as a high-molecular-weight complex containing nonco-valently bound dimers (Fig. 7A). Strikingly, viral RNA purified from Myr1E particles was almost entirely monomeric (Fig. 7A). Very little, if any, RNA was seen at the position expected FIG. 5. Detection of viral DNA after infection. (A) Virus particles produced from stably expressing cell lines were normalized, concentrated, and used to infect QT6 cells. Low-molecular-weight DNA was isolated and used as a tem-plate in PCRs with primers specific for minus-strand strong-stop DNA as de-scribed in Materials and Methods. In both panels, molecular size markers were run in the right lane (ladder). The PCR product amplified from minus-strand strong-stop DNA is predicted to be 86 bp in length with our primers. cl., clone. (B) Quantitative analysis of strong-stop DNA. Low-molecular-weight DNA iso-lated as described for panel A was serially diluted as indicated and amplified as described above. Virus particles used for infection were derived from indepen-dently derived cell lines (clones 1 and 2) stably expressing themyr1egenome.
VOL. 74, 2000 RNA DIMERIZATION DEFECT IN AN RSV MA MUTANT 169
on November 9, 2019 by guest
http://jvi.asm.org/
for dimeric RNA. Virus particles isolated from cell lines stably expressing mutant or wild-type virus were also examined for genomic RNA content (Fig. 7B). Again, the wild-type viral RNA was dimeric, and genomic RNAs from both Myr1E cell lines appeared as monomers (Fig. 7B, right). In contrast, Myr1E⫺ particles contained RNA in dimeric form that mi-grated at the same position as wild-type dimers (Fig. 7B, left). This is the expected result given that Myr1E⫺ has normal infectivity, and dimerization is believed to be critical for infec-tivity. In some experiments there is dramatically less total RNA visible in the Myr1E lanes of nondenaturing Northern blots compared to wild-type virus even though slot blot analysis reveals only a slight difference, suggesting that the Myr1E monomeric RNA is more susceptible to degradation during extraction than is the wild-type RNA.
DISCUSSION
In this report, we have characterized a mutant, called Myr1E, which efficiently produces virus particles that are de-fective in viral DNA synthesis following infection despite being normal in reverse transcription measured in an ERT assay. The lack of dimers in the RNA isolated from Myr1E particles raises interesting questions about the possible role of the MA sequence in viral RNA dimerization, the requirement for
dimerization prior to RNA packaging, and the ability of RT to transcribe monomeric RNA.
From electron microscopy images, the point of RNA dimer linkage in the RSV genome has been described as an approx-imately 50-nt sequence positioned around nt 511 as reported by one group (35) or around nt 466 (42). Other dimer-promot-ing sequences have been identified at nt 208 to 270 and 400 to 600 (6, 21, 35). An autocomplementary sequence at nt 258 to 274 identified by in vitro studies appears to contribute to dimerization of avian leukosis virus RNA (21), although the importance for this sequence in vivo has not been ascertained. The insertion mutation that gives rise to Myr1E begins imme-diately upstream of the normalgaginitiation site, which is at nt 380. Although the insertion lies between the regions known to be important for dimer formation in vivo, it does not directly overlap any of these sequences. In addition, there are two other reasons why it seems unlikely that the interference with dimerization resides entirely at the level of RNA structure. First, the myristate-lacking Myr1E⫺makes dimeric RNA and is fully infectious yet differs from noninfectious Myr1E at only one nucleotide (codon 2, GGA to GAA). It is unlikely that such a minor change would so severely interfere with dimer-ization. Second, we have found other, unrelated RSV MA mutants that also have altered dimerization (to be presented elsewhere). However, it remains of interest to determine whether the single nucleotide difference between Myr1E and Myr1E⫺could indeed have aciseffect on dimerization, be-cause this region has not been previously implicated in con-tributing to the dimer linkage structure.
[image:7.612.76.268.70.388.2]If it is not the RNA sequence ofmyr1ethat disrupts dimer formation, then the MA protein itself might influence dimer-ization intrans. The strikingly different phenotypes of Myr1E and Myr1E⫺provide insight into how MA might be involved. The two mutant proteins differ only at the second residue (Glu-to-Gly), but this difference allows myristylation of the Myr1E protein. Because the myristylated Src sequence is known to be a strong membrane-binding signal, the Myr1E MA protein might be tightly attached to the viral membrane, making MA unavailable for involvement in RNA dimerization after budding and virus maturation. Further support for this hypothesis is provided by earlier reports that the RSV MA protein has nucleic acid-binding properties (52). Although it has not been shown to specifically bind viral RNA with high FIG. 6. ERT activity. Permeabilized visions were incubated with nucleotides,
[image:7.612.315.550.73.226.2]and viral DNA was isolated and subjected to PCR analysis as for Fig. 5. (A) Intravirion minus-strand strong-stop DNA for particles isolated from stable cell lines. Molecular size markers were run in the right lane (ladder). (B) Serial 10-fold dilutions of ERT products to amplify sequences corresponding to minus-strand strong-stop (86-bp), first-minus-strand switch (117-bp), and second-minus-strand switch (300-bp) DNA products.
