Leukoencephalopathy
Leslie J. Marshall,* Michael W. Ferenczy, Elizabeth L. Daley,* Peter N. Jensen, Caroline F. Ryschkewitsch, Eugene O. Major
Laboratory of Molecular Medicine and Neuroscience, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland, USA
Progressive multifocal leukoencephalopathy (PML)-derived noncoding control region (NCCR) sequences permitted greater
early viral gene expression than kidney-associated NCCR sequences. This was driven in part by binding of the transcription
fac-tor Spi-B to unique PML-associated Spi-B binding sites. Spi-B is upregulated in developing B cells in response to natalizumab
therapy, a known risk factor for PML. Naturally occurring JCV sequence variation, together with drug treatment-induced
cellu-lar changes, may synergize to create an environment leading to an increased risk of PML.
T
he incidence of progressive multifocal leukoencephalopathy
(PML) has risen dramatically in recent years because of the
AIDS pandemic and the increased use of immunomodulatory
therapies (
1
). In particular, natalizumab (Tysabri; Biogen Idec), a
very effective multiple sclerosis (MS) treatment, has been highly
associated with PML (
2
).
Natalizumab treatment alters the expression profiles of
periph-eral blood mononuclear cells (PBMCs), including the expression
of the transcription factor Spi-B (
3
). Spi-B is required for normal
B-cell receptor signaling and maturation (
4–7
). Spi-B levels are
higher in cells permissive to JC virus (JCV) transcription and
rep-lication, and overexpression of Spi-B increases viral gene
expres-sion (
8
). Spi-B binds to target sites in the JCV noncoding control
region (NCCR) isolated from PML brain tissue but not in the
archetype NCCR commonly detected in the urine of
asymptom-atic healthy individuals (
8–11
). Importantly, mutation of these
sites in PML-associated NCCRs decreases Spi-B protein binding
and viral gene expression (
8
,
12
). Taken together, these results
suggest that Spi-B binding to sequences in the JCV NCCR have
functional effects on viral gene expression and may play a role in
the activation of JCV in peripheral blood cells (
13
).
JCV DNA has been detected in CD19
⫹B cells and CD34
⫹hema-topoietic progenitor cells (
14
,
15
) isolated from peripheral blood and
bone marrow. These CD34
⫹cells are strikingly increased in the
pe-ripheral blood of natalizumab-treated patients (
16
,
17
).
Immuno-magnetic separation was used to isolate CD3
⫹, CD19
⫹, and CD34
⫹cells from PBMCs of normal donors and MS patients treated with
natalizumab. Total RNA was isolated from each cell subset, and Spi-B
gene expression was measured by quantitative reverse transcription
(qRT)-PCR using the endogenous control PUM1 for normalization.
Spi-B gene expression was more variable in natalizumab patients. It
was upregulated up to 2-fold in CD19
⫹B cells in some patients
treated with natalizumab (
Fig. 1C
). Spi-B expression was increased
Received31 October 2013Accepted10 February 2014
Published ahead of print19 February 2014
Editor:R. M. Longnecker
Address correspondence to Michael W. Ferenczy, [email protected], or Eugene O. Major, [email protected].
* Present address: Leslie J. Marshall, Preclinical Microbicide and Prevention Research Branch, Prevention Sciences Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA; Elizabeth L. Daley, Johns Hopkins University, Department of Neurology, Baltimore, Maryland, USA.
L.J.M. and M.W.F. contributed equally to this work.
Supplemental material for this article may be found athttp://dx.doi.org/10.1128
/JVI.03221-13.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.03221-13
FIG 1Spi-B gene expression is upregulated in CD34⫹hematopoietic
precur-sors and CD19⫹B cells in response to long-term treatment with natalizumab.
