J
OURNAL OFV
IROLOGY,
0022-538X/00/$04.00⫹0
Oct. 2000, p. 9115–9124
Vol. 74, No. 19
Copyright © 2000, American Society for Microbiology. All Rights Reserved.
The LMP2A ITAM Is Essential for Providing B Cells with
Development and Survival Signals In Vivo
MARK MERCHANT, ROBERT G. CALDWELL,
ANDRICHARD LONGNECKER*
Department of Microbiology-Immunology, Northwestern University Medical School, Chicago, Illinois 60611
Received 23 March 2000/Accepted 5 July 2000
In Epstein-Barr virus-transformed B cells, known as lymphoblastoid cell lines (LCLs), LMP2A binds the
tyrosine kinases Syk and Lyn, blocking B-cell receptor (BCR) signaling and viral lytic replication. SH2
domains in Syk mediate binding to a phosphorylated immunoreceptor tyrosine-based activation motif (ITAM)
in LMP2A. Mutation of the LMP2A ITAM in LCLs eliminates Syk binding and allows for full BCR signaling,
thereby delineating the significance of the LMP2A-Syk interaction. In transgenic mice, LMP2A causes a
developmental alteration characterized by a block in surface immunoglobulin rearrangement resulting in
BCR-negative B cells. Normally B cells lacking cognate BCR are rapidly apoptosed; however, LMP2A
trans-genic B cells develop and survive without a BCR. When bred into the recombinase activating gene 1 null
(RAG
ⴚ/ⴚ) background, all LMP2A transgenic lines produce BCR-negative B cells that develop and survive in
the periphery. These data indicate that LMP2A imparts developmental and survival signals to B cells in vivo.
In this study, LMP2A ITAM mutant transgenic mice were generated to investigate whether the LMP2A ITAM
is essential for the survival phenotype in vivo. LMP2A ITAM mutant B cells develop normally, although
transgene expression is comparable to that in previously described nonmutated LMP2A transgenic B cells.
Additionally, LMP2A ITAM mutant mice are unable to promote B-cell development or survival when bred into
the RAG
ⴚ/ⴚbackground or when grown in methylcellulose containing interleukin-7. These data demonstrate
that the LMP2A ITAM is required for LMP2A-mediated developmental and survival signals in vivo.
Epstein-Barr virus (EBV) is a ubiquitous human oncogenic
herpesvirus associated with a number of proliferative diseases.
In lymphoid cell types, EBV causes infectious mononucleosis
in naive adolescents and is associated with African Burkitt’s
lymphoma, Hodgkin’s lymphoma, and adult T-cell leukemia
(40, 49). Furthermore, individuals with congenital or immune
deficiencies often develop EBV-associated
lymphoprolifera-tive diseases (66, 81). In the oral epithelia, EBV is associated
with nasopharyngeal carcinoma and oral hairy leukoplakia (4,
35, 63, 71). EBV has also been found in smooth muscle tumors
and in assorted lung, breast, gastric, and salivary carcinomas;
however, it is unclear what role the virus plays in these cancers
(2, 3, 11, 27, 31, 32, 43, 45, 64).
The ability for the virus to remain latent within host B cells
is a hallmark of EBV infection. During latency, EBV expresses
a set of nine well-studied viral proteins. Six proteins are termed
Epstein-Barr nuclear antigens (EBNAs), which include
EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and
EBNALP (40, 49). The remaining three proteins are the latent
membrane proteins 1 (LMP1), LMP2A, and LMP2B (40). B
cells transformed by EBV in vitro, termed lymphoblastoid cell
lines (LCLs), express all of these proteins, whereas most
EBV-related malignancies express only EBNA1, LMP1, and LMP2A
(65). In contrast to these cell types, primary B cells from
healthy individuals latently infected with EBV express a very
limited number of viral proteins, with LMP2A being the most
consistently detected transcript (1, 12, 20, 61, 74). While
EBNA1 has been shown to be necessary for maintaining the
viral episome (40, 80), LMP2A is thought to be important for
maintaining viral latency but not viral transformation (22, 48,
52–55, 57–59).
LMP2A consists of a long amino-terminal tail, 12
mem-brane-spanning domains, and a short carboxy-terminal tail
which aggregates in patches on the surface of latently infected
cells (22, 23, 50, 51, 58, 68). The amino-terminal tail of LMP2A
contains eight constitutively phosphorylated tyrosine residues
and several proline-rich regions which are critical for the
abil-ity of LMP2A to interact with cellular proteins (22, 24, 25,
57–59). Phosphorylation at tyrosine 112 is important for
asso-ciation between LMP2A and the protein tyrosine kinase (PTK)
Lyn (25). Similarly, phosphorylation at tyrosines 74 and 85 of
LMP2A, two key regulatory residues in the immunoreceptor
tyrosine-based activation motif (ITAM) of LMP2A, allows for
association and activation of the Syk PTK (24). Recent
evi-dence has suggested that ubiquitin ligases, such as Nedd4 and
AIP4, associate with the proline-rich PY motifs within LMP2A
in a phosphorylation-independent manner (37). These
LMP2A-cellular protein associations enable LMP2A to block
B-cell antigen receptor (BCR) cross-linking in LCLs, leading
to a model in which LMP2A maintains both B-cell quiescence
and viral latency.
To establish LMP2A as being critical for maintaining EBV
latency in vivo, we generated transgenic mice which express
LMP2A downstream of the
immunoglobulin (Ig)
heavy-chain enhancer and promoter (E
) (6, 7). Analysis of several
lines of these E
LMP2A transgenic mice has revealed that two
distinct LMP2A in vivo phenotypes exist. Two lines of mice
display an LMP2A
BCR⫺phenotype, best characterized by
transgenic line E (TgE), while three independent lines of
an-imals display an LMP2A
BCR⫹phenotype, best typified by
transgenic line 6 (Tg6). E
LMP2A
BCR⫺transgenic lines have
been shown by quantitative PCR to express more
LMP2A-specific message than E
LMP2A
BCR⫹transgenic lines.
How-ever, since there are no data addressing the timing of transgene
expression in each line during the B-cell developmental time
line, these animals are currently considered to represent two
distinct phenotypic variants. E
LMP2A
BCR⫺transgenic mice
* Corresponding author. Mailing address: Department of
Microbi-ology-Immunology, Northwestern University Medical School, 303 E.
Chicago Ave., Chicago, IL 60611. Phone: (312) 503-0467. Fax: (312)
503-1339. E-mail: r-longnecker@nwu.edu.
9115
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 1. Generation of LMP2A ITAM mutant transgenic mice. (A) LMP2A ITAM mutant transgene construct. The Eheavy-chain enhancer/promoter (wavy lines) was fused to the LMP2A ITAM mutant transgene containing both LMP2A cDNA sequences (striped lines) and EBV genomic sequences (stippled). The DNA, protein, and amino acid number (AA #) for the altered ITAM sequences are shown in the shaded inset at the bottom. A silent point mutation creates aKpnI site in the LMP2A ITAM mutant transgene, which aids in distinguishing ITAM mutant mice from LMP2A transgenic mice by PCR using OL129 and OL131 (upper inset) followed by
on November 9, 2019 by guest
http://jvi.asm.org/
display a B-cell developmental alteration characterized by a
bypass of Ig heavy-chain rearrangement resulting in an overall
loss of surface Ig (sIg) expression. PCR amplification of Ig
gene rearrangements in E
LMP2A
BCR⫺transgenic bone
mar-row cells demonstrated that rearrangement of the D-J
Hheavy
chain and V-J
Klight-chain genes occurs correctly; however,
there is a failure to complete the final V-DJ
Hrearrangement of
the heavy chain. B cells that lose a functioning BCR have been
shown to rapidly undergo apoptosis (44). Nonetheless,
E
LMP2A
BCR⫺B cells are capable of exiting the bone
mar-row and persisting in the periphery. E
LMP2A
BCR⫹lines
display a less prominent phenotype consisting of slight decreases
in mature B-cell numbers, while the resulting B cells appear
largely normal. When bred into a RAG-1-deficient (RAG
⫺/⫺)
background, both E
LMP2A
BCR⫺and E
LMP2A
BCR⫹lines
are capable of producing B cells that exit the bone marrow and
persist in the periphery. These observations were supported by
the growth of RAG
⫺/⫺E
LMP2A bone marrow cells in
meth-ylcellulose containing interleukin-7 (IL-7). These data indicate
that LMP2A drives B-cell development and survival in the
absence of normal BCR signals.
