Nucleic Acid Binding by Mason-Pfizer Monkey Virus CA Promotes
Virus Assembly and Genome Packaging
Tibor Füzik,aRu˚žena Píchalová,aFlorian K. M. Schur,dKarolína Strohalmová,bIvana Krˇížová,bRomana Hadravová,b Michaela Rumlová,b,cJohn A. G. Briggs,dPavel Ulbrich,aTomáš Rumla
Department of Biochemistry and Microbiology, University of Chemistry and Technology Prague, Prague, Czech Republica
; Institute of Organic Chemistry and Biochemistry IOCB Research Centre and Gilead Sciences, Academy of Sciences of the Czech Republic, Prague, Czech Republicb
; Department of Biotechnology, University of Chemistry and Technology Prague, Prague, Czech Republicc
; Structural and Computational Biology Unit and Molecular Medicine Partnership Unit, European Molecular Biology Laboratory, Heidelberg, Germanyd
ABSTRACT
The Gag polyprotein of retroviruses drives immature virus assembly by forming hexameric protein lattices. The assembly is
pri-marily mediated by protein-protein interactions between capsid (CA) domains and by interactions between nucleocapsid (NC)
domains and RNA. Specific interactions between NC and the viral RNA are required for genome packaging. Previously reported
cryoelectron microscopy analysis of immature Mason-Pfizer monkey virus (M-PMV) particles suggested that a basic region
(res-idues RKK) in CA may serve as an additional binding site for nucleic acids. Here, we have introduced mutations into the RKK
region in both bacterial and proviral M-PMV vectors and have assessed their impact on M-PMV assembly, structure, RNA
bind-ing, budding/release, nuclear traffickbind-ing, and infectivity using
in vitro
and
in vivo
systems. Our data indicate that the RKK
gion binds and structures nucleic acid that serves to promote virus particle assembly in the cytoplasm. Moreover, the RKK
re-gion appears to be important for recruitment of viral genomic RNA into Gag particles, and this function could be linked to
changes in nuclear trafficking. Together these observations suggest that in M-PMV, direct interactions between CA and nucleic
acid play important functions in the late stages of the viral life cycle.
IMPORTANCE
Assembly of retrovirus particles is driven by the Gag polyprotein, which can self-assemble to form virus particles and interact
with RNA to recruit the viral genome into the particles. Generally, the capsid domains of Gag contribute to essential
protein-protein interactions during assembly, while the nucleocapsid domain interacts with RNA. The interactions between the
nucleo-capsid domain and RNA are important both for identifying the genome and for self-assembly of Gag molecules. Here, we show
that a region of basic residues in the capsid protein of the betaretrovirus Mason-Pfizer monkey virus (M-PMV) contributes to
interaction of Gag with nucleic acid. This interaction appears to provide a critical scaffolding function that promotes assembly of
virus particles in the cytoplasm. It is also crucial for packaging the viral genome and thus for infectivity. These data indicate that,
surprisingly, interactions between the capsid domain and RNA play an important role in the assembly of M-PMV.
T
he retroviral structural polyprotein Gag contains three
con-served domains, matrix (MA), capsid (CA), and nucleocapsid
(NC). Gag plays the primary role in immature particle assembly
and viral genomic RNA (vRNA) recruitment and packaging.
Retroviruses assemble via two different morphogenetic
path-ways; the first, historically referred to as C-type, wherein particle
assembly occurs at the cell membrane, and the second, D-type,
assembling in perinuclear regions. The pathogenic human viruses,
HIV and human T-cell lymphotropic virus assemble via C-type
intermediates, whereas M-PMV is the prototypic D-type
retrovi-rus. The Gag polyprotein of M-PMV is first transported to an
intracytoplasmic pericentriolar site, where particle assembly
oc-curs (
1–3
). This targeting requires a cytoplasmic
targeting/reten-tion signal (CTRS) localized in the MA domain that mediates the
interaction of Gag with components of dynein to transport cargo
molecules toward the minus ends of the microtubules (
4
). After
assembly, the immature D-type particles are transported to the
plasma membrane, where budding occurs.
Gag nucleation and particle assembly is promoted by
interac-tions between CA domains and also by interacinterac-tions between NC
and both cellular and viral RNA. The importance of RNA for
assembly is well documented by
in vitro
assembly studies with
various retroviruses, e.g., Rous sarcoma virus (RSV), HIV, murine
leukemia virus (MLV), and M-PMV (
5–11
). Although cellular
RNA is sufficient to promote Gag assembly (
12
), formation of an
infectious retrovirus requires specific packaging of the viral
ge-nome (vRNA) into the assembling Gag particle. It is likely that the
initial recognition of the genomic nucleic acid is mediated by a few
Gag molecules that bring the vRNA to the assembly site (
13
). It has
been well documented both in HIV and in MLV that the selection
of vRNA for packaging into retroviral particles is mediated by
specific interaction between the highly structured
Psi
sequence at
Received22 December 2015Accepted15 February 2016
Accepted manuscript posted online24 February 2016
CitationFüzik T, Píchalová R, Schur FKM, Strohalmová K, Krˇížová I, Hadravová R,
Rumlová M, Briggs JAG, Ulbrich P, Ruml T. 2016. Nucleic acid binding by Mason-Pfizer monkey virus CA promotes virus assembly and genome packaging. J Virol 90:4593– 4603.doi:10.1128/JVI.03197-15.
Editor:W. I. Sundquist
Address correspondence to Pavel Ulbrich, [email protected], or Tomáš Ruml, [email protected].
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
on November 7, 2019 by guest
http://jvi.asm.org/
the 5
=
-untranslated region of unspliced vRNA and zinc-finger
motifs of the NC protein (
14–17
). Nonspecific interactions
be-tween RNA and other Gag domains such as MA may also
contrib-ute to recognition (
18
,
19
).
The current understanding is that the site at which the initial
Gag-vRNA interaction occurs is different for different
retrovi-ruses. For HIV, vRNA is exported from the nucleus by the
trans-activating factor Rev and subsequently interacts with Gag in the
cytoplasm (
20
). In RSV, it has been proposed that the primary
Gag-vRNA interaction may occur in the nucleus. This is
sup-ported by several pieces of evidence, including the presence of two
nuclear localization signals in NC and MA recognized by different
importins (
21
) and the detection of a large amount of RSV Gag in
the nuclei of cells treated with leptomycin B (LMB) (
22
), a specific
inhibitor of the karyopherin CRM1 (chromosome region
mainte-nance 1 receptor) nuclear export pathway. An RSV Gag mutant
that bypasses the nucleus packages vRNA less efficiently than the
wild type (wt), and both nuclear trafficking and vRNA packaging
is restored by the insertion of a heterologous nuclear localization
signal (
23
,
24
). It has been suggested that the formation of the RSV
Gag-vRNA complex induces a conformational change of Gag,
which leads to the exposure of the nuclear export signal, a
leucine-rich region within p10 (
25
). However, whether nuclear Gag
trig-gers the export of the full-length RSV vRNA remains an open
question, since even Gag proteins that do not enter the nucleus
generated infectious particles (
22
). The feline immunodeficiency
virus (FIV) Gag protein cycles through the nuclei of both human
and feline cells, and it has been proposed that encapsidation of FIV
vRNA may also initiate in the nucleus (
26
).
In the case of M-PMV Gag, a small proportion of Gag was
observed in nuclei of LMB-treated cells (
22
), suggesting a possible
interaction of Gag with vRNA within the nucleus. However, the
M-PMV vRNA contains a stem-loop structural motif, termed the
constitutive transport element (CTE), which can mediate nuclear
export of incompletely spliced genomic RNA (
27
,
28
) through
recognition by a cellular factor called TAP, which specifically
binds the CTE (
29
). The mode of nuclear export and the site of
Gag-vRNA interaction therefore remain unclear.
We described previously the local and global organization and
arrangements of Gag in
in vitro-assembled immature particles of
representatives of three retroviral genera, namely, HIV, RSV, and
M-PMV, using cryoelectron tomography (
30
). N-terminal
do-mains of CA were arranged into hexameric rings around large
holes with the CA domains forming dimers beneath this layer. In
HIV and RSV, strong rod-like densities formed by the spacer
pep-tide descended toward the particle center along the 6-fold axis. In
contrast, M-PMV lacks the extended rod-like densities
contrib-uted by the spacer region in other retroviruses, and the disordered
NC-RNA region is closer to CA than in other retroviruses (
31–33
).
The CA domain of M-PMV contains a stretch of three basic
resi-dues (RKK) located very close to its C terminus. Two RKK regions
provided by two neighboring CA molecules form a basic patch on
the underside of the CA layer in immature Gag arrays. We
previ-ously proposed that these residues may interact with nucleic acid,
explaining the proximity of the nucleic acid layer and CA in
M-PMV (
30
). Structural studies of
in vitro-assembled M-PMV tubes
show the presence of an extended “nucleic acid-like” filament of
density in the vicinity of these residues, suggesting that an
orga-nized nucleic acid structure interacts with this motif (
31–33
).
Here, we have explored the effects of mutating the RKK to AAA or
GPG on assembly, nucleic acid incorporation, structure, and virus
production.