CD3⫹, CD19⫹, and CD34⫹cells were isolated by immunomagnetic
separa-tion from PBMC samples from patients treated with natalizumab or normal donors. The remaining cells from the separation are labeled the negative frac-tion. Total RNA was isolated from each fraction of cells, and the Spi-B gene expression level was measured by qRT-PCR. Spi-B mRNA levels were normal-ized between samples by using the endogenous control PUM1 as a reference for the input template. Expression was normalized to levels in untreated
CD19⫹cells. The Spi-B mRNA level is expressed as a relative value calculated
by using a standard curve.
on November 7, 2019 by guest
http://jvi.asm.org/
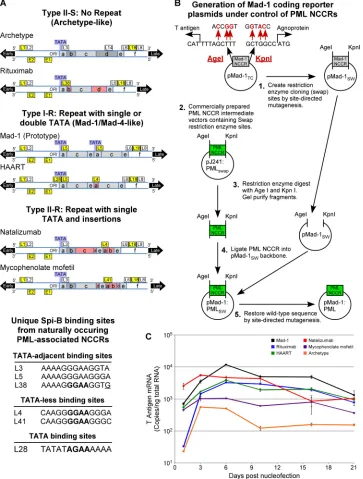
[image:1.585.41.287.395.622.2]FIG 2Schematic depiction of viral NCCR, cloning strategy, and early gene expression. (A) The NCCRs from PML patient tissues that are classified as type II-S no-repeat archetype like, type I-R single TATA box repeat Mad-4 like, and type II-R single TATA box repeats with insertions are represented. Conserved sequence blocks that contain deletions are red, and TATA boxes are blue. Sites that bind Spi-B protein in electrophoretic mobility shift assays are yellow, and sites that did not bind protein are white. The Spi-B binding sites grouped by location in reference to TATA boxes are listed. GGAA core binding sites targeted for mutation
analysis are bold, and the L38 3=G that creates an L3-like site and abrogates Spi-B binding when mutated is underlined. (B) An intermediate destination plasmid
termed a swap vector (pMad-1SW) was generated by creating restriction enzyme sites (swap sites) that overlap the start sites for the T antigen (AgeI) and
agnoprotein (KpnI) present on either side of the Mad-1 NCCR in the pM1TCplasmid. Plasmids containing the PML-derived NCCR with the same restriction sites
overlapping the start sites were generated by DNA 2.0 (pJ241:PMLswap). The pMad-1SWand pJ241:PMLswapplasmids were digested with KpnI and AgeI
restriction enzymes. The enzymatic products were separated by gel electrophoresis, and the destination vector and PML-derived NCCR inserts were purified
from the gel slices. The PML-derived NCCR inserts were ligated into the pMad-1SWdestination vector, and the restriction sites were restored to the wild-type
sequence by SDM. This cloning scheme allowed the insertion of the PML-derived NCCR sequences in frame with the T antigen and agnoprotein start sites within the Mad-1 coding sequence. (C) Plasmids encoding Mad-1 or archetype viral genome or the PML-derived NCCR–Mad-1 reporter genome were introduced into PDAs via nucleofection. Total RNA was isolated at the indicated time points, and T antigen mRNA expression (number of copies per nanogram of total RNA) was measured by qRT-PCR.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.112.475.69.548.2]tients (
Fig. 1D
). An increase in Spi-B expression in lymphocyte
sub-sets known to carry JCV may increase the chance of infection of the
central nervous system (CNS).
PML is caused by JCV lytic replication in glial cells in the CNS.