In this study, we used an LMP2A ITAM mutant, encoding
mutations of tyrosines 74 and 85 to phenylalanines [Y(74/
85)F], to create E
LMP2A
⌬
ITAM transgenic mice.
Genera-tion of these mice allows for investigaGenera-tion of whether the in
vivo developmental and survival signals observed in LMP2A
transgenic mice derive from the LMP2A ITAM. These mice
show no B-cell developmental alteration or survival phenotype
in any B-cell compartment. Furthermore, this transgene is
in-capable of driving B-cell development or survival when bred
into the RAG
⫺/⫺background in vivo or when grown in
meth-ylcellulose in vitro. These data show that LMP2A must have a
functional ITAM in order to enhance B-cell development and
survival. These results suggest that Syk activation by a
phos-phorylated ITAM is required for LMP2A to provide
develop-mental and survival signals in vivo.
MATERIALS AND METHODS
Mice.Constructions of the ELMP2A transgene and LMP2A ITAM mutant [Y(74/85)F] have been previously described (7, 24). The LMP2A⌬ITAM mutant transgene containing the double mutation Y(74/85)F was constructed as follows. TheEcoRI-SalI DNA fragment from pRH38, coding for the Y(74/85)F mutant amino-terminal tail of LMP2A, was cloned intoMunI-SalI-digested pRL202, which codes for the Eenhancer and promoter transgene construct, to generate pRG3. TheMscI-XhoI fragment from pRL209, which codes for the remainder of the LMP2A transgene, was inserted intoMscI-XhoI-digested pRG3 to generate pRG4. TheSacI-XhoI fragment of pRG4 containing the Epromoter/enhancer and LMP2A⌬ITAM transgene was used for standard microinjection into (B6⫻
CBA)F1single-cell fertilized eggs as described elsewhere (36). The resulting
offspring were screened by PCR and Southern blotting for the presence of LMP2A. The identified founders were bred to C57BL/6 or RAG⫺/⫺animals in the BL/6 background for all described experiments. The animals were housed at the Northwestern University Center for Experimental Animal Resources in accordance with university animal welfare guidelines. Wild-type C57BL/6 and RAG⫺/⫺animals were obtained from Jackson Laboratories.
Tail DNA isolation.DNA was prepared as previously described (6, 7). Briefly, a 1-cm piece of tail was digested in a 1.5-ml microcentrifuge tube overnight in 500
l of tail lysis buffer (100 mM Tris [pH 8], 100 mM NaCl, 5 mM EDTA, 0.2% sodium dodecyl sulfate [SDS], 100g of proteinase K/ml) at 55°C. Digested tail lysates were vortexed and centrifuged for 10 min, and supernatants were trans-ferred to a new 1.5-ml microcentrifuge tube. Supernatants were precipitated with 2 volumes of 95% ethanol and centrifuged at 4°C for 20 min; DNA pellets were air dried and resuspended in 200l of Tris-EDTA.
Southern hybridization.Ten micrograms of mouse genomic tail DNA was digested overnight at 37°C withBamHI (1⫻ NEB [New England Biolabs] BamHI buffer, 1⫻bovine serum albumin [BSA], 20 U ofBamHI [NEB]) and electrophoresed on a 0.8% agarose gel. The gel was then treated sequentially with 0.25 N HCl for 10 min and 0.4 M NaOH for 30 min followed by transfer of the gel onto GeneScreen Plus (NEN) using a vacuum blotter (Appligene). LMP2A probe was generated by digesting the LMP2A cDNA clone pRL34 with EcoRI, releasing the 2.0-kb cDNA in its entirety, followed by gel purification, ethanol precipitation, and resuspension to 20 ng/l. Then 1.0l (20 ng) of this DNA was labeled with [␣-32P]dCTP (Amersham-Pharmacia) using Ready-to-Go
(-dCTP) labeling beads (Amersham-Pharmacia) followed by a purification step to remove unincorporated [␣-32P]dCTP, using G-50 Probe-Quant spin columns
(Amersham-Pharmacia) according to the manufacturers recommendations. The blot was prehybridized for 1 h at 68°C in 30 ml of hybridization solution (6⫻SSC [0.9 M NaCl, 0.09 M sodium citrate], 1⫻Denhardt’s solution [0.02% BSA, 0.02% Ficoll, 0.02% polyvinylpyrrolidine], 62.5 mM Tris [pH 7.5], 0.05% SDS) with 100
g of denatured herring sperm DNA per ml. Roughly 106cpm of labeled probe
was denatured and added directly to 10 ml of 68°C hybridization solution; the blot was added, and the solution was allowed to hybridize overnight at 68°C. Following hybridization, the blot was washed according to the following regimen: twice with 2⫻SSC at room temperature, twice with 2⫻SSC–1% SDS, and twice with 0.1⫻SSX–1% SDS. The blot was then wrapped in Saran Wrap and exposed to film.
PCR.PCR for the presence of the LMP2A transgene was done as previously described (6, 7). Briefly, each 25.0-l reaction mixture contained 1⫻PCR buffer (Pharmacia), 0.2 mM each deoxynucleoside triphosphate, 1.0 U ofTaq polymer-ase (Pharmacia), 1M each oligonucleotide primer, 1⫻PCR cresol red loading dye (60% sucrose, 5 mM cresol red [Sigma]), and 1.0l of genomic tail DNA. The amplification cycle (15 s at 94°C, 30 s at 58°C, and 75 s at 72°C) was repeated 30 times, followed by a single 10-min extension at 72°C. A unique silentKpnI site was engineered between residues 74 and 85 to distinguish LMP2A ⌬ITAM mutant animals from nonmutated LMP2A transgenic animals (Fig. 1A). LMP2A amino-terminal primers (OL129 and OL131) were designed to amplify the re-gion of LMP2A containing the engineeredKpnI site. These primers were used, in combination with RAG internal control primers, in a PCR followed by over-night digestion withKpnI (1⫻NEB buffer 1, 1⫻BSA, 25.0l of PCR mixture, 1.0 U ofKpnI [NEB]) at 37°C. The oligonucleotide primers not previously described are as follows: LMP2A amino-terminal primer pair (370 bp), OL129 (TGGGTACGATGGCGGAAACAACTC) and OL131 (TGAAACACGAGGC GGCAATAGC); RAG (180 bp), OL107 (TGACTGTGGGAACTGCTGAAC TTT) and OL138 (CCGTGGACGCTAAAACCCAAAGTC); and Neo (280 bp), Neo-2 (CAACAGACAATCGGCTGCTCTGATG) and Neo-3 (CATTGC ATCAGCCATGATGGA).
Protein isolation.Spleens were dissected from animals, homogenized between the frosted edges of two microscope slides, and suspended into 10 ml of ice-cold 1⫻phosphate-buffered saline (PBS). Samples were centrifuged at 1,500 rpm for 10 min, supernatants were poured off, and red blood cells were lysed in 5 ml of red blood cell lysis buffer (150 mM NH4Cl, 17 mM Tris base [pH 7.2]) for 5 min.
[image:3.612.336.523.74.161.2]Cell suspensions were added to 5 ml of ice-cold 1⫻PBS through a nylon mesh. Bone marrow methylcellulose samples were prepared by washing methylcellulose cultures with 10 ml of 1⫻PBS, centrifuging them at 1,500 rpm for 10 min, and resuspending them in 10 ml of 1⫻PBS. Following cell counting, samples were FIG. 2. LMP2A expression in LMP2A ITAM mutant transgenic lines is sim-ilar to that in nonmutated LMP2A transgenic mice. Immunoblot analyses of LMP2A and p85 expression in splenic tissues from Tg⌬ITAM1, Tg⌬ITAM2, WT, TgE, and Tg6 animals are shown. The LMP2A and p85 subunit (PI-3K) bands are indicated by arrows. LMP2A-expressing EBV-transformed LCLs served as a positive control.