MATERIALS AND METHODS
Plasmids and viral constructs.All DNA manipulations were carried out according to standard subcloning techniques, and all plasmids were prop-agated inEscherichia coliDH5␣. All newly created DNA constructs were verified by DNA sequencing. Forin vitroassembly assays, the RKK muta-tions were introduced into pSIT⌬ProCANC M-PMV plasmid, which carries the gene for an M-PMV capsid and nucleocapsid fusion protein lacking the N-terminal proline (34) under the T7 promoter. The 201RKK203to AAA or GPG mutations in pSIT⌬ProCANC M-PMV were made by SLIM mutagenesis (35). To introduce mutations in the M-PMV proviral vector pSARM4 (36), we first used a helper vector (MHelppUC19) encoding M-PMV SacI-Eco72I fragment, which was prepared as described previously (37, 38). Mutations RKK/AAA and RKK/GPG were created by two-step PCR mutagenesis using primers car-rying appropriate mutations and suitable NotI and XmaI restriction sites, respectively. The obtained PCR products were digested with SacI-NotI/ NotI-Eco72I or SacI-XmaI/XmaI-Eco72I, and both fragments were li-gated into the MHelppUC19. After sequence verification, the mutated SacI-Eco72I fragments of the MHelppUC19, carrying the appropriate mutation, were inserted into pSARM4 or pSARM-EGFP. For the single-round infectivity assay, the M-PMV Env expression vector pTMO (39) and the pSARM-EGFP vector in which EGFP (enhanced green fluorescent protein) replaces theenvgene (40) were used. Further details of the clon-ing strategy and the full sequences of all PCR primers can be obtained from the authors upon request.
Cell growth and virus production.HEK 293T cells were grown in Dulbecco modified Eagle medium (DMEM; Sigma) supplemented with 10% fetal bovine serum (Gibco) and 1%L-glutamine (PAA Laboratories). Transfection of the HEK 293T cells was performed using FuGene HD transfection reagent (Roche Molecular Biochemicals) according to the manufacturer’s instructions. At 24 or 48 h posttransfection, virions in the culture media were harvested, filtered through a 0.45-m-pore-size filter, and centrifuged through a 20% sucrose cushion at 200,000⫻gfor 1 h in a Beckman SW41Ti rotor. M-PMV proteins were detected by Western blotting with a rabbit anti-M-PMV CA polyclonal antibody (41).
Bacterial expression and purification of⌬ProCANC, AAA, and GPG mutants.Luria-Bertani medium supplemented with ampicillin (100g/ ml) was inoculated with a 0.1% (vol/vol) overnight culture ofE. coli
BL21(DE3) carrying pSIT⌬ProCANC M-PMV or an AAA or a GPG mutant plasmid. The cells were cultivated at 37°C and 250 rpm until the culture reached the exponential growth phase (i.e., an optical density at 600 nm of 0.4 to 0.6). Protein expression was induced by the addition of 0.4 mM (final concentration) IPTG (isopropyl--D -thiogalactopyrano-side). The cells were harvested at 4 h postinduction by centrifugation at 5,000⫻gand stored at⫺20°C.
The purification of the proteins was performed according to previ-ously published protocols (5,42). Briefly, a high-salt containing buffer extraction was used to solubilize and release the protein from the cell lysate pellet. The extracted protein was subsequently purified by immobi-lized metal affinity chromatography on a Zn2⫹-charged column, followed
by ion-exchange chromatography on a phosphocellulose column, to re-move contaminating nucleic acids. Finally, the protein was dialyzed against storage buffer (50 mM phosphate, 500 mM NaCl, 1M ZnSO4, 0.05% mercaptoethanol [pH 7.5]), concentrated using ultrafiltration on Amicon Ultra-15 Ultracel 10K (Millipore, Ireland) concentrators to⬃1 mg/ml, and stored at⫺20°C. The protein concentration was determined by using a Bradford protein assay.
Analysis of VLP formation inE. coli.To determine whether the viral protein assembled inside the bacterial cells during the induced expression process, 1 ml of the cell culture at 4 h postinduction was pelleted, and the cells were resuspended in 350l of lysis buffer (50 mM Tris, 100 mM NaCl, 1% [wt/vol] octylthioglucoside, 1 mg/ml lysozyme [pH 8.0]). The
on November 7, 2019 by guest
http://jvi.asm.org/
suspension was incubated on a rotation mixer for 10 min at room tem-perature. Using this mild lysis process, the intact virus-like particles (VLPs) were released into the lysate and were subsequently analyzed using transmission electron microscopy (TEM) after negative staining.
In vitroassembly of purified⌬ProCANC proteins.Thein vitro as-sembly of purified wild-type and mutant⌬ProCANC M-PMV proteins was performed as previously described (5). Briefly, 60g of purified pro-tein in storage buffer was mixed with either MS2 phage genomic RNA or
phage genomic DNA at a 10:1 or 5:1 (wt/wt) ratio in 100l of total reaction volume. This mixture was dialyzed against the assembly buffer (50 mM Tris, 100 mM NaCl, 1M ZnSO4[pH 8.0]) for 2 h at room temperature. When the effect of reducing conditions on the assembly process was studied, the assembly mixture contained 60 mM dithiothre-itol (DTT), and the concentration of DTT in the dialysis buffer was 20 mM.
Gradient centrifugation.Thein vitroparticle assembly efficiency was determined by gradient centrifugation, followed by sodium dodecyl sul-fate-polyacrylamide gel electrophoresis (SDS-PAGE). The dialyzed as-sembly mixture was loaded on top of 10 to 55% linear OptiPrep (Axis-Shield, Oslo, Norway) gradient and centrifuged for 40 min at 215,000⫻g. The gradient was fractionated, and the individual fractions were analyzed by SDS-PAGE (12% gel). The gels were blue silver stained (43) and digi-tized using Uvitec Alliance 4.7 (Uvitec, United Kingdom), and the assem-bly efficiency was assessed by densitometric analysis of the lanes using the Fiji (ImageJ) software package (44). The protein content in the topmost fraction of the gradient represented the unassembled protein, whereas the protein content in fractions with an OptiPrep density of around 1.15 to 1.27 g/ml (wt/vol), i.e., fractions 6 to 9, represented the assembled VLPs (5,6,45).
EMSA.For the electrophoretic mobility shift assay (EMSA), 5 or 1.7
g of studied protein was mixed with 165 ng of 1-kb DNA ladder (Pro-mega, USA) in 10l of total volume of buffered environment (25 mM Tris, 250 mM NaCl, 0.5M ZnSO4[pH 8.0]), corresponding to a protein/ nucleic acid ratios of 30:1 (wt/wt) and 10:1 (wt/wt), respectively. The EMSAs were performed under reducing conditions, where the sample reaction mixture contained 60 mM DTT. The samples were incubated 45 min at room temperature. To prove that the nucleic acid shift was caused by the protein nucleic acid interaction, an equivalent reaction mixture was treated with proteinase K (5g/reaction) for 45 min at 37°C. All of the samples were analyzed by agarose gel electrophoresis (1% gel) at 8 V/cm. The gels were stained by ethidium bromide and digitized by UVIdoc HD2 (Uvitec).
Protein expression, radioactive labeling, and quantification of par-ticle release.The HEK 293T cells transfected with the appropriate DNA construct were grown for 48 h posttransfection, starved for 30 min in methionine- and cysteine-deficient DMEM (Sigma), and then pulse-la-beled for 30 min with Tran35S-label (M.G.P., Czech Republic) at 125
Ci/ml. The labeled cells were then chased in complete DMEM for 16 h. The cells from pulse and pulse-chase experiments were washed with phos-phate-buffered saline (PBS), lysed in 1 ml of lysis buffer (1% Triton X-100, 1% sodium deoxycholate, 0.05 M NaCl, 25 mM Tris [pH 8.0]) on ice for 30 min, and clarified by centrifugation at 14,000⫻gfor 2 min. The culture medium of the chased cells was filtered through a 0.45- m-pore-size filter, and SDS was added to a final concentration of 0.1%. Viral proteins were immunoprecipitated from the cell lysates and culture media with a polyclonal rabbit anti-M-PMV CA antibody (1:1,000 dilution) and separated by SDS-PAGE. Radiolabeled proteins were visualized using a Typhoon 9410 phosphorimager (Amersham Biosciences).
To quantify the particle release, the radiolabeled protein bands of35 S-pulse-labeled Gag (Pr78) and pulse-chase-labeled virion-associated CA (p27) were quantified using ImageQuant TL (Amersham Biosciences). The released viral proteins are shown as relative concentrations of CA correlated to the levels of intracellular Gag in individual samples.
Single-round infectivity assay.Infectivity of M-PMV wt and CA-CTD mutants was determined as described earlier (37,46). Briefly, HEK
293T cells were cotransfected with either wt or RKK/AAA mutant pSARM-EGFP expression vector, together with the glycoprotein expres-sion vector pTMO. At 48 h posttransfection, the culture supernatants were collected and filtered through a 0.45-m-pore-size filter, and each sample was normalized for capsid protein content by quantitative West-ern blotting (37). The volume of culture supernatant used to infect HEK 293T cells was adjusted to 4 ml with complete DMEM, and the cells were incubated for an additional 48 h. The cells were fixed with 4% formalde-hyde, and the number of GFP-positive cells was determined using flow cytometry (BD FACSAria).