While many people are infected with JCV, PML is categorized as a
rare disease (
18
). NCCRs detected in the urine of asymptomatic
healthy individuals are relatively conserved and referred to as
“ar-chetype” (
19
). However, NCCR sequences change over the course
of active infection, leading to PML, and are highly variable
be-tween PML patients. Depending on the exact sequence, NCCRs
contain binding sites for various transcription factors, including
Spi-B (
9
,
10
) (
Fig. 2A
). We asked whether transcriptionally
im-portant Spi-B binding sites may be present in patients treated with
other disease-modifying therapies, including a rheumatoid
arthri-tis patient treated with rituximab (Rituxan; Genentech), a
sys-temic lupus erythematosus patient treated with mycophenolate
mofetil (CellCept; Genentech), and an HIV/AIDS patient treated
with highly active antiretroviral therapy (HAART). The NCCR
from one of the natalizumab PML reference cases was also isolated
and sequenced (
Fig. 2A
;
Table 1
) (
20
). Viral DNA was isolated as
previously described (
21
,
22
). The NCCR was amplified by PCR
directly from the DNA isolate and sequenced by the Division of
Intramural Research Sequencing Facility, NINDS. The primers
and probes used are described in
Table 2
.
Spi-B binding to the JCV L4 site identical to that in the
natali-zumab patient was previously demonstrated (
8
). By similar EMSA
methods, Spi-B was found to bind the JCV L28 site in the HAART
NCCR, the JCV L38 site in the rituximab NCCR, and the L41 site
in the mycophenolate mofetil NCCR (data not sown).
In order to create viral plasmid DNA containing identical gene
coding sequences under the direction of the PML-derived NCCR
sequences, a system using the pMad-1
TCJCV plasmid (
23
) was
established (
Fig. 2B
). With the QuikChange site-directed
mu-tagenesis (SDM) kit (Agilent Technologies), an intermediate
des-tination plasmid (swap vector, pMad-1
SW) was generated by
cre-ating restriction enzyme “swap” sites that overlap the start sites for
the T antigen (AgeI) and agnoprotein (KpnI) present on either
side of the Mad-1 NCCR. Plasmids containing a PML-derived
NCCR with the same restriction sites overlapping the start sites
were generated by DNA 2.0. The Mad-1 swap vector and
PML-derived NCCR plasmids (pJ241:PML
swap) were digested with the
KpnI and AgeI restriction enzymes, separated by gel
electropho-resis, and purified. PML-derived NCCR inserts were ligated into
the Mad-1 swap destination vector, and restriction sites were
re-stored to the wild-type sequence by SDM as described above. This
generated in-frame insertions of the PML-derived NCCRs within
the Mad-1 coding sequence.
These plasmids were transfected by nucleofection
electropora-tion (Lonza) into progenitor-derived astrocytes (PDAs) (
24
) as
previously described (
8
,
12
). Early (T antigen) RNA expression
was measured by qRT-PCR as previously described (
Fig. 2C
) (
8
,
12
). The Mad-1 (
25
) NCCR directed early RNA expression at the
highest level over 3 weeks, and the archetype NCCR drove
ap-proximately 100-fold less expression. Patient-derived NCCR
sequences drove early RNA expression at intermediate levels,
generally at least 10-fold higher than those obtained with the
archetype, as was also seen with HIV/AIDS NCCRs with a
fluorescent-reporter system (
26
).
Spi-B binding sites were mutated to sequences shown to
abro-1
Characteristics
of
the
patients
included
in
this
study
Therapy
Age
(yr)
Gender
Anatomical
site
JCV
DNA
Yr
of
PML
diagnosis
NCRR
type
Previous
disease-modifying
treatments
PML
therapy
Miscellaneous
HAART,
started
in
1998
NA
a
NA
CSF
3,975
copies/ml
1999
I-R
None
None/HAART
Antiherpetic
antivirals
for
VZV
d
retinitis
and
CMV
e
in
urine
sclerosis
Natalizumab,
37
doses,
2002–2005
46
Female
Brain
b
⬎
3.5
⫻
10
8
copies
in
1
section
2005
II-R
2000–2005,
beta
1a
interferon;
2001–2005,
methylprednisolone;
2001–
2003,
2004–2005,
donepezil;
2003–2004,
galantamine
None;
diagnosed
14
February
2005,
deceased
24
February
2005
Continued
beta
1a
interferon
during
natalizumab
treatment
arthritis
Rituximab,
5
courses,
May
2006–
September
2008
73
Female
CSF
9,138
copies/ml
2008
II-S
2002–2003,
etanercept;
2005-NA,
adalimumab;
before
July
2006–30
October
2008,
methotrexate
Mirtazapine,
mefloquine,
2nd
JCV
test
19
March
2009
c
lupus
Mycophenolate
mofetil
NA
Female
CSF
27,150
copies/ml
2008
II-R
NA
NA
not
available.
autopsy
sample.