KpnI digestion. The LMP2A cDNA probe shown spans sequences throughout the LMP2A transgene.BamHI (B) sites are indicated. TheBamHI site in parentheses may be lost upon integration. (B) PCR/KpnI digest identification of LMP2A ITAM mutant transgenic mice. Tail genomic DNA sequences from LMP2A ITAM mutant, nonmutated LMP2A, and WT littermate control animals were amplified using OL129-OL131 and RAG positive control primers, followed by digestion withKpnI (K) or no enzyme (NE). Identities of the amplified products and their subsequent digested fragments are shown to the left.X DNA digested withHaeIII was used as a size standard (right-most lane). (C) Southern hybridization analysis of LMP2A ITAM mutant lines. Tail DNAs from Tg⌬ITAM1, Tg⌬ITAM2, TgE, and WT animals were digested withBamHI, run on an 0.8% agarose gel, and probed for LMP2A sequences by Southern hybridization. Size standards are indicated at the right.
V
OL. 74, 2000
LMP2A
⌬ITAM TRANSGENIC MICE
9117
on November 9, 2019 by guest
http://jvi.asm.org/
centrifuged at 1,500 rpm for 5 min, and cell pellets were lysed with the addition of enough Triton X lysis buffer (1% Triton X-100, 20 mM Tris [pH 8.0], 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM sodium vanadate, 10 mM sodium fluoride, 1⫻phenylmethylsulfonyl fluoride, 1⫻pepstatin, 1⫻leupeptin) to bring samples to 200⫻106cells/ml for spleen samples or 100⫻106cells/ml for
methylcellulose samples. Lysates were allowed to rotate at 4°C for 15 min before being vortexed and cleared of nonsoluble proteins by centrifugation three times for 20 min. Cleared lysates were mixed 1:1 with 2⫻SDS sample buffer (150 mM Tris-HCl [pH 6.8], 2% SDS, 2% glycerol, 1.43 M-mercaptoethanol) before being loaded.
Immunoblotting.For immunoblot analysis, 2.5⫻106cell equivalents of spleen
cell equivalents and 0.25⫻106cell equivalents of control LCLs were heated at
70°C for 10 min, loaded onto a 1.5 mM 7% polyacrylamide gels, and run at 100 V for 1.5 h. Gels were electroblotted to Immobilon-P (Millipore) according to the manufacturer’s recommendations. Blots were stained with Ponceau S reagent (Sigma) to ensure equal loading and then blocked in 4% milk in 1⫻TBST (10 mM Tris [pH 8.0], 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature. Blots were then washed once for 15 min and twice for 5 min each with 1⫻TBST. Rat monoclonal anti-LMP2A antibody (14B7-1-1; 2.8g/ml) or rabbit anti-phosphatidylinositol 3-kinase (PI3-K) p85 antibody (1g/ml; Upstate Biotech-nology) was diluted in 1% milk–1⫻TBST to 1:2,500 and 1:5,000, respectively, and allowed to incubate with blots overnight at 4°C. Blots were again washed once for 15 min and twice for 5 min each with 1⫻TBST. Horseradish peroxidase (HRP)-conjugated goat anti-rat (Jackson ImmunoResearch Laboratories) or HRP-conjugated sheep anti-rabbit (New England Biolabs) secondary antibodies were diluted in 1⫻TBST to 1:10,000 and 1:5,000, respectively, and added to the appropriate blots for 30 min at room temperature. Blots were washed once for 15 min and four times for 5 min each with 1⫻TBST followed by detection with Western blotting Luminol reagent (Santa Cruz Biotechnology, Inc.) according to manufacturer’s recommendations; the blots were exposed to film.
Flow cytometry.Spleens were dissected from animals, homogenized between the frosted edges of two microscope slides, and suspended into 10 ml of ice-cold FACS (fluorescence-activated cell sorting) buffer (1⫻PBS, 10 mM HEPES, 1% fetal calf serum, 0.01% sodium azide). The femurs and tibia were dissected from mice, and the bone marrow was rinsed out into 15-ml tubes with a total of 10 ml of ice-cold serum-free RPMI 1640 (Gibco). Spleen samples were centrifuged at 1,500 rpm for 10 min, and supernatants were poured off. The splenic red blood cells were lysed in 5 ml of red blood cell lysis buffer (150 mM NH4Cl, 17 mM Tris
base [pH 7.2]) for 5 min and were then added to 5 ml of ice-cold FACS buffer through a nylon mesh. Following cell counting, samples were centrifuged at 1,500 rpm for 5 min, and cells were resuspended to 40⫻106/ml in ice-cold FACS
buffer; 2⫻106cells per spleen stain or 4⫻106cells per bone marrow stain were
used in 100l of antibody stains. Samples were incubated with 0.5g of Fc block (Pharmingen) per stain for 15 min at 4°C, followed by addition of staining antibodies for 30 min at 4°C in the dark. The following antibody dilutions were made in FACS buffer and used in various combinations to stain cells: IgM-fluorescein isothiocyanate (FITC) (1:60) and CD19-phycoerythrin (PE) (1:1,200). Following staining, samples were washed once in 2 ml of 1⫻PBS and
were routinely fixed in 1% paraformaldehyde–1⫻PBS at 4°C overnight in the dark. All antibodies were purchased from Pharmingen. Samples were run on a Becton Dickinson FACScan, and data were analyzed using CellQuest software.
Methylcellulose cultures.Bone marrow cells were collected in serum-free RPMI 1640 as outlined above, counted, centrifuged at 1,500 rpm for 10 min, and resuspended to 10⫻106/ml. A total of 2⫻106bone marrow cells were seeded
in 3 ml of murine preB Methocult containing IL-7 (Stem Cell Technologies). Cells were vortexed for 30 s to evenly distribute cells in the methylcellulose and then evenly plated into two 60-mm-diameter dishes according to the manufac-turer’s recommendations. Cultures were grown for 7 days at 37°C in 5% CO2.
Colonies were photographed and scored for morphology and number before being harvested for use in flow cytometry and immunoblots.
RESULTS
Generation of LMP2A ITAM mutant transgenic mice.
The
E
LMP2A Y(74/85)F mutant ITAM transgene was
con-structed as outlined in Materials and Methods, released from
the parent plasmid by digestion with
Sac
I and
Xho
I, purified,
and microinjected into (B6
⫻
CBA)F
1mouse oocytes. From
these injections, 12 offspring were produced, 2 of which were
found to be transgenic by PCR (see Materials and Methods)
and designated Tg
⌬
ITAM1 and Tg
⌬
ITAM2.
[image:4.612.61.552.73.285.2]Founder DNA was subjected to restriction enzyme and
se-quence analysis to determine whether the newly constructed
transgenic lines contained the proper mutated sequences. The
Y(74/85)F ITAM mutant LMP2A sequence contains an
engi-neered silent point mutation creating a unique
Kpn
I site in the
amino-terminal portion of the transgene (Fig. 1A), enabling
ITAM mutant founders to be distinguished from previously
described nonmutated LMP2A transgenic mice. Genomic tail
DNA from the Tg
⌬
ITAM1 founder, the Tg
⌬
ITAM2 founder,
an LMP2A transgenic line E (TgE) animal, and a wild-type
(WT) littermate control animal were subjected to PCR using
primers OL129 and OL131, amplifying the amino-terminal
portion of LMP2A containing the unique
Kpn
I site. Following
PCR amplification, the reactions were subjected to digestion
with
Kpn
I or an equivalent reaction with no enzyme. DNA
from both Tg
⌬
ITAM1 and Tg
⌬
ITAM2 produced PCR
prod-ucts of 370 bp, equal to those produced in nonmutated LMP2A
transgenic mice (Fig. 1B). When these products were digested
FIG. 3. Tg⌬ITAM1 and Tg⌬ITAM2 mice show no B-cell developmental defect. (A) Spleen (SP) tissues were immunostained with CD19-PE and IgM-FITC
antibodies; flow cytometry dot plots are shown. Two distinct cell types are highlighted with boxes, and the respective percentages of gated cells within those regions are denoted. (B) Bone marrow (BM) cells were immunostained as above; the boxes highlight pre-B and mature B cells, and the respective percentages within those regions are denoted.
on November 9, 2019 by guest
http://jvi.asm.org/
with
Kpn
I, only products from Tg
⌬
ITAM1 and Tg
⌬
ITAM2
were digested to produce two smaller products of 197 and 173
bp; PCR products from TgE remained undigested (Fig. 1B).