Viral RNA isolation and quantitative RT-PCR.Reverse transcrip-tion-PCR (RT-PCR) was performed as described previously (37). Briefly, the HEK 293T cells were transfected with wt or RKK/AAA mutant proviral construct. At 48 h posttransfection, the virus-containing medium was filtered and centrifuged, and the M-PMV CA content in the viral pellets was normalized by semiquantitative Western blot analysis. Encapsidated RNA was isolated using the QIAamp viral RNA minikit (Qiagen) accord-ing to the manufacturer’s instructions. The amount of isolated RNA was quantified by measuring theA260. To specifically quantitate the genomic RNA, RT using RevertAid H Minus M-MuLV reverse transcriptase (Fer-mentas) of isolated total RNA (of the CA-normalized amount) was per-formed. Subsequently, 2l of each RT product was used for real-time PCR using a Light Cycler 480 II Real-Time PCR system (Roche) and DyNAmo Hot Start SYBR green mix (Finnzymes). Three pairs of MPMV-CA derived primers—CA1ss (GTGGAATCTGTAGCGGACAA) and CA1as (ATTACCGGCTTGTTGGTTTC), CA2ss (GAAACCAACAA GCCGGTAAT) and CA2as (GAGCAAACAATCCTGGATCA), and CA3ss (TATTGGGCCCTCTTATCAGC) and CA3as GCAACACCCTCC TTTCTCTT—were used for each wt and RKK/AAA sample. Three con-trol samples containing (i) total RNA isolated from wt and RKK/AAA mutant, (ii) cDNA prepared from mock-infected cells, and (iii) water were used for real-time quantitative PCR. The viral genomic RNA content in individual samples was determined in three independent experiments. Nucleus isolation.HEK 293T cells transiently expressing the wild-type or RKK/AAA mutant M-PMV proviral constructs grown in 100-mm plastic dishes were incubated with leptomycin B (LMB; final concentra-tion, 20 nM) at 24 h posttransfection for 30, 60, and 120 min. The cells were then washed with PBS, and nuclear and cytosolic fractions were isolated using a Nuclei EZ Prep Nuclei Isolation kit (Sigma-Aldrich) ac-cording to the manufacturer’s protocol. Individual samples of total cell lysate, and nuclear and cytosolic fractions were then analyzed by Western blotting with the following antibodies: rabbit anti-M-PMV CA (the pres-ent study), rabbit anti-lamin A (L1293; Sigma-Aldrich), rabbit anti-cyclo-philin A (sc-20360; Santa Cruz), and monoclonal anti--actin (clone AC-74; Sigma-Aldrich).
TEM sample preparation and analysis.HEK 293T cells transiently expressing the wild-type or mutant M-PMV proviral constructs grown in 100-mm plastic dishes were washed with PBS, scraped into a microtube, and prefixed with freshly prepared 2.5% glutaraldehyde in 0.1 M cacody-late buffer (pH 7.5). After a washing step with 0.1 M cacodycacody-late buffer (pH 7.5), the cells were postfixed in 1% osmium tetroxide, dehydrated in an ethanol series (30, 50, 70, 80, 90, and 100%), and embedded in fresh Agar 100 epoxy resin. Ultrathin sections (70 nm) of cells were cut with a dia-mond knife on a Leica UC6 ultramicrotome (Leica Microsystems, Wet-zlar, Germany). The thin sections were collected on Parlodion-coated microscopy grids and contrasted using saturated uranyl acetate and lead citrate. For each sample, we analyzed approximately 30 infected cells.
In vitro-assembled VLPs were deposited on carbon-coated copper grids for 3 to 6 min. The grid was washed twice on a drop of deionized water for 20 s and stained with sodium silicotungstic acid (4%, pH 7.4) for 30 s. The excess stain was wicked off using a filtration paper, and the samples were dried in air and analyzed using a JEM-1010 transmission electron microscope (Jeol, Japan) operated at 80 kV and equipped with a Megaview III CCD (charge-coupled device) camera. The images were processed using the AnalySIS software suit (Olympus, Japan).
on November 7, 2019 by guest
http://jvi.asm.org/
Cryoelectron tomography and subtomogram averaging.In vitro -as-sembled mutant⌬ProCANC M-PMV RKK/AAA was diluted in PBS con-taining 10-nm colloidal gold and transferred to glow-discharged C-Flat 2/2 Holey carbon grids in a high-humidity chamber. Cryogrid prepara-tion was performed using a manual plunging device (EMBL, Heidelberg, Germany). Grids were blotted from the back, frozen in liquid ethane, and then stored under liquid nitrogen conditions until imaging.
Data acquisition and image processing were performed as previously described (32). In brief, tilt series were imaged on a FEI Titan Krios elec-tron microscope operated at 200 keV, with a GIF2002 post-column energy filter (using a slit width of 20 eV) and a 2k⫻2k Gatan Multiscan 795 CCD camera. Low-magnification montages for search and navigation were ac-quired using SerialEM (47). Tilt series were then acquired at appropriate positions using FEI tomography software version 4 in automated batch mode. The nominal magnification was 33,000, giving a calibrated pixel size of 2.87 Å. The tilt range was from⫺45° to⫹60° in 3° steps, collecting first from 0° to⫺45° and then from 3° to 60°, with a total dose of⬃40 e Å⫺2being applied to each tilt series. Defoci ranged between⫺1.5 and
⫺3.5m. Tomograms were reconstructed using the IMOD software suite (48).
Subtomogram averaging was performed as described previously (32) using MATLAB scripts derived from the TOM (49) and AV3 (50) pack-ages. The Dynamo software package was used for generation of masks and for FSC (Fourier shell correlation) calculations (51). Initial alignment was performed on 3⫻binned data. Processing was carried out entirely inde-pendent for two half data sets. Each half data set contained roughly the same number of tubes and an equal distribution of defoci. To obtain an initial structure, one tomogram with a defocus of⫺3.5m was chosen for each half-data set. Extracted subtomograms from this tomogram were assigned initial angles based only upon the geometry of the tubes and were averaged to generate a smooth starting reference. The subtomograms from this tomogram were then iteratively aligned and averaged in six dimensions against the reference as described previously (50). After the structure stabilized it was used as starting reference for its respective half-data set. Subsequently, all subtomograms within each half-half-data set were aligned and averaged against their respective independent starting refer-ence for six iterations. After the first two iterations, a cross-correlation-based cleaning was performed to remove subtomograms that contained no density corresponding to the M-PMV⌬ProCANC protein layer. No symmetry was applied in the alignments performed with binned, non-CTF-corrected data.
The defocus of each tomogram was measured by fitting theoretical contrast transfer function (CTF) curves to averaged power spectra from 512 square pixel tiles generated from all images in a tilt series using MATLAB scripts. CTF correction was performed using the program “CTF phase flip” implemented in IMOD (52).
Subvolumes with a size of 310 Å3were extracted from unbinned, CTF-corrected tomograms at the positions determined in the 3⫻binned
align-ments. The subtomograms were subjected to two further alignment iter-ations in which an additional 2-fold symmetry was applied. The two final references were aligned, averaged, and multiplied with a Gaussian filtered mask. Subsequent comparison of the two final references (averaged from 52,566 and 53,038 asymmetric units in each of the half-data sets, respec-tively) by FSC indicated a resolution of 10.9 Å. The final structure was sharpened applying a negative B-factor of⫺1,500 Å2, while filtering to the resolution determined at the 0.143 FSC threshold. The wt M-PMV
⌬ProCANC tube structure used for comparison was EMD-2089 (31). Visualization of tomograms or electron microscopy density maps was performed using either IMOD (48), UCSF chimera (53), or Amira4 (FEI Visualization Sciences Group) with the electron microscopy toolbox (54).
RESULTS
To study the role of the basic RKK region, we prepared mutants in
which the RKK amino acids were replaced either by a triple alanine
(RKK/AAA mutant), or by a GPG (RKK/GPG mutant) sequence.
The reason for selecting the latter mutation was that in HIV-1 CA
the GPG sequence is located at a position corresponding to that of
RKK in M-PMV and thus could have the same functional role.
The RKK/AAA and RKK/GPG mutations were introduced into
both bacterial expression and M-PMV proviral vectors.
Mutation of the RKK region influences assembly
in vitro
.
We
first used a bacterial system to study the role of the RKK basic
region in the assembly of immature particles. The RKK/AAA and
RKK/GPG mutations were introduced into truncated M-PMV
Gag (
⌬
ProCANC), and its ability to assemble in bacterial cells was
assessed using TEM. In contrast to wt
⌬
ProCANC protein that
forms tubular and spherical VLPs in
E. coli
cells, none of the RKK
mutant proteins formed any particles, indicating that the RKK
sequence is important for the assembly of VLPs in bacterial cells.