684
copies/ml.
varicella-zoster
virus.
cytomegalovirus.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.327.535.69.724.2]TABLE 2 Primers and probes used in this study Name 5 = –3 = sequence b Purpose Changes JCT 1 AGAGTGTTGGGATCCTGTGTTTT Forward primer to detect JCV T antigen RNA NA a JCT 2 GAGAAGTGGGGATGAAGACCTGTTT Reverse primer to detect JCV T antigen RNA NA JCT 1.1 FAM-TCATCACTGGCAAACATTTCTTCATGGC-TAMRA Probe to detect JCV T antigen RNA NA JRR25 CATGGATTCCTCCCTATTCAGCA Sequencing of NCCRs from early side NA JRR28 TCACAGAAGCCTTACGTGACAGC Sequencing of NCCRs from late side NA MAD-1 SWAP Agno FOR GTTTTGGCTTGTCACCAGGTACCCATGGTTCTTCGCCAGC Generation of pMad-1 SW by inserting KpnI site on late side of NCCR AGCTTT ¡ GGTACC MAD-1 SWAP Agno REV GCTGGCGAAGAACCATGGGTACCTGGTGACAAGCCAAAAC Generation of pMad-1 SW by inserting KpnI site on late side of NCCR AGCTTT ¡ GGTACC MAD-1 SWAP Tag FOR GATTCCTCCCTATTCAGCACTTTGTCCATTTTACCGGTTTGCAGCAAAAAATTCCT Generation of pMad-1 SW by inserting AgeI site on early side of NCCR GCTGGC ¡ ACCGGT MAD-1 SWAP Tag REV AGTAATTTTTTGCTGCAAACCGGTAAAATGGACAAAGTGCTGAATAGGGAGGAATC Generation of pMad-1 SW by inserting AgeI site on early side of NCCR GCTGGC ¡ ACCGGT 506 SWAP Agno FOR AGCTGTTTTGGCTTGTCACCAACTGGCCATGGTTCTTCGCCAGC Repair of late side KpnI site to wild-type sequence GGTACC ¡ ACTGGC 506 SWAP Agno REV GCTGGCGAAGAACCATGGCCAGTTGGTGACAAGCCAAAACAGCT Repair of late side KpnI site to wild-type sequence GGTACC ¡ ACTGGC 506 SWAP Tag FOR GATTCCTCCCTATTCAGCACTTTGTCCATTTTGGCTTTTTGTAGCAAAAATTTAT TGCAAAAAAGG Repair of early side AgeI site to wild-type sequence ACCGGT ¡ GGCTTT 506 SWAP Tag REV CCTTTTTTGCAATAAATTTTTGCTACAAAAAGCCAAAATGGACAAAGTGCTGAAT AGGGAGGAATC Repair of early side AgeI site to wild-type sequence ACCGGT ¡ GGCTTT 482 SWAP Agno FOR TTTTGGCTGTCACCAGCTGCCCATGGTTCTTCGCC Repair of late side KpnI site to wild-type sequence GGTACC ¡ GCTGCC 482 SWAP Agno REV GGCGAAGAACCATGGGCAGCTGGTGACAGCCAAAA Repair of late side KpnI site to wild-type sequence GGTACC ¡ GCTGCC 482 SWAP Tag FOR GATTCCTCCCTATTCAGCACTTTGTCCATTTAGCTTTTTGCAGCAAAAAATTACT Repair of early side AgeI site to wild-type sequence ACCGGT ¡ AGCTTT 482 SWAP Tag REV AGTAATTTTTTGCTGCAAAAAGCTAAAATGGACAAAGTGCTGAATAGGGAGGAATC Repair of early side AgeI site to wild-type sequence ACCGGT ¡ AGCTTT 459 SWAP Agno FOR GTTTTGGCTTGTCACCAGCTGGCCATGGTTCTTCGCCAGC Repair of late side KpnI site to wild-type sequence GGTACC ¡ GCTGGC 459 SWAP Agno REV