These data show that the Tg
⌬
ITAM1 and Tg
⌬
ITAM2 animals
contain the expected
Kpn
I site engineered into the transgene.
Sequence analysis of OL129-OL131 PCR products from each
line confirmed the presence of the expected sequence in each
line (data not shown).
LMP2A sequence was detected in
Bam
HI-digested genomic
tail DNAs from Tg
⌬
ITAM1, Tg
⌬
ITAM2, TgE, and WT
litter-mate control animals by Southern blot analysis (Fig. 1C). Each
transgenic sample hybridizes to LMP2A-specific bands,
indi-cating the presence of the transgenes in each line. These
hy-bridization patterns have been stable for over four generations
(data not shown).
LMP2A is expressed similarly in LMP2A ITAM mutant
transgenic lines and nonmutated LMP2A transgenic mice.
B-cell transgene expression was detected in Tg
⌬
ITAM1 and
Tg
⌬
ITAM2 splenocytes by immunoblot analysis. Whole spleen
lysates from age-matched 4- to 8-week-old Tg
⌬
ITAM1,
Tg
⌬
ITAM2, TgE, Tg6, and WT control animals were
immu-noblotted using an anti-LMP2A antibody (14B7-1-1) and an
antibody against the p85 subunit of PI-3K, to control for
load-ing. Tg
⌬
ITAM1 and Tg
⌬
ITAM2 express LMP2A to similar
levels as both TgE and Tg6 animals (Fig. 2). A higher
nonspe-cific band is consistently seen in all mouse samples tested but
not in the human LCL positive control lane, presumably due to
cross-reactivity of the secondary goat anti-rat HRP-labeled
antibody with mouse proteins. Blots were stripped and
immu-noblotted against PI-3K to show that protein loading was
roughly equivalent in all lanes. No obvious differences in onset
of transgene expression or in the general pattern described
above were observed when expression was tested in young
animals (data not shown).
LMP2A ITAM mutant transgenic mice display no B-cell
developmental alteration.
Spleen and bone marrow samples
from Tg
⌬
ITAM1 and Tg
⌬
ITAM2 were analyzed by flow
cy-tometry to determine whether LMP2A ITAM mutant mice
display a B-cell developmental phenotype similar to that seen
in previously described nonmutated LMP2A lines TgE and
Tg6. Spleen and bone marrow samples from each transgenic
line as well as WT littermate controls were analyzed by flow
cytometry for mouse sIgM (IgM-FITC) and the pan-B-cell
marker CD19 (CD19-PE) (Fig. 3). WT animals displayed
nor-mal levels of CD19
⫹IgM
⫹B cells in the spleen (52%) and
bone marrow (20%). In contrast, TgE animals displayed the
previously reported developmental alteration, characterized by
a severe reduction in the numbers of CD19
⫹IgM
⫹B cells in
the spleen (5%) and bone marrow (1%). Similarly, Tg6
ani-mals showed the previously reported phenotype of slight
de-creases in the numbers of CD19
⫹, IgM
⫹B-cell numbers in the
spleen (30%) and bone marrow (9%). Both Tg
⌬
ITAM1 and
Tg
⌬
ITAM2 mice displayed no B-cell developmental defects
and had no significant differences in the spleen (58 and 50%
versus 52%, respectively) or bone marrow (20 and 19% versus
20%, respectively) compared to WT littermate controls. No
defects were observable in any T-cell compartment in either
LMP2A
⌬
ITAM mutant or nonmutated LMP2A transgenic
mice (data not shown).
LMP2A ITAM mutant mice cannot support development or
survival in the RAG
ⴚ/ⴚbackground.
Previous reports have
divided LMP2A transgenic animals into E
LMP2A
BCR⫺and
E
LMP2A
BCR⫹classes (6, 7). E
LMP2A
BCR⫺transgenic
mice can be clearly distinguished by loss of IgM expression on
their B cells when analyzed by flow cytometry. However, the
E
LMP2A
BCR⫹transgenic B cells appear similar to their WT
littermate controls by the same analysis. When both classes of
LMP2A transgenics are bred into the RAG
⫺/⫺background,
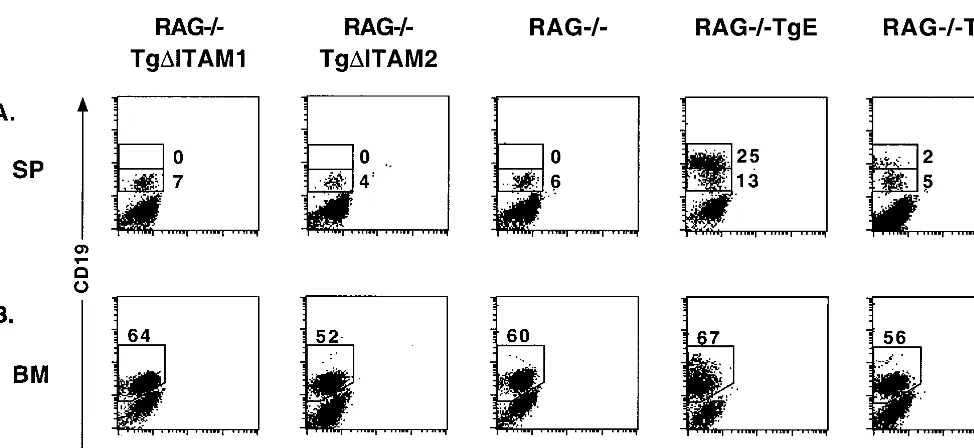
both have a readily observable phenotype. RAG
⫺/⫺animals
are unable to rearrange Ig genes to form Ig, the key protein of
the BCR, thus blocking B-cell development at the pre-B cell
stage. This results in a complete absence of mature B cells in
RAG
⫺/⫺animal bone marrow and peripheral lymphoid
or-gans. When bred into the RAG
⫺/⫺background, both classes of
LMP2A transgenic animals generate a greater number of
CD19
⫹B cells in the bone marrow and peripheral lymphoid
organs. These data indicate that LMP2A imparts a
develop-mental and survival signal in all classes of LMP2A transgenic
animals.
To determine whether the Tg
⌬
ITAM1 and Tg
⌬
ITAM2
pro-teins could provide B cells with developmental or survival
signals in the absence of RAG-1, both lines were bred into the
RAG
⫺/⫺background. Spleen and bone marrow samples were
prepared from RAG
⫺/⫺Tg
⌬
ITAM1, RAG
⫺/⫺Tg
⌬
ITAM2,
RAG
⫺/⫺TgE, RAG
⫺/⫺Tg6, and RAG
⫺/⫺control animals for
analysis by flow cytometry. Both RAG
⫺/⫺TgE and RAG
⫺/⫺Tg6 animals show enhanced survival of B cells, as indicated by
the accumulation of CD19
hi⫹B cells in the spleen (25 and 2%,
respectively) as well as an increase in CD19 expression in the
bone marrow (Fig. 4). In contrast to these results, neither
RAG
⫺/⫺Tg
⌬
ITAM1 nor RAG
⫺/⫺Tg
⌬
ITAM2 animals show
any enhancement of CD19 expression or B-cell survival in
the spleen or bone marrow and are indistinguishable from
RAG
⫺/⫺littermate controls.
LMP2A ITAM mutant transgenic bone marrow cells grow
comparably to WT littermate controls in methylcellulose
con-taining IL-7.