FIG. 7. Nondenaturing Northern blot analysis of viral RNA. Genomic RNA was isolated from virus particles produced by transient transfection of proviral plasmids (A) or from two independently arising stably producing cell lines (clones [cl.] 1 and 2) (B). The positions of dimers (D) and monomers (M) are indicated by arrows.
on November 9, 2019 by guest
http://jvi.asm.org/
affinity, within the confines of a virus particle, the concentra-tion of MA may be high enough to allow MA to play a role in dimerization. The tighter-than-normal membrane-binding hy-pothesis also suggests that MA needs to be in close proximity to the genomic RNA for it to have a direct effect on dimer formation; however, for RSV it is unknown whether any MA molecules actually associate with the viral core. Perhaps just a few MA molecules might be required to influence dimerization of the RNA because the area of dimer linkage is only a small region of the genome. Small amounts of MA protein could be very difficult to detect within the ribonucleoprotein complex in the interior of the virion.
Our findings in no way suggest a role for the RSV MA domain in viral RNA packaging (i.e., the specific selection of viral RNA for incorporation into virions). Earlier studies have shown that MA is not needed for RNA incorporation and that deleting the N-terminal half of MA results in a minimal reduc-tion in the amount of genomic RNA incorporated into virus-like particles (less than a 25% decrease compared with Myr1) (50). However, dimerization of the RNA was not examined in that study.
One interesting interpretation of our data is that Myr1E packages a lower amount of RNA because it incorporates monomers rather than dimers. If so, then the processes of dimerization and RNA packaging would have to be separable functions. We hasten to point out that our data do not rule out the possibility that dimers are initially packaged into particles but are unstable due to failure of the dimers to mature into stable structures. This has been proposed as the explanation for rapidly harvested virus, NC mutants, and protease-deficient viruses that contain dimeric RNA with increased electro-phoretic mobility and decreased thermostability (9, 19, 24–26, 40). However, in contrast to those studies, we did not detect any dimers for Myr1E under standard low-temperature RNA extraction conditions identical to those used by others (25).
If indeed only monomers are present in Myr1E particles, then the efficient ERT activity suggests that dimerization might not be required for reverse transcription. However, we cannot rule out that the RNA within Myr1E particles is dimeric under the gentle permeabilizing conditions of the ERT reaction, even though only monomers are seen following extraction of the viral RNA. Likewise, it is puzzling that reverse transcription is so severely impaired after infection. These findings suggests two main alternative conclusions: (i) the Myr1E viral RNA is able to be reverse transcribed in the protected milieu of the virion but not in the environment of the cell, perhaps due to instability of the dimeric structure; or (ii) the problem with dimer formation has nothing to do with the inability of Myr1E to perform reverse transcription after infection; rather, the Myr1E MA protein might be unable to participate in a differ-ent step early in infection. We hope to be able to distinguish between these possibilities by examining other noninfectious mutants involving the MA sequence for dimer formation and postentry RT activity.
The interpretations above have implied an active role for MA at a step after budding; however, it is important to point out an alternative and equally plausible model. It could be that the wild-type MA protein is not directly involved in dimer formation or stability or in the initiation of proviral synthesis after infection, but rather the altered and possibly misshapen Myr1E MA protein interferes with these functions, which are performed by other viral factors. A defect in genomic RNA dimerization in Myr1E does not necessarily mean that the normal MA protein provides this function in the wild-type virus.
On the other hand, we do find it compelling that there is
remarkable similarity in the three-dimensional structures of all retroviral MA proteins, each having four major alpha helices separated by flexible loops, with the first two helices overlap-ping the second two (14, 28, 38, 39). The structures of these evolutionarily diverse MA proteins are conserved even though the mechanisms that they use for membrane binding are not (e.g., the requirements for myristate and the clustering of basic residues differ for RSV and HIV) (55, 63). This observation suggests that there are other shared and essential functions among MA proteins of different viruses. The conserved MA structure might have a role during another step in replication, perhaps one involved in RNA dimerization or efficient proviral DNA synthesis.