GCTGGCGAAGAACCATGGCCAGCTGGTGACAAGCCAAAAC Repair of late side KpnI site to wild-type sequence GGTACC ¡ GCTGGC 459 SWAP Tag FOR GATTCCTCCCTATTCAGCACTTTGTCCATTTTAGCTTTTTGTAGCAAAAAATTAGT Repair of early side AgeI site to wild-type sequence ACCGGT ¡ AGCTTT 459 SWAP Tag REV ACTAATTTTTTGCTACAAAAAGCTAAAATGGACAAAGTGCTGAATAGGGAGGAATC Repair of early side AgeI site to wild-type sequence ACCGGT ¡ AGCTTT 399 SWAP Agno FOR GTTTTGGCTTGTCACCAGCTGGCCATGGTTCTTCGCCAGC Repair of late side KpnI site to wild-type sequence GGTACC ¡ GCTGGC 399 SWAP Agno REV GCTGGCGAAGAACCATGGCCAGCTGGTGACAAGCCAAAAC Repair of late side KpnI site to wild-type sequence GGTACC ¡ GCTGGC 399 SWAP Tag FOR GATTCCTCCCTATTCAGCACTTTGTCCATTTTAGCTTTTTGTAGCAAAAAATTAGT Repair of early side AgeI site to wild-type sequence ACCGGT ¡ AGCTTT 399 SWAP Tag REV ACTAATTTTTTGCTACAAAAAGCTAAAATGGACAAAGTGCTGAATAGGGAGGAATC Repair of early side AgeI site to wild-type sequence ACCGGT ¡ AGCTTT 506 L38 3 = FOR TATATATAAAAAAAAGGGAAGGTAATGGCTGCCAGCCAAGCATGAG Abrogation of Spi-B binding to rituximab L38 site by mutation of 3 = end G ¡ T 506 L38 3 = REV CTCATGCTTGGCTGGCAGCCATTACCTTCCCTTTTTTTTATATATA Abrogation of Spi-B binding to rituximab L38 site by mutation of 3 = end G ¡ T
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.55.525.64.727.2]506
L38
CORE
FOR
TCCTGTATATATAAAAAAAAGCCAAGGGGATGGCTGCCAGCC
Abrogation
of
Spi-B
binding
to
rituximab
L38
site
by
mutation
of
core
binding
site
GG
¡
CC
506
L38
CORE
REV
GGCTGGCAGCCATCCCCTTGGCTTTTTTTTATATATACAGGA
Abrogation
of
Spi-B
binding
to
rituximab
L38
site
by
mutation
of
core
binding
site
GG
¡
CC
399
L4
FOR
GTAAACAAAGCACAAGGCCAAGGGAGGAGCTGGCTA
Abrogation
of
Spi-B
binding
to
Natalizumab
L4
site
by
mutation
of
core
binding
site
GG
¡
CC
399
L4
REV
TAGCCAGCTCCTCCCTTGGCCTTGTGCTTTGTTTAC
Abrogation
of
Spi-B
binding
to
Natalizumab
L4
site
by
mutation
of
core
binding
site
GG
¡
CC
482
L41
FOR
CAAGTAAACAAAGCACAAGGCCAAAGGCTAAAACTGGATGGC
Abrogation
of
Spi-B
binding
to
mycophenolate
mofetil
L41
site
by
mutation
of
core
binding
site
GG
¡
CC
482
L41
REV
GCCATCCAGTTTTAGCCTTTGGCCTTGTGCTTTGTTTACTTG
Abrogation
of
Spi-B
binding
to
mycophenolate
mofetil
L41
site
by
mutation
of
core
binding
site
GG
¡
CC
459
L28
FOR
GCCTCGGCCTCCTGTATATCCAAAAAAAAGGGAAGGGATG
Abrogation
of
Spi-B
binding
to
HAART
L28
site
by
mutation
of
core
binding
site
AG
¡
CC
459
L28
REV
CATCCCTTCCCTTTTTTTTGGATATACAGGAGGCCGAGGC
Abrogation
of
Spi-B
binding
to
HAART
L28
site
by
mutation
of
core
binding
site
AG
¡
CC
459