Growth of bone marrow cells in methylcellulose
medium containing IL-7 has allowed for an additional measure
of the developmental effect of LMP2A on B cells. When bone
marrow cells from Tg
⌬
ITAM1, Tg
⌬
ITAM2, TgE, Tg6, and
WT littermates were cultured for 7 days in methylcellulose
medium, each line produced B-cell colonies (Fig. 5A). Cells
collected from methylcellulose cultures from each transgenic
line were subjected to flow cytometry analysis (Fig. 5B). The
B-cell outgrowths from these cultures reflect the general trend
observed in bone marrow samples in vivo. TgE methylcellulose
cultures are predominantly (98%) CD19
⫹IgM
⫺, while Tg6
methylcellulose cultures show a slight reduction in the number
of CD19
⫹IgM
⫹cells (11% versus 30%) and an increase in the
number of CD19
⫹IgM
⫺(88% versus 68%) compared to WT
controls. In contrast, both Tg
⌬
ITAM1 and Tg
⌬
ITAM2
meth-ylcellulose cultures contain similar percentages of CD19
⫹IgM
⫹(31 and 26% versus 30%, respectively) and CD19
⫹IgM
⫺(65 and 70% versus 68%, respectively) cells compared to
WT cultures (Fig. 5B). The number of B-cell colonies formed
in each plate correlates with the strength of the developmental
and survival signal that LMP2A imparts. TgE bone marrow
produces nearly twice as many colonies on average (Student’s
t
test,
P
⫽
0.0017) compared to Tg
⌬
ITAM1, Tg
⌬
ITAM2, and
WT littermate controls (Fig. 5C). These data represent counts
collected from four separate experiments in which Tg6 bone
marrow was not cultured and is therefore not represented
in the graph. These results indicate that Tg
⌬
ITAM1 and
Tg
⌬
ITAM2 mutant LMP2A protein does not provide any
colony-forming enhancement signal as seen in nonmutated
LMP2A transgenic bone marrow when cultured in vitro.
In the RAG
ⴚ/ⴚbackground, the LMP2A ITAM mutant
transgene is unable to support development or survival of
bone marrow cells in methylcellulose.
When bone marrow
from any RAG
⫺/⫺LMP2A transgenic mouse is cultured in
methylcellulose media, a substantial enhancement of
col-ony number can be observed (6, 7). Therefore, RAG
⫺/⫺V
OL. 74, 2000
LMP2A
⌬ITAM TRANSGENIC MICE
9119
on November 9, 2019 by guest
http://jvi.asm.org/
Tg
⌬
ITAM1, RAG
⫺/⫺Tg
⌬
ITAM2, RAG
⫺/⫺TgE, RAG
⫺/⫺Tg6, and RAG
⫺/⫺littermate control bone marrow samples
were cultured in IL-7-containing methylcellulose. As
ex-pected, both RAG
⫺/⫺TgE and RAG
⫺/⫺Tg6 bone marrow
cultures generated significant numbers of colonies in
methyl-cellulose (Fig. 5D). Both RAG
⫺/⫺Tg
⌬
ITAM1 and RAG
⫺/⫺Tg
⌬
ITAM2 did not form any significant numbers of colonies,
similar to their RAG
⫺/⫺littermate control animals. Flow
cy-tometry analysis of recovered RAG
⫺/⫺TgE and RAG
⫺/⫺Tg6
bone marrow cells showed that nearly all were CD19
⫹IgM
⫺as
expected (data not shown). Too few cells were recovered from
RAG
⫺/⫺Tg
⌬
ITAM1, RAG
⫺/⫺Tg
⌬
ITAM2, and RAG
⫺/⫺an-imals to determine surface marker phenotype. These results
show that Tg
⌬
ITAM1 and Tg
⌬
ITAM2 animals lack any
LMP2A-derived developmental or survival signal.
DISCUSSION
Both in vitro and in vivo systems have been used to
deter-mine the function of LMP2A in EBV latency. In vitro studies
using EBV-transformed LCLs have shown that LMP2A is
highly tyrosine phosphorylated and forms aggregates in the
plasma membrane of latently infected cells. Associated with
LMP2A are the Src family PTKs, the Syk PTK, and Nedd4
family ubiquitin ligases (24, 25, 37, 57). Of particular interest to
these studies is the specific association of the LMP2A ITAM
with the Syk PTK (24). In vitro, these LMP2A protein
com-plexes result in a complete block in BCR-mediated signal
transduction (24, 25, 57, 58). In vivo studies have provided
additional information into LMP2A function. In transgenic
mice, LMP2A provides both developmental and survival
sig-nals to B lymphocytes (6, 7).
This study was designed to determine the importance of the
LMP2A ITAM in the LMP2A-mediated developmental and
survival signal observed in the previously described LMP2A
transgenic mice (6, 7). Comparing ITAM defective LMP2A
transgenic mice with the previously constructed LMP2A
trans-genic lines, we demonstrate the importance of the LMP2A
ITAM in providing the LMP2A-mediated developmental and
survival signal. Previous studies have described two distinct
LMP2A transgenic phenotypes. Two of five genetically distinct
LMP2A lines have a dramatic phenotype (LMP2A
BCR⫺)
char-acterized by the development and survival of B lymphocytes
lacking a BCR in an immunocompetent murine background
(7). Three other lines exhibit a more subtle phenotype
(LMP2A
BCR⫹) when analyzed in an immunocompetent
back-ground. However, when all LMP2A transgenic animals are
bred into the RAG-null background, LMP2A allows for both
the developmental and survival of B lymphocytes in peripheral
immune organs (6, 7).
[image:6.612.60.547.70.294.2]In this study, we analyzed two different LMP2A ITAM
mu-tant transgenic lines, both of which were unable to promote
B-cell development or survival in either an
immunocompe-tent or RAG-null background. Although it is possible that
these ITAM-defective LMP2A lines may not express enough
LMP2A to generate the previously characterized LMP2A
phe-notypes, this seems unlikely for two reasons. First, as
demon-strated, both ITAM-defective lines generated in this study
ex-press approximately equal levels of LMP2A protein in B
lymphocytes compared to the previously described LMP2A
transgenic lines. Second, of the five nonmutated LMP2A
trans-genic lines analyzed to date, all have the developmental and
survival phenotype when bred into the RAG-null background.
The requirement for an intact LMP2A ITAM to provide B
cells with a developmental and survival signal in vivo implicates
Syk as being a major target for LMP2A. Many studies have
demonstrated a vital role for Syk in B-cell development and
activation (13, 73, 76). Syk
⫺/⫺mice display perinatal lethality,
indicating that Syk plays a vital role in organismal development
(13, 76). However, when irradiated WT mice are reconstituted
with fetal liver cells from Syk
⫺/⫺mice, the reconstituted
lym-phoid cells exhibit a severe B-cell developmental phenotype.
These mice are characterized by a block of B-cell development
at the pro-B-to-pre-B cell transition, resulting in a lack of
FIG. 4. LMP2A ITAM mutant mice do not impart a survival signal to RAG⫺/⫺B cells. (A) Spleen (SP) tissues were immunostained with CD19-PE and IgM-FITC
antibodies; flow cytometry dot plots are shown. Two distinct cell types are highlighted with boxes in the plots, and the respective percentages of gated cells within those
regions are denoted. (B) Bone marrow (BM) cells were immunostained as above; the boxed inset shows the percentage of CD19⫹cells arising in the bone marrow,
and the respective percentages of gated cells within those regions are denoted.
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 5. LMP2A ITAM mutant bone marrow B cells grow similarly to WT cells in methylcellulose media and are incapable of enhancing survival or development of RAG ⫺ / ⫺ B cells. (A) Tg ⌬ ITAM1, Tg ⌬ ITAM2, WT, TgE, and Tg6 bone marrow cells were cultured in methylcellulose medium containing IL-7 for 7 days; pictures of typical resulting colonies are shown. (B) Methylcellulose-cultured bone marrow cells were immunostained with CD19-PE and IgM-FITC antibodies; flow cytometry dot plots are shown. Two distinct cell types are highlighted with boxes, and the respective percentages of gated cells within those regions are denoted. (C) The colonies arising in Tg ⌬ ITAM1, Tg ⌬ ITAM2, WT, and TgE bone marrow methylcellulose cultures were counted and plotted in a histogram. The increase in colony number seen in TgE bone marrow cultures is statistically significant (Student’s t test, P ⫽ 0.0017). (D) RAG ⫺ / ⫺ Tg ⌬ ITAM1, RAG ⫺ / ⫺ Tg ⌬ ITAM2, RAG ⫺ / ⫺ , RAG ⫺ / ⫺ TgE, and RAG ⫺ / ⫺ Tg6 bone marrow cells were cultured in methylcellulose medium containing IL-7 for 7 days; pictures of typical resulting colonies are shown.