We next purified the expressed proteins and studied their
abil-ity to assemble into VLPs
in vitro
using TEM. Our previous results
showed that the wt
⌬
ProCANC of M-PMV could form
in vitro
either spherical (
5
) or tubular (
31
) particles, depending on the
assembly conditions. The RKK/GPG
⌬
ProCANC mutant protein
did not assemble into any particles
in vitro
under any assembly
conditions tried (see Materials and Methods). In the presence of
phage DNA at reducing conditions, the RKK/AAA mutant of
⌬
ProCANC protein formed tubular VLPs that were similar to
those formed by the wt protein (
Fig. 1
) but were typically longer
(up to several microns) than those of the wt protein. This mutant
assembled also in the presence of MS2 phage RNA, although at
both reducing and nonreducing conditions it formed only a few
regular spherical particles (as seen in
Fig. 2C
), alongside fragments
D
C
B
A
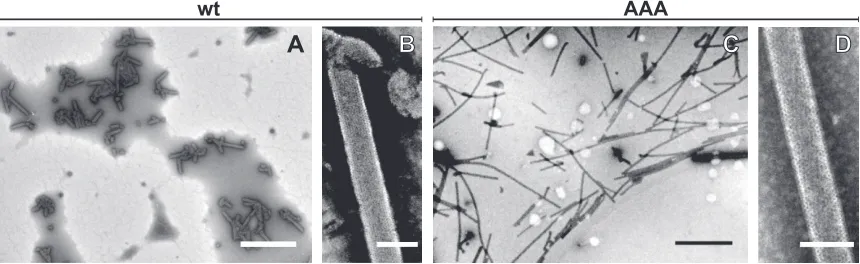
wt
AAA
FIG 1TEM analysis of negatively stainedin vitroassembled M-PMV particles. wt⌬ProCANC (A and B) and RKK/AAA mutant⌬ProCANC (C and D) particles assembledin vitrounder reducing conditions in the presence ofphage DNA. Scale bars: A and C, 2m; B and D, 100 nm.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.79.509.68.200.2]of particles and aggregated protein. Particle assembly was
depen-dent on the presence of nucleic acid, an observation consistent
with our previous finding that the efficiency of wt
⌬
ProCANC
VLP assembly is significantly facilitated by the presence of nucleic
acids (
5
).
To assess the efficiency of
in vitro
VLP assembly, we performed
OptiPrep gradient ultracentrifugation of particles assembled
un-der reducing conditions from both wt and mutant
⌬
ProCANC
proteins in the presence of either MS2 RNA or
phage DNA,
followed by SDS-PAGE analysis of individual fractions and by
densitometric analysis of the bands (
Fig. 2
). All VLPs assembly
reactions, together with ultracentrifugations and protein
quanti-tation analyses, were performed in triplicate from independent
protein isolations. Independent of the type of added nucleic acid,
the
in vitro
assembly of wt
⌬
ProCANC protein proceeded
effi-ciently. The proportion of the assembled protein ranged between
ca. 60% and ca. 50% in the presence MS2 RNA and
DNA,
re-spectively (
Fig. 2A
). For the RKK/GPG mutant, for which no
par-ticles were observed by TEM, the assembly efficiency was
negligi-ble, and no visible bands corresponding to assembled particles
were observed in the SDS-PAGE gel. The yield of the RKK/AAA
mutant particles was significantly lower than that of wild type
under both conditions (i.e., with MS2 RNA or
DNA) and, unlike
the wild-type particles, it was more dependent on the amount of
nucleic acid added. Increasing the amount of MS2 RNA in the
assembly mixture from 6 to 12
g enhanced the VLP assembly
efficiency of RKK/AAA mutant from 6.4% to 21% while the wt
protein assembled most efficiently when 6
g of MS2 RNA was
added (
Fig. 2A
). On the other hand, increased concentration of
the
DNA slightly reduced the assembly efficiency of the RKK/
AAA mutant from ca. 21%
⫾
1% to ca. 16%
⫾
2% (
Fig. 2A
),
which presumably reflects the redistribution of particles in the
gradient as a result of aggregation. We inspected the gradient
frac-tions using TEM to confirm the presence of assembled particles in
the relevant fractions (
Fig. 2B
to
E
). As expected, spherical or
tubular assembled particles were observed in fractions 6 to 9.
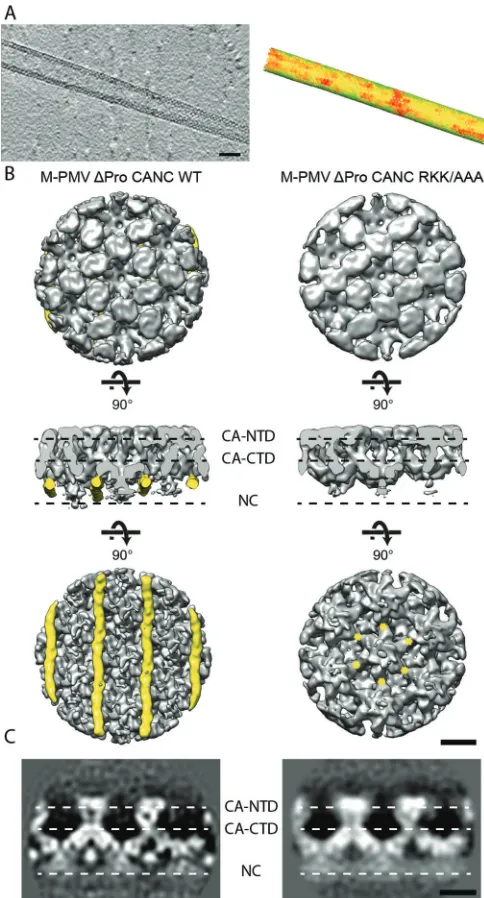
Structure of
in vitro
-assembled RKK/AAA VLPs.
We next
assessed whether the RKK/AAA mutation affects the structure of
the assembled tubular particles.
⌬
ProCANC M-PMV RKK/AAA
tubes assembled
in vitro
under reducing conditions in the
pres-ence of
DNA were subjected to cryoelectron tomography.
Con-sistent with the negative-stain electron microscopy results, long
tubular arrays were observed (
Fig. 3A
). Along the surface of the
tubes hexagonal patches were visible, similar to those observed in
cryoelectron tomograms of wt
⌬
ProCANC tubes (
32
). We
per-formed subtomogram averaging and obtained an
⬃
11-Å
resolu-tion structure (
Fig. 3B
). We compared the mutant structure with
the available wt
⌬
ProCANC tube structure (
31
). No differences
were seen in the capsid region (
Fig. 3B
), indicating that the RKK/
AAA mutation does not influence the tertiary or quaternary
struc-ture of the capsid domains. Differences were observed in the
re-gion underlying the C-terminal part of capsid (
Fig. 3B
and
C
),
where the nucleocapsid and nucleic acids are located. Here, the
RKK/AAA mutant lacks a filamentous density that was present in
the wt tube (
Fig. 3B
, yellow). The dimensions of this density are
approximately that of a nucleic acid double helix. We previously
suggested that this density was nucleic acid recruited by the RKK
motif in M-PMV that is not present in the HIV CA sequence nor
in the cryoelectron microscopy structure (
30
,
31
). The absence of
this structure in the RKK/AAA mutant strongly supports this
hy-pothesis.
The RKK region plays role in nucleic acid binding.
The
ob-servations presented so far suggest a role for the RKK basic region
in binding of nucleic acids. To investigate whether the RKK/GPG
and RKK/AAA mutations alter nucleic acid binding, we mixed the
studied proteins with a 1-kb DNA ladder in two different ratios
(protein/DNA ratios of 30:1 [wt/wt] and 10:1 [wt/wt]) and
per-formed an EMSA. The ratios of protein to nucleic acid were based
on the optimization of reaction conditions, where the DNA was
bound efficiently to wt and mutant protein. None of the tested
proteins (wt, RKK/AAA, or RKK/GPG) showed any traces of
con-taminating nucleic acids (
Fig. 4
, lanes PC), and no shifts were
observed when the equivalent reaction mixtures were also treated
with proteinase K (
Fig. 4
, lane Pr), indicating that any observed
shifts are caused exclusively by the interaction of the protein with
added nucleic acid. At a protein/DNA ratio of 30:1 the wt protein
1 2 3 4 5 6 7 8 9 10 11 12
15% 55%
12 μg
61±5%
6 μg
64±4%
6 μg
6±4%
12 μg
21±5%
12 μg
47±5%
6 μg
50±7%
6 μg
21±1%
12 μg
16±2%
wt unassembled 0%
λ DNA
MS2 RNA
wt
AAA
wt
AAA
A
B
wt, MS2 RNAC
AAA, MS2 RNAD
wt, λ DNAE
AAA, λ DNAFIG 2SDS-PAGE and TEM analysis of OptiPrep gradient fractions. (A) A dialyzed assembly mixture was ultracentrifuged through a 15 to 55% OptiPrep equilibrium gradient, and collected fractions were analyzed by SDS-PAGE. The amount of proteins in the lanes was assessed densitometrically. The percentage (means with standard deviations,n⫽3) represents the relative amounts of wt and RKK/AAA mutant proteins assembled into particles (fractions 6 to 9) in the presence of either MS2 phage RNA orphage DNA. (B, C, D, and E) TEM analysis of OptiPrep gradient fractions 7 containing wt and RKK/AAA mutant protein particles assembled in the presence of 12g of MS2 RNA (B and C, respectively) and wt and RKK/AAA mutant protein particles assembled in the presence of 6
g ofDNA (D and E, respectively). Scale bars, 100 nm.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.78.509.70.226.2]quantitatively bound DNA content from the reaction mixture
(
Fig. 4
, left panel, wt lane P), whereas at a protein/DNA ratio of
10:1 some DNA was not bound by the protein and remained free
in the solution (
Fig. 4
, right panel, wt, lane P). The RKK/AAA and
RKK/GPG mutants showed a much lower ability to bind DNA at
both ratios (
Fig. 4
, AAA and GPG, lanes P), the reduction was
particularly severe for the RKK/GPG mutant. The
higher-molec-ular-weight fragments from the 1-kb ladder showed greater
reten-tion than those of the lower-molecular-weight fragments
(com-pare
Fig. 4
, lanes P). The EMSA indicates that mutations in the
RKK region indeed lead to reduced DNA binding that correlates
with a reduced efficiency of particle assembly.