L4
FOR
GTAAACAAAGCACAAGGCCAAGGGATGGCTGCCAGC
Abrogation
of
Spi-B
binding
to
HAART
L4
site
by
mutation
of
core
binding
site
GG
¡
CC
459
L4
REV
GCTGGCAGCCATCCCTTGGCCTTGTGCTTTGTTTAC
Abrogation
of
Spi-B
binding
to
HAART
L4
site
by
mutation
of
core
binding
site
GG
¡
CC
aNA,
not
applicable.
bFAM,
6-carboxyfluorescein;
TAMRA,
6-carboxytetramethylrhodamine.
Underlining
indicates
nucleotides
targeted
for
site-directed
mutagenesis.
on November 7, 2019 by guest
http://jvi.asm.org/
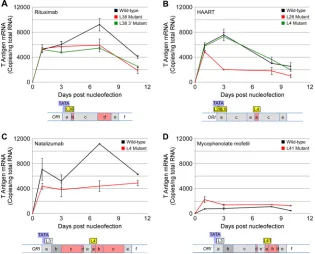
gate Spi-B binding (
8
,
12
). Mutation of the core of the L38 binding
site of the rituximab NCCR, as well as mutation of the 3
=
end of the
L38 site—resulting in an archetype-like sequence (L3) that cannot
bind Spi-B (
8
)—reduced early gene expression (
Fig. 3A
).
Muta-tion of the L28 binding site of the HAART NCCR decreased early
gene expression, while mutation of the L4 binding site did not
(
Fig. 2B
). Mutation of the L4 binding site in the natalizumab
NCCR, however, did reduce early gene expression (
Fig. 2D
).
Mu-tation of the L4 site of the Mad-4 variant NCCR completely
abro-gated early gene expression (
8
). Therefore, the promoter context
of Spi-B sites influences their effect on viral gene expression.
Fur-ther illustrating the complexity of dissecting the regulation of JCV
gene expression, mutation of the core L41 Spi-B binding site of the
mycophenolate mofetil NCCR resulted in a slight increase over
the already low early gene expression. This mutation results in a
nuclear factor I (NFI) binding site. NFI-X, present at high levels in
PDAs, has been shown to drive JCV early gene expression (
8
,
24
,
27–29
). We additionally sequenced a number of PML patient
NCCRs and searched for putative Spi-B sites (see Tables S1 and S2
and Fig. S1 in the supplemental material). While not all patients
had new Spi-B sites, all had a deletion in the D segment of the
sequence, deleting a nonfunctional Spi-B site. Deletion of the D
DNA segment is a feature of almost every PML-type NCCR. A
clinical test has been developed to identify JCV NCCR sequences
with complete or partial deletions in the D segment (
30
). Almost
all PML patients have changes in the NCCR, and almost every
patient has unique changes (see Fig. S1 in the supplemental
ma-terial). Many have newly acquired, functional Spi-B binding sites.
These may reflect underlying risk factors, or there may be multiple
changes that make JCV more likely to cause PML.