V
OL. 74, 2000
LMP2A
⌬ITAM TRANSGENIC MICE
9121
on November 9, 2019 by guest
http://jvi.asm.org/
mature B cells as well as a partial impairment of T-cell
devel-opment.
Syk has been firmly established as a critical factor for the
generation of both the Ca
2⫹influx and mitogen-activated
pro-tein kinase (MAPK) signals that follow BCR cross-linking (for
reviews, see references 8, 41, and 42). For production of the
Ca
2⫹response, Syk is critical for phosphorylation and
activa-tion of a number of downstream targets, including PI-3K,
BLNK, and phospholipase
␥
2 (PLC-
␥
2) (10, 26, 38, 73).
Following sIg cross-linking, Syk phosphorylates and activates
PI-3K, which initiates the production of biologically active
phos-phoinositides that act as second messengers (16, 21).
Interest-ingly, activation of PI-3K is also critical for activation of the
antiapoptotic protein Akt, which may be a target for LMP2A in
enhancing cell survival (17, 29). Btk associates with the PI-3K
product phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P
3],
allowing for autophosphorylation and activation (47, 69, 78).
Phosphorylated BLNK has been shown to provide docking
sites for downstream signaling molecules, thus allowing
them to aggregate and become activated (26). BLNK binds
both activated Btk and PLC-
␥
2, thus allowing Btk and Syk to
phosphorylate and activate PLC-
␥
2 (19, 33, 38, 72). Once
ac-tivated, PLC-
␥
2 mediates the production of diacylglycerol and
inositol trisphosphate from the PI-3K product PI(4,5)P
2(34).
Diacylglycerol is critical for activation of protein kinase C,
whereas inositol trisphosphate causes a release of Ca
2⫹from
intracellular stores (56, 62). Influx of Ca
2⫹leads to activation
of the calmodulin-dependent protein kinase II and the
calm-odulin-activated serine/theonine phosphatase calcineurin (70,
77). One of the factors activated by an influx of calcium is the
transcription factor NF-AT, which is important for
upregula-tion of a number of cytokines and immune effector molecules
(15, 79).
A more direct generation of MAPK signals by Syk
down-stream of the BCR has also been well established. BLNK
phosphorylation by Syk leads to binding of guanine nucleotide
exchange factors Vav and the Grb2 and Nck adapter proteins
through SH2-phosphotyrosine associations (26). Docking of
these effectors to BLNK allow for activation of the Ras and
Rac GTPases (5, 9, 18, 46). Phosphorylation cascades though
the ERK, JNK, and p38 arms of the MAPK pathways result in
activation of a number of transcription factors such as Fos, Jun,
and members of the Ets family (14, 30, 60, 75). Studies using
Syk-null DT40 chicken B cells have shown that Syk is critical
for activation of the ERK1 and JNK1 arms of the MAPK
family (39).
The LMP2A ITAM mutant mouse data suggest that LMP2A
may be manipulating Syk to direct the activation of a
MAPK-and/or a Ca
2⫹-mediated signal in order to enhance B-cell
sur-vival and maintain latency. In so doing, LMP2A may be
mim-icking a constitutively active BCR or a docking protein, such as
BLNK, thus directing signals away from the actual BCR while
replacing the essential signals normally arising from a
func-tional BCR with LMP2A-derived survival signals. Future
stud-ies will map the pathways downstream of the LMP2A
ITAM-Syk interaction critical for LMP2A to block BCR signal
transduction and produce survival signals. Of particular
inter-est are pathways that lead to activation or upregulation of
antiapoptotic proteins such as Akt. Other signaling proteins
acting downstream of Syk that are important for B-cell
devel-opment, such as Btk and BLNK, also clearly warrant further
study using our transgenic mouse models. These studies should
provide a clearer understanding of the transmission of LMP2A
signals and how these signals affect B-cell development,
sur-vival, and EBV latency.
ACKNOWLEDGMENTS
R.L. is supported by Public Health Service grants CA62234 and
CA73507 from the National Cancer Institute and DE13127 from the
National Institute of Dental and Craniofacial Research. R.L. is a
Scholar of the Leukemia Society of America.
We are grateful for the contributions from members of the
Long-necker and Spear laboratories at Northwestern University.
REFERENCES
1.Babcock, G. J., L. L. Decker, M. Volk, and D. A. Thorley-Lawson.1998. EBV persistence in memory B cells in vivo. Immunity9:395–404.
2.Begin, L. R., J. Eskandari, J. Joncas, and L. Panasci.1987. Epstein-Barr virus related lymphoepithelioma-like carcinoma of lung. J. Surg. Oncol.
36:280–283.
3.Bonnet, M., J. M. Guinebretiere, E. Kremmer, V. Grunewald, E. Benhamou, G. Contesso, and I. Joab.1999. Detection of Epstein-Barr virus in invasive breast cancers. J. Natl. Cancer Inst.91:1376–1381.
4.Bouzid, M., D. Djennaoui, J. Dubreuil, A. Bouguermouh, D. Ellouz, J. Abdelwahab, G. Decaussin, and T. Ooka.1994. Epstein-Barr virus genotypes in NPC biopsies from North Africa. Int. J. Cancer56:468–473.
5.Buday, L., S. E. Egan, V. P. Rodriguez, D. A. Cantrell, and J. Downward.
1994. A complex of Grb2 adaptor protein, Sos exchange factor, and a 36-kDa membrane-bound tyrosine phosphoprotein is implicated in ras activation in T cells. J. Biol. Chem.269:9019–9023.
6.Caldwell, R. G., R. C. Brown, and R. Longnecker.2000. Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of ELMP2A trans-genic mice. J. Virol.74:1101–1113.
7.Caldwell, R. G., J. B. Wilson, S. J. Anderson, and R. Longnecker.1998. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity9:405–411.
8.Campbell, K. S.1999. Signal transduction from the B cell antigen-receptor. Curr. Opin. Immunol.11:256–264.
9.Cantrell, D.1998. Lymphocyte signalling: a coordinating role for Vav? Curr. Biol.8:R535–R538.
10. Carter, R. H., D. J. Park, S. G. Rhee, and D. T. Fearon.1991. Tyrosine phosphorylation of phospholipase C induced by membrane immunoglobulin in B lymphocytes. Proc. Natl. Acad. Sci. USA88:2745–2749.
11. Chan, J. K., T. T. Yip, W. Y. Tsang, Y. F. Poon, C. S. Wong, and V. W. Ma.
1994. Specific association of Epstein-Barr virus with lymphoepithelial carci-noma among tumors and tumorlike lesions of the salivary gland. Arch. Pathol. Lab. Med.118:994–997.
12. Chen, F., J. Z. Zou, and L. di Renzo.1995. A subpopulation of normal B cells latently infected with Epstein-Barr virus resembles Burkitt lymphoma cells in expressing EBNA-1 but not EBNA-2 or LMP1. J. Virol.69:3752–3758. 13. Cheng, A. M., B. Rowley, W. Pao, A. Hayday, J. B. Bolen, and T. Pawson.
1995. Syk tyrosine kinase required for mouse viability and B-cell develop-ment. Nature378:303–306.
14. Chiles, T. C., J. L. Liu, and T. L. Rothstein.1991. Cross-linking of surface Ig receptors on murine B lymphocytes stimulates the expression of nuclear tetradecanoyl phorbol acetate-response element-binding proteins. J. Immu-nol.146:1730–1735.
15. Choi, M., R. D. Brines, M. J. Holman, G. G. Klaus.1994. Induction of NF-AT in normal B lymphocytes by anti-immunoglobulin or CD40 ligand in conjunction with IL-4. Immunity1:179–187.