The RKK region regulates the efficiency of intracytoplasmic
particle assembly.
To investigate whether the effect of mutations
in the RKK region on nucleic acid binding and particle assembly
observed
in vitro
are reflected in changes in the viral life cycle, HEK
293T cells were transfected with proviral wt pSARM4 plasmid and
the corresponding RKK/AAA and RKK/GPG mutants.
Transfec-tion with all of the constructs led to the expression of similar
amounts of Gag, Gag-Pro, and Gag-Pro-Pol polyproteins,
indi-cating that the level of expression was not affected by the
muta-tions (
Fig. 5A
). At 48 h posttransfection, the expression and
processing of the wt and mutant M-PMVs were assessed by
pulse-chase assay and Western blot analysis of transfected cells.
We measured the amount of protein in the supernatant (after a
16-h chase) relative to the amount of cell-associated proteins
(be-fore chase) in order to estimate the amount of virus released (
Fig.
5C
). The efficiency of release for the RKK/AAA mutant was only
ca. 45% that of the wt. Both the wt and the RKK/AAA virions
contained processed CA protein, indicating that proteolytic
pro-cessing of capsid was not affected by the RKK/AAA mutation (
Fig.
5B
). In the RKK/GPG mutant the ratio of CA protein detected in
the medium was
⬍
20% that of the wt.
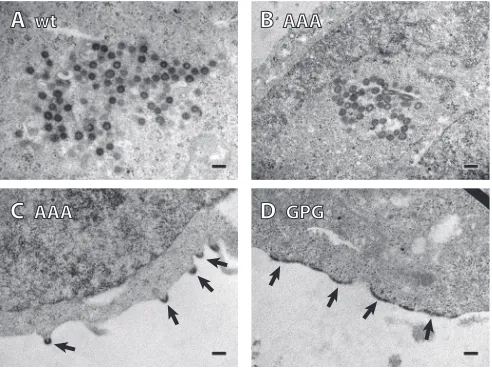
To analyze the assembly pathway of the virions, we performed
TEM analysis of thin sections of transfected HEK 293T cells (
Fig.
6
). We inspected about 30 infected cells of each wt or mutant
viruses and semiquantitatively assessed the virus particle
morpho-genetic types. Transfection with wt M-PMV gives rise to large
numbers of intracellularly assembled D-type particles in the
peri-centriolar region. We counted approximately 100 intracellular
FIG 3Cryoelectron tomography and subtomogram averaging analysis of M-PMV⌬ProCANC RKK/AAA tubes. (A, left) Slice through a cryoelec-tron tomogram containing a⌬ProCANC RKK/AAA tube. The protein den-sity is black. Scale bar, 50 nm. (A, right) Corresponding subtomogram averaging output lattice map to the tube represented in panel A. Hexagons are placed on the positions determined during alignment, resolving the orientations of the hexameric unit cells of the proteins along the tube. Hexagon colors denote cross-correlation of the respective subtomogram with the reference (green, high; red, low). (B) Comparison of the structures in wt⌬ProCANC (left) (EMD-2089) and ⌬ProCANC RKK/AAA tubes (right). Isosurface representations of the two structures, both filtered to 11 Å, are shown from the outside (top), from a horizontal view (middle), and from the inside of the tube (bottom). The filamentous densities present only in wt tubes are colored yellow. Yellow asterisks mark the approximate positions of the RKK sequences in the central hexamer. Scale bar, 50 Å. (C) Slices through the electron density, indicating the positions of the protein domains in an orientation corresponding to the middle panel in panel B. The protein density is white.
1kbPC P Prwt PC P PrAAA PC P PrGPG 30 : 1
1kbPC P Prwt PCAAAP PrPC P PrGPG 10 : 1
250 500 750 1000 1500 2000 3000 10 000 5000 bp
FIG 4EMSA results for wt⌬ProCANC and RKK/AAA and RKK/GPG mutant proteins. The EMSAs were performed at 30:1 (wt/wt) and 10:1 (wt/wt) pro-tein/DNA ratios under reducing conditions. PC, protein control (no nucleic acid added); 1kb, 1-kbp DNA ladder; P, protein-DNA interaction mixture; Pr, Proteinase K-treated protein-DNA interaction mixture.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.40.282.66.515.2] [image:6.585.303.543.67.213.2]particles which were exclusively D-type. In the case of the RKK/
AAA mutant, we observed not only D-type pericentriolar
assem-bly but also a considerable amount of particles assembling at the
plasma membrane (C-type assembly) (
Fig. 6B
and
C
,
respec-tively). In 30 cells we counted 58 intracellular D-type particles and
29 partially assembled C-type particles budding from the plasma
membrane. The intracellular RKK/AAA M-PMV particles had a
spherical shape similar to that of the wt M-PMV (compare
Fig. 6A
and
B
). Released virions were also observed. The GPG mutant
failed to form immature particles inside the cells; however, a large
amount of electron-dense material (presumably Gag)
accumu-lated underneath the cell plasma membrane (
Fig. 6D
) (
55
). This
compact layer on the membrane did not form any released virus
particles or budding structures. Based on this analysis (see further
discussion below), we concluded that the CA protein detected in
the medium of the RKK/GPG mutant most likely represents free
protein and not virus particles.
The infectivity of the released particles was tested by
single-round infectivity assay (
Fig. 7A
). The infectivity of the RKK
mu-tants was below the detection limit (
⬍
1%) compared to that of the
wt M-PMV, indicating that the mutation affected some key step of
the infectivity process.
The above-mentioned results of particle release and TEM
anal-ysis (
Fig. 5C
and
6
, respectively) indicate that mutation of the RKK
motif leads to a reduction in particle release efficiency and to a
partial relocalization of the assembly site from the pericentriolar
region to the plasma membrane. The mutant particles are released
from the cells but show severely abolished infectivity.
The RKK region is important for genomic RNA
incorpora-tion.
To assess whether mutation of the RKK motif influences
nucleic acid incorporation in infected cells, we determined the
total RNA and the genomic RNA content of the released particles.
We found that the total amounts of RNA (measured
spectropho-tometrically at
A
260) incorporated into the mutant and wt virions
were comparable (wt [100%
⫾
5%] and RKK/AAA mutant [118%
⫾
18%]). To quantify genomic RNA incorporated into released
M-PMV particles, the isolated total RNA from the wt and RKK/AAA
mutant was reverse transcribed and analyzed by real-time qPCR
using three various CA-specific primer pairs. No positive signals
(i.e., threshold cycle [C
T]
⬎
38) were determined in the control
samples in which cDNA isolated from mock-infected HEK 293T
cells, isolated total RNA, or water were used as a template for
real-time quantitative PCR. Surprisingly, significant differences
between the RKK/AAA mutant and the wt particles were observed
by specific quantification (quantitative RT-PCR) of the genomic
RNA (
Fig. 7B
). The RKK/AAA mutant particles contained only
mock wt AAA GPG mock wt AAA GPG
CA Gag
Gag-P ro Gag-P
ro-P ol
pulse cells chase virions
A
B
C
particle releaseFIG 5Synthesis, release, and processing of M-PMV wild-type and RKK mutants. The HEK 293T cells were transfected with wt, RKK/AAA, and RKK/GPG mutant proviral DNAs. Viral proteins were metabolically labeled with [35S]cysteine-methionine mix for 30 min and then chased for 16 h. (A) M-PMV CA
(p27)-related polyproteins Gag (Pr78), Gag-Pro (Pr95), and Gag-Pro-Pol (Pr180) were then immunoprecipitated from the cells by rabbit anti-M-PMV CA antibody, separated by SDS-PAGE, and analyzed by using a Typhoon phosphorimager. (B) Released M-PMV CA (p27) was immunoprecipitated from the culture medium by rabbit anti-M-PMV CA antibody at 16 h after the chase, separated by SDS-PAGE, and analyzed by using a Typhoon phosphorimager. (C) Quantification of M-PMV wt and RKK mutants release. The band intensities of35S-pulse-labeled Gag (Pr78) and released CA (p27) were calculated. The relative
percentage of CA released into the culture media was corrected for intracellular expression of individual samples. The released viral proteins are shown as the average relative concentration of CA correlated to the level of intracellular Gag in individual samples. Error bars represent standard errors of the mean calculated from two independent experiments.