ACKNOWLEDGMENTS
We thank Tim Nelle for contributing the S106A mutants and other unpublished results.
Support for this research was provided by The Pennsylvania State University College of Medicine and by grants from the NIH to L.J.P. (RO1 CA76534 and K11 AI01148) and J.W.W. (RO1 CA47482) and from the American Cancer Society to J.W.W. (FRA-427) and L.J.P. (IRG-196A).
REFERENCES
1.Aronoff, R., A. M. Hajjar, and M. L. Linial.1993. Avian retroviral encapsi-dation: reexamination of functional 5⬘RNA sequences and the role of the nucleocapsid Cys-His motifs. J. Virol.67:178–188.
2.Aronoff, R., and M. L. Linial.1991. Specificity of retroviral packaging. J. Vi-rol.65:71–80.
3.Bennett, R. B., and J. W. Wills.1999. Conditions for copackaging Rous sarcoma virus and murine leukemia virus Gag proteins during retroviral budding. J. Virol.73:2045–2051.
4.Bennett, R. P., T. D. Nelle, and J. W. Wills.1993. Functional chimeras of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Vi-rol.67:6487–6498.
5.Berkowitz, R., J. Fisher, and S. P. Goff.1996. RNA packaging, p. 177–218.In H.-G. Krausslich (ed.), Morphogenesis and maturation of retroviruses. Springer-Verlag, Berlin, Germany.
6.Bieth, E., C. Gabus, and J.-L. Darlix.1990. A study of the dimer formation of Rous sarcoma virus RNA and its effect on viral protein synthesis in vitro. Nucleic Acids Res.18:119–127.
7.Boone, L. R., and A. Skalka.1980. Two species of full-length cDNA are synthesized in high yield by melittin-treated avian retrovirus particles. Proc. Natl. Acad. Sci. USA77:847–851.
8.Boone, L. R., and A. M. Skalka.1981. Viral DNA synthesis in vitro by avian retrovirus particles permeabilized with melittin. J. Virol.37:109–116. 9.Bowles, N. E., P. Damay, and P.-F. Spahr.1993. Effects of rearrangements
and duplications of the Cys-His motifs of Rous sarcoma virus nucleocapsid protein. J. Virol.67:623–631.
10. Buss, J. E., M. P. Kamps, and B. M. Sefton.1984. Myristic acid is attached to the transforming protein of Rous sarcoma virus during or immediately after synthesis and is present in both soluble and membrane-bound forms of the protein. Mol. Cell. Biol.4:2697–2704.
11. Cannon, P. M., S. Matthews, N. Clark, E. D. Byles, O. Iourin, D. J. Hockley, S. Kingsman, and A. J. Kingsman.1997. Structure-function studies of the human immunodeficiency virus type 1 matrix protein, p17. J. Virol.71:3474– 3483.
12. Carriere, C., B. Gay, N. Chazal, N. Morin, and P. Boulanger.1995. Sequence requirements for encapsidation of deletion mutants and chimeras of human immunodeficiency virus type 1 Gag precursor into retrovirus-like particles. J. Virol.69:2366–2377.
13. Casella, C. R., L. J. Raffinni, and A. T. Panganiban.1997. Pleotropic mu-tations in the HIV-1 matrix protein that affect diverse steps in replication. Virology228:294–306.
14. Christensen, A. M., M. A. Massiah, B. G. Turner, W. I. Sundquist, and M. F. Summers.1996. Three-dimensional structure of the HTLV-II matrix protein and comparative analysis of matrix proteins from the different classes of pathogenic human retroviruses. J. Mol. Biol.264:1117–1131.
15. Craven, R. C., A. E. Leure-duPree, R. A. Weldon, and J. W. Wills.1995. Genetic analysis of the major homology region of the Rous sarcoma virus Gag protein. J. Virol.69:4213–4227.
16. Crawford, S., and S. P. Goff.1984. Mutations ingagproteins P12 and P15 of Moloney leukemia virus block early stages of infection. J. Virol.49:909–917. 17. Cross, F. R., E. A. Garber, D. Pellman, and H. Hanafusa.1984. A short sequence in the p60srcN terminus is required for p60srcmyristylation and
membrane association and for cell transformation. Mol. Cell. Biol.4:1834– 1842.
VOL. 74, 2000 RNA DIMERIZATION DEFECT IN AN RSV MA MUTANT 171
on November 9, 2019 by guest
http://jvi.asm.org/
18.Dong, J., J. W. Dubay, L. G. Perez, and E. Hunter.1992. Mutations within the proteolytic cleavage site of the Rous sarcoma virus glycoprotein define a requirement for dibasic residues for intracellular cleavage. J. Virol.66:865– 874.