16. Corvera, S., and M. P. Czech.1998. Direct targets of phosphoinositide 3-kinase products in membrane traffic and signal transduction. Trends Cell Biol.8:442–446.
17. Craxton, A., A. Jiang, T. Kurosaki, and E. A. Clark.1999. Syk and Bruton’s tyrosine kinase are required for B cell antigen receptor-mediated activation of the kinase Akt. J. Biol. Chem.274:30644–30650.
18. Crespo, P., X. R. Bustelo, D. S. Aaronson, O. A. Coso, M. Lopez-Barahona, M. Barbacid, and J. S. Gutkind.1996. Rac-1 dependent stimulation of the JNK/SAPK signaling pathway by Vav. Oncogene13:455–460.
19. DeBell, K. E., B. A. Stoica, M. C. Veri, A. Di Baldassarre, S. Miscia, L. J. Graham, B. L. Rellahan, M. Ishiai, T. Kurosaki, and E. Bonvini.1999. Functional independence and interdependence of the Src homology domains of phospholipase C-␥1 in B-cell receptor signal transduction. Mol. Cell. Biol.
19:7388–7398.
20. Decker, L. L., L. D. Klaman, and D. A. Thorley-Lawson.1996. Detection of the latent form of Epstein-Barr virus DNA in the peripheral blood of healthy individuals. J. Virol.70:3286–3286.
21. Duronio, V., M. P. Scheid, and S. Ettinger.1998. Downstream signalling events regulated by phosphatidylinositol 3-kinase activity. Cell Signal.10:
233–233.
22. Fruehling, S., R. Caldwell, and R. Longnecker.1997. LMP2 function in EBV Latency. EBV Rep.4:141–159.
23. Fruehling, S., S. K. Lee, R. Herrold, B. Frech, G. Laux, E. Kremmer, F. A. Grasser, and R. Longnecker.1996. Identification of latent membrane pro-tein 2A (LMP2A) domains essential for the LMP2A dominant-negative effect on B-lymphocyte surface immunoglobulin signal transduction. J. Virol.
70:6216–6226.
on November 9, 2019 by guest
http://jvi.asm.org/
24.Fruehling, S., and R. Longnecker.1997. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology235:241–251.
25. Fruehling, S., R. Swart, K. M. Dolwick, E. Kremmer, and R. Longnecker.
1998. Tyrosine 112 of latent membrane protein 2A is essential for protein tyrosine kinase loading and regulation of Epstein-Barr virus latency. J. Virol.
72:7796–7806.
26. Fu, C., C. W. Turck, T. Kurosaki, and A. C. Chan.1998. BLNK: a central linker protein in B cell activation. Immunity9:93–103.
27. Fukayama, M., Y. Hayashi, Y. Iwasaki, J. Chong, T. Ooba, T. Takizawa, M. Koike, S. Mizutani, M. Miyaki, and K. Hirai.1994. Epstein-Barr virus-associated gastric carcinoma and Epstein-Barr virus infection of the stom-ach. Lab. Investig.71:73–81.
28. Ghaffari-Tabrizi, N., B. Bauer, A. Villunger, G. Baier-Bitterlich, A. Altman, G. Utermann, F. Uberall, and G. Baier.1999. Protein kinase C, a selective upstream regulator of JNK/SAPK and IL-2 promoter activation in Jurkat T cells. Eur. J. Immunol.29:132–142.
29. Gold, M., M. P. Scheid, L. Santos, M. Dang-Lawson, R. A. Roth, L. Mat-suuchi, V. Duronio, and D. L. Krebs.1999. The B cell antigen receptor activates the Akt (protein kinase B)/glycogen synthase kinase-3 signaling pathway via phosphatidylinositol 3-kinase. J. Immunol.163:1894–1905. 30. Grant, P. A., C. B. Thompson, and S. Pettersson. 1995. IgM
receptor-mediated transactivation of the IgH 3⬘enhancer couples a novel Elf-1-AP-1 protein complex to the developmental control of enhancer function. EMBO J.14:4501–4513.
31. Gulley, M. L., D. R. Pulitzer, P. A. Eagan, and B. G. Schneider.1996. Epstein-Barr virus infection is an early event in gastric carcinogenesis and is independent of bcl-2 expression and p53 accumulation. Hum. Pathol.27:20– 27.
32. Harn, H. J., L. I. Ho, W. H. Chung, J. J. Lin, H. S. Lee, and W. H. Lee.1995. Epstein-Barr virus-associated typical gastric carcinoma detected by in situ hybridization and polymerase chain reaction. J. Clin. Gastroenterol.20:253– 254.
33. Hashimoto, S., A. Iwamatsu, M. Ishiai, K. Okawa, T. Yamadori, M. Mat-sushita, Y. Baba, T. Kishimoto, T. Kurosaki, and S. Tsukada.1999. Identi-fication of the SH2 domain binding protein of Bruton’s tyrosine kinase as BLNK—functional significance of Btk-SH2 domain in B-cell antigen recep-tor-coupled calcium signaling. Blood94:2357–2364.
34. Hempel, W. M., R. C. Schatzman, and A. L. DeFranco. 1992. Tyrosine phosphorylation of phospholipase C-gamma 2 upon cross-linking of mem-brane Ig on murine B lymphocytes. J. Immunol.148:3021–3027.
35. Hitt, M. M., M. J. Allday, T. Hara, L. Karran, M. D. Jones, P. Busson, T. Tursz, I. Ernberg, and B. E. Griffin.1989. EBV gene expression in an NPC-related tumour. EMBO J.8:2639–2651.
36. Hogan, B., R. Beddington, F. Constantini, and E. Lacy.1994. Manipulating the mouse embryo, a laboratory manual, 2nd ed. Cold Spring Harbor Lab-oratory Press, Cold Spring Harbor, N.Y.
37. Ikeda, M., A. Ikeda, L. C. Longan, and R. Longnecker.2000. The Epstein-Barr virus (EBV) latent membrane protein 2A (LMP2A) PY motif recruits WW domain-containing ubiquitin-protein ligases. Virology268:178–191. 38. Ishiai, M., M. Kurosaki, R. Pappu, K. Okawa, I. Ronko, C. Fu, M. Shibata,
A. Iwamatsu, A. C. Chan, and T. Kurosaki.1999. BLNK required for cou-pling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity10:117–125. 39. Jiang, A., A. Craxton, T. Kurosaki, E. A. Clark.1998. Different protein tyrosine kinases are required for B cell antigen receptor-mediated activation of extracellular signal-regulated kinase, c-Jun NH2-terminal kinase 1, and p38 mitogen-activated protein kinase. J. Exp. Med.188:1297–1306. 40. Kieff, E.1996. Epstein-Barr virus and its replication, p. 1109–1162.InB. N.
Fields, D. M. Knipe, and P. M. Howley (ed.), Fundamental virology, Lip-pincott-Raven, Philadelphia, Pa.
41. Kurosaki, T.1999. Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol.17:555–592.
42. Kurosaki, T.1998. Molecular dissection of B cell antigen receptor signaling. Bioorg. Med. Chem. Lett.1:515–527.
43. Labrecque, L. G., D. M. Barnes, I. S. Fentiman, and B. E. Griffin.1995. Epstein-Barr virus in epithelial cell tumors: a breast cancer study. Cancer Res.55:39–45.
44. Lam, K., R. Kuhn, and K. Rajewsky.1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell90:1073–1083.
45. Lanier, A. P., S. R. Clift, G. Bornkamm, W. Henle, H. Goepfert, and N. Raab Traub.1991. Epstein-Barr virus and malignant lymphoepithelial lesions of the salivary gland. Arctic Med. Res.50:55–61.
46. Li, N., A. Batzer, R. Daly, V. Yajnik, E. Skolnik, P. Chardin, S. D. Bar, B. Margolis, and J. Schlessinger.1993. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature363:85–88.
47. Li, Z., M. I. Wahl, A. Eguinoa, L. R. Stephens, P. T. Hawkins, and O. N. Witte. 1997. Phosphatidylinositol 3-kinase-gamma activates Bruton’s ty-rosine kinase in concert with Src family kinases. Proc. Natl. Acad. Sci. USA
94:13820–13825.