A
wt
C
AAA
D
GPG
B
AAA
FIG 6TEM analysis of thin sections of M-PMV-infected HEK 293T cells. (A) wt virus. The virus assembled inside the cytoplasm (D-type particles). (B and C) RKK/AAA mutant. The particles were assembled in the cytoplasm (D-type) and also on the cell membrane (C-type); the particles are marked with arrows. (D) RKK/GPG mutant. Large amounts of protein, presumably Gag, accumu-lated at the cell membrane (indicated by arrows). Scale bars, 200 nm.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.100.487.67.233.2] [image:7.585.41.287.482.667.2]about 9% (
⫾
1.5%) of genomic RNA in comparison to the wt. The
data represent three independent transfections, where the wt
val-ues were arbitrarily assigned as 100%.
The RKK region influences nuclear trafficking of Gag.
It was
previously observed that mutation of basic amino acid patches in
the pp24 domain of M-PMV Gag affects its localization to nuclear
pores (
56
). We therefore next assessed whether the genome
pack-aging defect observed in the RKK/AAA mutant might also relate to
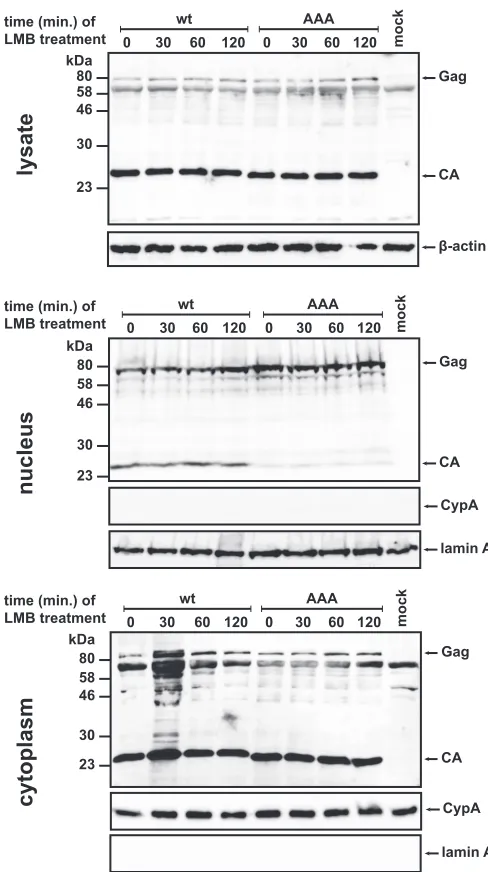
altered nuclear trafficking of Gag. To delay fast export of proteins
from the nucleus, we treated the cells at 24 h posttransfection with
LMB. This inhibitor of CRM1-dependent nuclear export was
se-lected because it was suggested to affect M-PMV Gag exit from the
nucleus (
22
). Analysis of LMB-treated cells thus should reveal
even transient presence of Gag inside the nuclei. Nuclear and
cy-tosolic fractions of cells transfected with wt or RKK/AAA mutant
M-PMV vector were analyzed for the presence of Gag (
Fig. 8
).
The level of wt M-PMV Gag in the nuclear fractions gradually
increased with increasing length of LMB treatment over a 120-min
time frame. For the RKK/AAA mutant, the initial amount of Gag
in the nucleus was notably higher than for the wt but did not
increase during longer incubations. These results are consistent
with a reduced rate of nuclear export for the RKK/AAA mutant of
Gag. A reduced export rate would lead to increased
accumula-tion of RKK/AAA Gag during the 24-h period of
posttransfec-tion before LMB treatment. Upon reaching saturaposttransfec-tion, no
in-crease in nuclear RKK/AAA was observed when export was
inhibited by LMB.
Another remarkable observation is the difference in the
pres-ence of the wt and RKK/AAA mutant CA proteins inside the cell
nucleus (
Fig. 8
). Although nuclear localization is not surprising
for the wt CA protein since it was also reported for the HIV-1 CA
protein in primary human macrophages and HeLa cells at the
early stages of infection (
57
,
58
), the AAA mutation of the RKK
motif in the M-PMV CA protein almost completely abolished the
import of the CA protein into the nucleus (
Fig. 8
). The detectable
amounts of M-PMV CA in the nucleus may originate from CA
present in a preintegration complex in cells newly infected by the
released particles. The absence of CA in the nuclei of cells
trans-fected with the RKK mutant is consistent with the fact that the
RKK/AAA mutant virus lacking the vRNA is noninfectious and
thus unable to introduce mature CA into the cells.
A
B
FIG 7Relative infectivity and genomic RNA incorporation into M-PMV wt and RKK mutant viruses. (A) The relative infectivity of RKK mutants was determined by a single-round assay. HEK 293T cells were cotransfected with wild-type or RKK mutant pSARM-EGFP and pTMO vectors. At 48 h post-transfection, the virus particles from the culture medium were filtered and normalized for CA (p27) by quantitative Western blotting. Equivalent amounts of virions were used to infect fresh HEK 293T cells. At 48 h postin-fection, the cells were harvested, and the numbers of GFP-positive cells were determined by flow cytometry (BD FACSAria). The mean percentage of three independent infectivity measurements (with calculated standard deviations) for each mutant relative to the wild-type is shown. (B) Mean relative RNA contents with standard deviations (n⫽3) of the wt and RKK/AAA mutant viruses are shown. Viral RNA was isolated from purified viral particles released into the culture media at 48 h posttransfection. After reverse transcription of normalized samples (see Materials and Methods), real-time PCR was used to quantify the amount of incorporated RNA.
time (min.) of LMB treatment
β-actin Gag
CA 80
58 46
30
23 kDa
0 30 60 120 0 30 60 120 mock
wt AAA
lysate
time (min.) of LMB treatment
cytoplasm
80 58 46
30
23 kDa
Gag
CA 0 30 60 120 0 30 60 120 mock
wt AAA
lamin A CypA time (min.) of
LMB treatment
nucleus
lamin A 80
58 46
30
23 kDa
Gag
CA 0 30 60 120 0 30 60 120 mock
wt AAA
CypA
FIG 8Western blot analysis of LMB-treated transfected HEK 293T cells. At 24 h posttransfection of the HEK 293T cells with the wild-type or RKK/AAA mutant M-PMV proviral constructs, LMB at a final concentration of 20 nM was added to the cells. Incubation proceeded for 30, 60, and 120 min, and then nuclear and cytosolic fractions were isolated. Individual samples of total cell lysate and nuclear and cytosolic fractions were then analyzed by Western blot-ting with the following antibodies: rabbit M-PMV CA, rabbit anti-lamin-A, rabbit anti-cyclophilin A (CypA), and mouse monoclonal anti- -actin.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.297.541.63.499.2] [image:8.585.41.289.69.216.2]DISCUSSION
Role of RKK in nucleic acid binding and virus assembly.
The
mutation of the RKK region to either AAA or GPG had
wide-ranging effects on Gag assembly, nucleic acid incorporation, and
virus production. In the case of mutation to GPG, assembly,
nu-cleic acid incorporation and virus production were essentially
abolished. We speculate that the insertion of a proline residue in
this region may cause structural defects in CA which interfere with
its proper function.
Mutation of RKK to AAA was still permissive for
in vitro
as-sembly. This allowed us to test our previous hypothesis—that the
extended filament of density underlying CA in
in vitro-assembled
⌬
ProCANC tubes represents an ordered nucleic acid structure
bound to the basic RKK region (
30
). Indeed, structural analysis
showed that this filament is lost in the RKK/AAA mutant,
indicat-ing that the nucleic acid is either absent in the particles or is no
longer bound to the RKK motif. Consistent with this observation,
the efficiency of nucleic acid binding by the mutant protein is
reduced. We observed that the mutation also leads to significantly
reduced efficiency of Gag assembly, suggesting that the
interac-tions between RKK and nucleic acid may facilitate the assembly.
This could be achieved either by promoting CA dimerization or
further oligomerization into a hexameric lattice. Since the wt
⌬
ProCANC assembles into particles of similar morphology both
in
E. coli
and
in vitro, the failure of the
⌬
ProCANC RKK/AAA
mutant to assemble in
E. coli
suggests that RKK-mediated CA
oligomerization may be particularly important in crowded
cellu-lar environments. Together, these observations are consistent
with a model in which nucleic acid binding by the RKK motif
creates a scaffold promoting Gag assembly.
When placed in the context of the infectious virus, the RKK/
AAA mutation leads to a ca. 50% drop in virus particle release,
which is consistent with the assembly defect observed
in vitro.
Strikingly, the mutation also influences the site of assembly:
whereas wild-type M-PMV assembles D-type particles in the
peri-centriolar region (
1
,
59
), in the AAA mutant, approximately
one-third of the observed particles appeared as C-type particles
assem-bling at the plasma membrane. In the GPG mutant, dense layers of
accumulated protein are seen underlying the plasma membrane,
which are not seen in noninfected cells. Based on the thickness of
these layers, and because similar structures have previously been
shown to be formed by assembly-incompetent Gag polyproteins
(
55
,
60
,
61
), we presume that these extra layers observed under the
plasma membrane are formed by accumulated mutant Gag
pro-tein.