19.Dupraz, P., S. Oertle, C. Meric, P. Damay, and P. F. Spahr.1990. Point mutations in the proximal Cys-His box of Rous sarcoma virus nucleocapsid protein. J. Virol.64:4978–4987.
20.Erdie, C. R., and J. W. Wills.1990. Myristylation of Rous sarcoma virus Gag protein does not prevent replication in avian cells. J. Virol.64:5204–5208. 21. Fosse, P., N. Motte, A. Roumier, C. Gabus, D. Muriaux, J. L. Darlix, and J.
Paoletti.1996. A short autocomplementary sequence plays an essential role in avian sarcoma-leukosis virus RNA dimerization. Biochemistry35:16601– 16609.
22. Franke, E. K., H. E. H. Yuan, K. L. Bossolt, S. P. Goff, and J. Luban.1994. Specificity and sequence requirements for interactions between various ret-roviral Gag proteins. J. Virol.68:5300–5305.
23. Freed, E. O., G. Englund, and M. A. Martin.1995. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol.69:3949–3954.
24. Fu, W., R. Gorelick, and A. Rein.1994. Characterization of human immu-nodeficiency virus type 1 dimeric RNA from wild-type and protease-deficient virions. J. Virol.68:5013–5018.
25. Fu, W., and A. Rein.1993. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol.67:5443–5449.
26. Gorelick, R., D. J. Chabot, A. Rein, L. E. Henderson, and L. O. Arthur.1993. The two zinc fingers in the human immunodeficiency virus type 1 nucleo-capsid protein are not functionally equivalent. J. Virol.67:4027–4036. 27. Gottlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine.1991.
Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA88:3195–3199.
28. Hill, C. P., D. Worthylake, D. P. Bancroft, A. M. Christensen, and W. I. Sundquist.1996. Crystal structure of the trimeric human immunodeficiency virus type 1 matrix protein. Proc. Natl. Acad. Sci. USA93:3099–3104. 29. Hirt, B. 1967. Selective extraction of polyomavirus DNA from infected
mouse cell cultures. J. Mol. Biol.26:365–369.
30. Kamps, M. P., J. E. Buss, and B. M. Sefton.1985. Mutation of NH2-terminal glycine of p60src prevents both myristoylation and morphological transfor-mation. Proc. Natl. Acad. Sci. USA14:4625–4628.
31. Katz, R. A., R. W. Terry, and A. Skalka.1986. A conserved cis-acting sequence in the 5⬘ leader of avian sarcoma virus RNA is required for packaging. J. Virol.59:163–167.
32. Kiernan, R. E., A. Ono, G. Englund, and E. O. Freed.1998. Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J. Virol.72:4116–4126.
33. Kunkel, T. A., K. Bebenek, and J. McClary.1991. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol.204:125–139. 34. Lai, M. M.1976. Phosphoproteins of Rous sarcoma viruses. Virology74:
287–301.
35. Lear, A. L., M. Haddrick, and S. Heapy.1995. A study of the dimerization of Rous sarcoma virus RNA in vitro and in vivo. Virology212:47–57. 36. Lee, T., X. B. Tang, L. M. Cimakasky, J. E. Hildreth, and X. F. Yu.1997.
Mutations in the matrix protein of human immunodeficiency virus type 1 inhibit surface expression and virion incorporation of viral envelope glyco-proteins in CD4⫹T lymphocytes. J. Virol.71:1443–1452.
37. Mammano, F., E. Kondo, J. G. Sodroski, A. Bukovsky, and H. G. Gottlinger.
1995. Rescue of human immunodeficiency virus type 1 matrix mutants by envelope glycoproteins with short cytoplasmic tails. J. Virol.69:3824–3830. 38. Matthews, S., M. Mikhailov, A. Burny, and P. Roy.1996. The solution structure of the bovine leukaemia virus matrix protein and similarity with lentiviral matrix proteins. EMBO J.15:3267–3274.
39. McDonnell, J. M., C. B. Wilson, W. Zhou, A. Wolven, T. D. Nelle, J. W. Wills, M. D. Resh, and D. Cowburn.1998. Solution structure and dynamics of the bioactive retroviral M domain from Rous sarcoma virus. J. Mol. Biol.279:
921–928.
40. Meric, C., E. Gouilloud, and P.-F. Spahr.1988. Mutations in Rous sarcoma virus nucleocapsid protein p12 (NC): deletions of Cys-His boxes. J. Virol.
62:3328–3333.