48. Longnecker, R.1994. Biochemical and genetic studies of Epstein-Barr virus
latent membrane protein 2. Leukemia8:S46–S50.
49. Longnecker, R.1998. Molecular biology of Epstein-Barr virus, p. 133–172.In Dennis McCance (ed.), Human tumor viruses, American Society for Micro-biology, Washington, D.C.
50. Longnecker, R., B. Druker, T. M. Roberts, and E. Kieff.1991. An Epstein-Barr virus protein associated with cell growth transformation interacts with a tyrosine kinase. J. Virol.65:3681–3692.
51. Longnecker, R., and E. Kieff.1990. A second Epstein-Barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J. Virol.64:2319–2326.
52. Longnecker, R., and C. L. Miller.1996. Regulation of Epstein-Barr virus latency by latent membrane protein 2. Trends Microbiol.4:38–42. 53. Longnecker, R., C. L. Miller, X.-Q. Miao, A. Marchini, and E. Kieff.1992.
The only domain which distinguishes Epstein-Barr virus latent membrane 2A (LMP2A) from LMP2B is dispensable for lymphocyte infection and growth transformation in vitro, and LMP2A is therefore nonessential. J. Vi-rol.66:6461–6469.
54. Longnecker, R., C. L. Miller, X. Q. Miao, B. Tomkinson, and E. Kieff.1993. The last seven transmembrane and carboxy-terminal cytoplasmic domains of Epstein-Barr virus latent membrane protein 2 (LMP2) are dispensable for lymphocyte infection and growth transformation in vitro. J. Virol.67:2006– 2013.
55. Longnecker, R., C. L. Miller, B. Tomkinson, X. Q. Miao, and E. Kieff.1993. Deletion of DNA encoding the first five transmembrane domains of Epstein-Barr virus latent membrane proteins 2A and 2B. J. Virol.67:5068–5074. 56. McConnell, F. M., S. B. Shears, P. J. Lane, M. S. Scheibel, and E. A. Clark.
1992. Relationships between the degree of cross-linking of surface immuno-globulin and the associated inositol 1,4,5-trisphosphate and Ca2⫹signals in human B cells. Biochem. J.284:447–455.
57. Miller, C. L., A. L. Burkhardt, J. H. Lee, B. Stealey, R. Longnecker, J. B. Bolen, and E. Kieff.1995. Integral membrane protein 2 of Epstein-Barr virus regulates reactivation from latency through dominant negative effects on protein-tyrosine kinases. Immunity2:155–166.
58. Miller, C. L., J. H. Lee, E. Kieff, and R. Longnecker.1994. An integral membrane protein (LMP2) blocks reactivation of Epstein-Barr virus from latency following surface immunoglobulin crosslinking. Proc. Natl. Acad. Sci. USA91:772–776.
59. Miller, C. L., R. Longnecker, and E. Kieff.1993. Epstein-Barr virus latent membrane protein 2A blocks calcium mobilization in B lymphocytes. J. Vi-rol.67:3087–3094.
60. Mittlestadt, P. R., and A. L. DeFranco.1993. Induction of early response genes by cross-linking membrane Ig on B lymphocytes. J. Immunol.150:
4822–4832.
61. Miyashita, E. M., B. Yang, G. J. Babcock, and D. A. Thorley-Lawson.1997. Identification of the site of Epstein-Barr virus persistence in vivo as a resting B cell. J. Virol.71:4882–4891.
62. Padeh, S., A. Levitzki, A. Gazit, G. B. Mills, and C. M. Roifman.1991. Activation of phospholipase C in human B cells is dependent on tyrosine phosphorylation. J. Clin. Investig.87:1114–1118.
63. Raab-Traub, N.1992. Epstein-Barr virus and nasopharyngeal carcinoma. Semin. Cancer Biol.3:297–307.
64. Raab-Traub, N., P. Rajadurai, K. Flynn, and A. P. Lanier.1991. Epstein-Barr virus infection in carcinoma of the salivary gland. J. Virol.65:7032– 7036.
65. Rickinson, A. B., and E. Kieff.1996. Epstein-Barr virus, p. 2397–446.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Lippincott-Raven Publishers, Philadelphia, Pa.
66. Riddler, S. A., M. C. Breinig, and J. L. McKnight.1994. Increased levels of circulating Epstein-Barr virus (EBV)-infected lymphocytes and decreased EBV nuclear antigen antibody responses are associated with the develop-ment of posttransplant lymphoproliferative disease in solid-organ transplant recipients. Blood84:972–984.
67. Sakata, N., H. Kawasome, N. Terada, P. Gerwins, G. L. Johnson, E. W. Gelfand.1999. Differential activation and regulation of mitogen-activated protein kinases through the antigen receptor and CD40 in human B cells. Eur. J. Immunol.29:2999–3008.
68. Sample, J., D. Liebowitz, and E. Kieff.1989. Two related Epstein-Barr virus membrane proteins are encoded by separate genes. J. Virol.63:933–937. 69. Scharenberg, A., and J. P. Kinet.1998. PtdIns-3,4,5-P3: a regulatory nexus
between tyrosine kinases and sustained calcium signals. Cell94:5–8. 70. Schreiber, S. L., and G. R. Crabtree.1992. The mechanism of action of
cyclosporin A and FK506. Immunol. Today13:136–142.
71. Sugihara, K., H. Reupke, A. Schmidt Westhausen, H. D. Pohle, H. R. Gelder-blom, and P. A. Reichart.1990. Negative staining EM for the detection of Epstein-Barr virus in oral hairy leukoplakia. J. Oral Pathol. Med.19:367– 370.
72. Takata, M., and T. Kurosaki.1996. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J. Exp. Med.184:31–40.
73. Takata, M., H. Sabe, A. Hata, T. Inazu, Y. Homma, T. Nukada, H. Yamamura, and T. Kurosaki.1994. Tyrosine kinases Lyn and Syk regulate B
V
OL. 74, 2000
LMP2A
⌬ITAM TRANSGENIC MICE
9123
on November 9, 2019 by guest
http://jvi.asm.org/
cell receptor-coupled Ca2⫹mobilization through distinct pathways. EMBO J.13:1341–1349.
74.Tierney, R. J., N. Steven, L. S. Young, and A. B. Rickinson.1994. Epstein-Barr virus latency in blood mononuclear cells: analysis of viral gene tran-scription during primary infection and in the carrier state. J. Virol.68:7374– 7385.
75.Tilzey, J. F., T. C. Chiles, and T. L. Rothstein.1991. Jun-B gene expression mediated by the surface immunoglobulin receptor of primary B lymphocytes. Biochem. Biophys. Res. Commun.175:77–83.
76.Turner, M., P. J. Mee, P. S. Costello, O. Williams, A. A. Price, L. P. Duddy, M. T. Furlong, R. L. Geahlen, and V. L. Tybulewicz.1995. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk. Nature378:298–302.
77. Valentine, M. A., A. J. Czernik, N. Rachie, H. Hidaka, C. L. Fisher, J. C. Cambier, and K. Bomsztyk.1995. Anti-immunoglobulin M activates nuclear
calcium/calmodulin-dependent protein kinase II in human B lymphocytes. J. Exp. Med.182:1943–1949.
78. Varnai, P., K. I. Rother, and T. Balla.1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton’s tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem.274:10983– 10989.
79. Venkataraman, L., D. A. Francis, Z. Wang, J. Liu, T. L. Rothstein, and R. Sen.1994. Cyclosporin-A sensitive induction of NF-AT in murine B cells. Immunity1:189–196.
80. Yates, J. L., N. Warren, and B. Sugden.1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature313:
812–815.
81. Young, L., C. Alfieri, K. Hennessy, H. Evans, C. O’Hara, K. C. Anderson, J. Ritz, R. S. Shapiro, A. Rickinson, E. Kieff, and J. I. Cohen.1989. Expression of Epstein-Barr virus transformation-associated genes in tissues of patients with EBV lymphoproliferative disease. N. Engl. J. Med.321:1080–1085.
on November 9, 2019 by guest
http://jvi.asm.org/