Trafficking of M-PMV Gag to the pericentriolar assembly site
is dependent on the interaction between a CTRS signal in MA with
the Tctex-1 component of dynein (
4
). We consider it unlikely that
mutation of RKK influences this interaction because the MA site is
distal to CA. We prefer the following alternative explanation for
the relocation of assembly. The assembly of C-type retroviruses at
the plasma membrane is promoted by CA-CA interactions,
NC-RNA-NC interactions, and MA-membrane-MA interactions. In
the case of M-PMV, a D-type retrovirus, assembly in the
cyto-plasm is promoted by CA-CA, NC-RNA-NC, and
nu-cleic acid-CA(RKK) interactions. In the case where the
CA(RKK)-nucleic acid-CA(RKK) interaction is abolished by mutation of the
RKK, the cytosolic assembly efficiency is reduced, and part of Gag
is transported to the PM, where MA-membrane-MA interactions
promote C-type assembly.
Role of RKK in genome packaging and nuclear transport.
Surprisingly, while the RKK/AAA mutation does not affect the
total amount of RNA being incorporated into virus particles, it
dramatically reduces the packaging of genomic RNA. This is most
likely the cause of the observed reduced infectivity. Specific
pack-aging of genomic RNA in retroviruses is typically mediated by NC
(
62–64
). While some studies have indicated that interactions
be-tween MA and nucleic acid may modulate this effect (
18
,
19
), a
role for CA in genome packaging has not been described. We
cannot rule out that the RKK motif influences packaging by direct
interaction with sequences in the genomic RNA, but this seems
unlikely. A number of other hypotheses seem more likely. The
RKK motif may help to stabilize the Gag-vRNA complex, or it may
contribute to a structural arrangement of the NC region which is
required for packaging. The reduction in genome packaging may
be a secondary effect resulting from the relocalization of viral
as-sembly from the pericentriolar region to the plasma membrane.
Alternatively, packaging may be hindered by a change in the
nu-clear transport of Gag.
Nuclear trafficking of Gag has previously been shown to be
important for genomic RNA incorporation for RSV (
24
,
25
). In
the case of M-PMV, Gag associates with nuclear pores (
56
) and
may enter the nucleus (
22
). We found both RKK/AAA and
wild-type Gag in the nucleus. The phenowild-type we observed for the RKK/
AAA Gag is consistent with reduced efficiency of nuclear export.
Because the RSV nuclear export signal consists mainly of nonpolar
amino acids in the p10 domain (
65
), we expect that the export of
Gag from the nucleus is not directly inhibited by the mutation of
the polar RKK region, but it is rather mediated by the Gag-vRNA
interactions and their simultaneous export from the nucleus. This
could lead to a reduction in genomic RNA export and packaging
into virus particles. Alternatively, if nuclear export is promoted by
a Gag-vRNA complex, then reduced export might result indirectly
from the RKK/AAA mutation interfering with genomic RNA
binding. In either case, the reduction in genomic RNA
incorpora-tion and the altered nuclear trafficking would be related
pheno-types.
In summary, our data suggest that the RKK region of CA
mod-ulates interactions between Gag and the viral genomic RNA in a
manner that is also accompanied with changes in nuclear
traffick-ing. The nucleic acid recruited by RKK serves a scaffolding
func-tion that promotes Gag assembly. We speculate that D-type
ret-roviruses such as M-PMV may require an additional assembly
scaffold to replace the function of the plasma membrane in the
assembly of C-type retroviruses.
ACKNOWLEDGMENTS
This study was supported by the Grant Agency of the Czech Republic (14-15326S), by the LH12011 project and NPU I sustainability projects LO1302 and LO1304 from the Czech Ministry of Education, and by Deut-sche Forschungsgemeinschaft grant BR 3635/2-1 to J.A.G.B.
We thank Tanmay Bharat for preparation of the cryoelectron mi-croscopy grids and Wim Hagen for assistance with cryoelectron to-mography.
This study was technically supported by Frank Thommen and EMBL IT-services.
on November 7, 2019 by guest
http://jvi.asm.org/
FUNDING INFORMATION
Work in the laboratory of John A. G. Briggs was funded by Deutsche Forschungsgemeinschaft (DFG) (BR 3635/2-1). This work, including the efforts of Tomas Ruml, was funded by the Grant Agency of the Czech Republic (14-15326S) and the Czech Ministry of Education (NPU I sus-tainability projects LO1302 and LO1304).
REFERENCES
1.Choi G, Park S, Choi B, Hong S, Lee J, Hunter E, Rhee SS. 1999. Identification of a cytoplasmic targeting/retention signal in a retroviral Gag polyprotein. J Virol73:5431–5437.
2.Sfakianos JN, Hunter E.2003. M-PMV capsid transport is mediated by Env/Gag interactions at the pericentriolar recycling endosome. Traffic
4:671– 680.http://dx.doi.org/10.1034/j.1600-0854.2003.00126.x. 3.Sakalian M, Hunter E.1999. Separate assembly and transport domains
within the Gag precursor of Mason-Pfizer monkey virus. J Virol73:8073– 8082.
4.Vlach J, Lipov J, Rumlová M, Veverka V, Lang J, Srb P, Knejzlík Z, Pichová I, Hunter E, Hrabal R, Ruml T.2008. D-retrovirus morphoge-netic switch driven by the targeting signal accessibility to Tctex-1 of dy-nein. Proc Natl Acad Sci U S A105:10565–10570.http://dx.doi.org/10 .1073/pnas.0801765105.
5.Ulbrich P, Haubova S, Nermut MV, Hunter E, Rumlova M, Ruml T.
2006. Distinct roles for nucleic acid in in vitro assembly of purified Mason-Pfizer monkey virus CANC proteins. J Virol80:7089 –7099.http://dx.doi .org/10.1128/JVI.02694-05.
6.Campbell S, Vogt VM.1997. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: identification of the p10 do-main as a morphological determinant in the formation of spherical parti-cles. J Virol71:4425– 4435.
7.Gross I, Hohenberg H, Kräusslich HG.1997. In vitro assembly proper-ties of purified bacterially expressed capsid proteins of human immuno-deficiency virus. Eur J Biochem249:592– 600.http://dx.doi.org/10.1111/j .1432-1033.1997.t01-1-00592.x.
8.Hadravová R, de Marco A, Ulbrich P, Stokrová J, Dolezal M, Pichová I, Ruml T, Briggs JAG, Rumlová M.2012. In vitro assembly of virus-like particles of a gammaretrovirus, the murine leukemia virus XMRV. J Virol
86:1297–1306.http://dx.doi.org/10.1128/JVI.05564-11.
9.Ma YM, Vogt VM.2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J Virol78:52– 60.
http://dx.doi.org/10.1128/JVI.78.1.52-60.2004.
10. Pastré D, Hamon L, Landousy F, Sorel I, David M-O, Zozime A, Le Cam E, Piétrement O.2006. Anionic polyelectrolyte adsorption on mica mediated by multivalent cations: a solution to DNA imaging by atomic force microscopy under high ionic strengths. Langmuir22:6651– 6660.
http://dx.doi.org/10.1021/la053387y.
11. Datta SAK, Zuo X, Clark PK, Campbell SJ, Wang Y-X, Rein A.2011. Solution properties of murine leukemia virus gag protein: differences from HIV-1 gag. J Virol85:12733–12741.http://dx.doi.org/10.1128/JVI .05889-11.
12. Rulli SJ, Hibbert CS, Mirro J, Pederson T, Biswal S, Rein A. 2007. Selective and nonselective packaging of cellular RNAs in retrovirus parti-cles. J Virol81:6623– 6631.http://dx.doi.org/10.1128/JVI.02833-06. 13. Jouvenet N, Simon SM, Bieniasz PD.2009. Imaging the interaction of
HIV-1 genomes and Gag during assembly of individual viral particles. Proc Natl Acad Sci U S A106:19114 –19119.http://dx.doi.org/10.1073 /pnas.0907364106.
14. Dorfman T, Luban J, Goff SP, Haseltine WA, Göttlinger HG.1993. Mapping of functionally important residues of a cysteine-histidine box in the human immunodeficiency virus type 1 nucleocapsid protein. J Virol
67:6159 – 6169.
15. Dannull J, Surovoy A, Jung G, Moelling K.1994. Specific binding of HIV-1 nucleocapsid protein to PSI RNA in vitro requires N-terminal zinc finger and flanking basic amino acid residues. EMBO J13:1525–1533. 16. Aldovini A, Young RA.1990. Mutations of RNA and protein sequences
involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J Virol64:1920 –1926.
17. Gorelick RJ, Henderson LE, Hanser JP, Rein A.1988. Point mutants of Moloney murine leukemia virus that fail to package viral RNA: evidence for specific RNA recognition by a “zinc finger-like” protein sequence. Proc Natl Acad Sci U S A85:8420 – 8424.http://dx.doi.org/10.1073/pnas.85.22 .8420.
18. Alfadhli A, Still A, Barklis E.2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol83:
12196 –12203.http://dx.doi.org/10.1128/JVI.01197-09.
19. Ott DE, Coren LV, Gagliardi TD.2005. Redundant roles for nucleocap-sid and matrix RNA-binding sequences in human immunodeficiency vi-rus type 1 assembly. J Virol79:13839 –13847.http://dx.doi.org/10.1128 /JVI.79.22.13839-13847.2005.