41. Moscovici, C., M. G. Moscovici, H. Jimenez, M. M. C. Lai, M. J. Hayman,
and P. K. Vogt.1977. Continuous tissue culture cell lines derived from chemically induced tumors of Japanese quail. Cell11:95–103.
42. Murti, K. G., M. Boundurant, and A. Tereba.1981. Secondary structural features in the 70S RNAs of Moloney murine leukemia virus and Rous sarcoma virus as observed by electron microscopy. J. Virol.37:411–419. 43. Nelle, T. D., M. F. Verderame, J. Leis, and J. W. Wills.1998. The major site
of phosphorylation within the Rous sarcoma virus MA protein is not re-quired for replication. J. Virol.72:1103–1107.
44. Nelle, T. D., and J. W. Wills.1996. A large region within the Rous sarcoma virus matrix protein dispensable for budding and infectivity. J. Virol.70:
2269–2276.
45. Parent, L. J., C. B. Wilson, M. D. Resh, and J. W. Wills.1996. Evidence for a second function of the MA sequence in the Rous sarcoma virus Gag protein. J. Virol.70:1016–1026.
46. Pellman, D., E. A. Garber, F. R. Cross, and H. Hanafusa.1985. An N-terminal peptide from p60srccan direct myristylation and plasma membrane
localization when fused to heterologous proteins. Nature (London)346:84– 86.
47. Perez, L. G., G. L. Davis, and E. Hunter.1987. Mutants of the Rous sarcoma virus envelope glycoprotein that lack the transmembrane anchor and cyto-plasmic domains: analysis of intracellular transport and assembly into viri-ons. J. Virol.61:2981–2988.
48. Reicin, A., A. Ohagen, L. Yin, S. Hoglund, and S. P. Goff.1996. The role of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps in the replication cycle. J. Virol.70:8645–8652.
49. Resh, M. D., and H. P. Ling.1990. Identification of a 32K plasma membrane protein that binds to the myristylated amino-terminal sequence of p60v-src. Nature (London)346:84–86.
50. Sakalian, M., J. W. Wills, and V. M. Vogt.1994. Efficiency and selectivity of RNA packaging by Rous sarcoma virus Gag deletion mutants. J. Virol.
68:5969–5981.
51. Sorge, J., W. Ricci, and S. H. Hughes.1983.cis-acting RNA packaging locus in the 115-nucleotide direct repeat of Rous sarcoma virus. J. Virol.48:667– 675.
52. Steeg, C. M., and V. M. Vogt.1990. RNA-binding properties of the matrix protein (p19gag) of avian sarcoma and leukemia viruses. J. Virol.64:847–855.
53. Stoltzfus, C. M., and P. N. Snyder.1975. Structure of B77 sarcoma virus RNA: stabilization of RNA after packaging. J. Virol.16:1161–1170. 54. Swanstrom, R., and J. W. Wills.1997. Synthesis, assembly, and processing of
retroviral proteins, p. 263–334.InJ. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
55. Verderame, M. F., T. D. Nelle, and J. W. Wills.1996. The membrane-binding domain of the Rous sarcoma virus Gag protein. J. Virol.70:2664–2668. 56. Weldon, R. A., Jr., C. R. Erdie, M. G. Oliver, and J. W. Wills.1990.
Incor-poration of chimeric Gag protein into retroviral particles. J. Virol.64:4169– 4179.
57. Weldon, R. A., Jr., and J. W. Wills.1993. Characterization of a small (25-kilodalton) derivative of the Rous sarcoma virus Gag protein competent for particle release. J. Virol.67:5550–5561.
58. Wills, J. W., C. E. Cameron, C. B. Wilson, Y. Xiang, R. P. Bennett, and J. Leis.1994. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol.68:6605–6618.
59. Wills, J. W., and R. C. Craven.1991. Form, function, and use of retroviral Gag proteins. AIDS5:639–654.
60. Wills, J. W., R. C. Craven, and J. A. Achacoso.1989. Creation and expression of myristylated forms of Rous sarcoma virus Gag protein in mammalian cells. J. Virol.63:4331–4343.
61. Young, J. A. T., P. Bates, K. Willert, and H. E. Varmus.1990. Efficient incorporation of human CD4 protein into avian leukosis virus particles. Science250:1421–1423.
62. Yu, X., X. Yuan, Z. Matsuda, T.-H. Lee, and M. Essex.1992. The matrix protein of human immunodeficiency virus type 1 is required for incorpora-tion of viral enveloped protein into mature virions. J. Virol.66:4966–4971. 63. Zhou, W., L. J. Parent, J. W. Wills, and M. D. Resh.1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phos-pholipids. J. Virol.68:2556–2569.