20. Fritz CC, Green MR.1996. HIV Rev uses a conserved cellular protein export pathway for the nucleocytoplasmic transport of viral RNAs. Curr Biol6:848 – 854.http://dx.doi.org/10.1016/S0960-9822(02)00608-5. 21. Butterfield-Gerson KL, Scheifele LZ, Ryan EP, Hopper AK, Parent LJ.
2006. Importin-beta family members mediate alpharetrovirus gag nuclear entry via interactions with matrix and nucleocapsid. J Virol80:1798 – 1806.http://dx.doi.org/10.1128/JVI.80.4.1798-1806.2006.
22. Baluyot MF, Grosse SA, Lyddon TD, Janaka SK, Johnson MC.2012. CRM1-dependent trafficking of retroviral Gag proteins revisited. J Virol
86:4696 – 4700.http://dx.doi.org/10.1128/JVI.07199-11.
23. Lochmann TL, Bann DV, Ryan EP, Beyer AR, Mao A, Cochrane A, Parent LJ.2013. NC-mediated nucleolar localization of retroviral gag proteins. Virus Res 171:304 –318. http://dx.doi.org/10.1016/j.virusres .2012.09.011.
24. Garbitt-Hirst R, Kenney SP, Parent LJ.2009. Genetic evidence for a connection between Rous sarcoma virus gag nuclear trafficking and genomic RNA packaging. J Virol 83:6790 – 6797.http://dx.doi.org/10 .1128/JVI.00101-09.
25. Gudleski N, Flanagan JM, Ryan EP, Bewley MC, Parent LJ. 2010. Directionality of nucleocytoplasmic transport of the retroviral Gag pro-tein depends on sequential binding of karyopherins and viral RNA. Proc Natl Acad Sci U S A 107:9358 –9363. http://dx.doi.org/10.1073/pnas .1000304107.
26. Kemler I, Saenz D, Poeschla E.2012. Feline Immunodeficiency Virus Gag Is a Nuclear Shuttling Protein. J Virol86:8402– 8411.http://dx.doi .org/10.1128/JVI.00692-12.
27. Tabernero C, Zolotukhin AS, Valentin A, Pavlakis GN, Felber BK.1996. The posttranscriptional control element of the simian retrovirus type 1 forms an extensive RNA secondary structure necessary for its function. J Virol70:5998 – 6011.
28. Ernst RK, Bray M, Rekosh D, Hammarskjöld ML.1997. A structured retroviral RNA element that mediates nucleocytoplasmic export of in-tron-containing RNA. Mol Cell Biol 17:135–144. http://dx.doi.org/10 .1128/MCB.17.1.135.
29. Grüter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E.1998. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol Cell1:649 – 659.http://dx.doi.org/10.1016/S1097-2765(00)80065-9. 30. de Marco A, Davey NE, Ulbrich P, Phillips JM, Lux V, Riches JD,
Fuzik T, Ruml T, Kräusslich H-G, Vogt VM, Briggs JAG. 2010. Conserved and variable features of Gag structure and arrangement in immature retrovirus particles. J Virol84:11729 –11736.http://dx.doi .org/10.1128/JVI.01423-10.
31. Bharat TAM, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JAG. 2012. Structure of the immature retroviral capsid at 8 Å resolution by cryo-electron microscopy. Nature
487:385–389.http://dx.doi.org/10.1038/nature11169.
32. Schur FKM, Hagen WJH, de Marco A, Briggs JAG.2013. Determination of protein structure at 8.5Å resolution using cryo-electron tomography and sub-tomogram averaging. J Struct Biol184:394 – 400.http://dx.doi .org/10.1016/j.jsb.2013.10.015.
33. Schur FK, Hagen WJ, Rumlová M, Ruml T, Müller B, Kräusslich HG, Briggs JAG.2014. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature517:505–508.http://dx.doi.org/10 .1038/nature13838.
34. Rumlova-Klikova M, Hunter E, Nermut MV, Pichova I, Ruml T.2000. Analysis of Mason-Pfizer monkey virus Gag domains required for capsid assembly in bacteria: role of the N-terminal proline residue of CA in di-recting particle shape. J Virol74:8452– 8459.http://dx.doi.org/10.1128 /JVI.74.18.8452-8459.2000.
35. Chiu J, March PE, Lee R, Tillett D. 2004. Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approach-ing 100% efficiency in 4 h. Nucleic Acids Res32:e174.http://dx.doi.org/10 .1093/nar/gnh172.
36. Song C, Hunter E.2003. Variable sensitivity to substitutions in the N-terminal heptad repeat of Mason-Pfizer monkey virus transmembrane
on November 7, 2019 by guest
http://jvi.asm.org/
protein. J Virol77:7779 –7785.http://dx.doi.org/10.1128/JVI.77.14.7779 -7785.2003.
37. Krˇízová I, Hadravová R, Štokrová J, Günterová J, Doležal M, Ruml T, Rumlová M, Pichová I. 2012. The G-patch domain of Mason-Pfizer monkey virus is a part of reverse transcriptase. J Virol86:1988 –1998.http: //dx.doi.org/10.1128/JVI.06638-11.
38. Bohmová K, Hadravová R, Stokrová J, Tuma R, Ruml T, Pichová I, Rumlová M.2010. Effect of dimerizing domains and basic residues on in vitro and in vivo assembly of Mason-Pfizer monkey virus and human immunodeficiency virus. J Virol84:1977–1988.http://dx.doi.org/10.1128 /JVI.02022-09.
39. Brody BA, Kimball MG, Hunter E.1994. Mutations within the trans-membrane glycoprotein of Mason-Pfizer monkey virus: loss of SU-TM association and effects on infectivity. Virology202:673– 683.http://dx.doi .org/10.1006/viro.1994.1389.
40. Newman RM, Hall L, Connole M, Chen G-L, Sato S, Yuste E, Diehl W, Hunter E, Kaur A, Miller GM, Johnson WE.2006. Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5␣. Proc Natl Acad Sci U S A103:19134 –19139.http://dx.doi.org /10.1073/pnas.0605838103.
41. Rumlová M, Benedíková J, Cubínková R, Pichová I, Ruml T.2001. Comparison of classical and affinity purification techniques of Mason-Pfizer monkey virus capsid protein: the alteration of the product by an affinity tag. Protein Expr Purif23:75– 83.http://dx.doi.org/10.1006/prep .2001.1488.
42. Vorácˇková I, Suchanová S, Ulbrich P, Diehl WE, Ruml T.2011. Puri-fication of proteins containing zinc finger domains using immobilized metal ion affinity chromatography. Protein Expr Purif79:88 –95.http://dx .doi.org/10.1016/j.pep.2011.04.022.
43. Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Car-nemolla B, Orecchia P, Zardi L, Righetti PG.2004. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Elec-trophoresis25:1327–1333.http://dx.doi.org/10.1002/elps.200305844. 44. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M,
Pietz-sch T, PreibiPietz-sch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A.2012. Fiji: an open-source platform for biological-image analysis. Nat Methods9:676 – 682.http://dx.doi.org/10.1038/nmeth.2019.
45. Klikova M, Rhee SS, Hunter E, Ruml T.1995. Efficient in vivo and in vitro assembly of retroviral capsids from Gag precursor proteins expressed in bacteria. J Virol69:1093–1098.
46. Stansell E, Apkarian R, Haubova S, Diehl WE, Tytler EM, Hunter E.
2007. Basic residues in the Mason-Pfizer monkey virus gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J Vi-rol81:8977– 8988.http://dx.doi.org/10.1128/JVI.00657-07.
47. Mastronarde DN.2005. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol152:36 –51.
http://dx.doi.org/10.1016/j.jsb.2005.07.007.
48. Kremer JR, Mastronarde DN, McIntosh JR.1996. Computer visualiza-tion of three-dimensional image data using IMOD. J Struct Biol116:71– 76.http://dx.doi.org/10.1006/jsbi.1996.0013.
49. Nickell S, Förster F, Linaroudis A, Del Net W, Beck F, Hegerl R, Baumeister W, Plitzko JM.2005. TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol149:227–234.http://dx.doi .org/10.1016/j.jsb.2004.10.006.
50. Förster F, Medalia O, Zauberman N, Baumeister W, Fass D. 2005. Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc Natl Acad Sci U S A102:4729 – 4734.http://dx .doi.org/10.1073/pnas.0409178102.
51. Castaño-Díez D, Kudryashev M, Arheit M, Stahlberg H.2012. Dynamo: a flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. J Struct Biol178:139 –151.http://dx.doi.org/10.1016/j.jsb.2011.12.017. 52. Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN.
2009. CTF determination and correction for low dose tomographic tilt series. J Struct Biol168:378 –387.http://dx.doi.org/10.1016/j.jsb.2009.08 .016.
53. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE.2004. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem25:1605–1612.http: //dx.doi.org/10.1002/jcc.20084.
54. Pruggnaller S, Mayr M, Frangakis AS.2008. A visualization and segmen-tation toolbox for electron microscopy. J Struct Biol164:161–165.http: //dx.doi.org/10.1016/j.jsb.2008.05.003.
55. Strohalmová-Bohmová K, Spiwok V, Lepšík M, Hadravová R, Krˇížová I, Ulbrich P, Pichová I, Bednárová L, Ruml T, Rumlová M.2014. Role of Mason-Pfizer mo