In a subset of natalizumab patients, Spi-B expression is greatly
increased in the CD19
⫹and CD34
⫹compartments. Spi-B
bind-ing sites found in naturally occurrbind-ing JCV NCCRs from PML
pa-tients with a variety of underlying conditions and
immunomodu-latory treatments are important for early viral transcription. These
sites may work in concert with increased Spi-B expression in
cel-lular compartments where JCV is found and may help explain the
increased risk of PML during natalizumab therapy.
Nucleotide sequence accession numbers.
The NCCR DNA
sequences described here have been deposited in GenBank and
assigned accession numbers
KF788287
to
KF788290
, and those in
the supplemental material have been assigned accession numbers
KJ001213
to
KJ001223
.
ACKNOWLEDGMENTS
We thank Ludwig Kappos and Raija Lindberg of the University Hospital Basel, Basel, Switzerland, for kindly providing natalizumab-treated pa-tient blood. We thank James Nagle of the DNA sequencing facility at the National Institute of Neurological Disorders and Stroke (NINDS) for aid in sequencing viral NCCRs. We thank all of the members of the Labora-tory of Molecular Medicine and Neuroscience (LMMN) at the NINDS for their hard work, support, and valuable input.
FIG 3Early viral gene expression from patient and Spi-B-mutated sequences. Cells were nucleofected with wild-type or Spi-B-mutated PML variant NCCRs in the pMad-1 coding region plasmid. Total RNA was harvested, and T antigen mRNA expression (number of copies per nanogram of total RNA) was measured by qRT-PCR at the indicated time points. (A) Mutation of the L38 Spi-B binding site in the rituximab patient NCCR reduces early viral gene expression in PDAs.
The red line represents a GG-to-CC mutation in the core of the L38 Spi-B binding site. The green line represents a G-to-A mutation at the 3=end of the L38 Spi-B
binding site, which results in the creation of an L3-like site (archetype) incapable of binding Spi-B protein. (B) Mutation of the L28 Spi-B binding site in the HAART patient NCCR reduces early viral gene expression, but mutation of the L4 Spi-B binding site does not. The red line represents a GG-to-CC mutation in the core of the L4 Spi-B binding site. The green line represents a GG-to-CC mutation in the core of the L28 Spi-B binding site. (C) Mutation of the L4 Spi-B binding site in the natalizumab patient NCCR reduces early viral gene expression in PDAs. The red line represents a GG-to-CC mutation in the core of the L4 Spi-B binding site. (D) Mutation of the L41 Spi-B binding site in the mycophenolate mofetil patient NCCR slightly increases early viral gene expression in PDAs. The red line represents a GG-to-CC mutation in the core of the L41 Spi-B binding site, which creates an NFI binding site.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.135.450.68.322.2]lowship from the NIH Office of AIDS Research. The LMMN is supported by the Division of Intramural Research of the NINDS.
REFERENCES
1.Major EO.2010. Progressive multifocal leukoencephalopathy in patients
on immunomodulatory therapies. Annu. Rev. Med.61:35– 47.http://dx
.doi.org/10.1146/annurev.med.080708.082655.
2.Marshall LJ, Major EO.2010. Molecular regulation of JC virus tropism: insights into potential therapeutic targets for progressive multifocal
leu-koencephalopathy. J. Neuroimmune Pharmacol. 5:404 – 417.http://dx
.doi.org/10.1007/s11481-010-9203-1.
3.Lindberg RL, Achtnichts L, Hoffmann F, Kuhle J, Kappos L. 2008. Natalizumab alters transcriptional expression profiles of blood cell
sub-populations of multiple sclerosis patients. J. Neuroimmunol.194:153–
164.http://dx.doi.org/10.1016/j.jneuroim.2007.11.007.
4.Garrett-Sinha LA, Su GH, Rao S, Kabak S, Hao Z, Clark MR, Simon
MC.1999. PU.1 and Spi-B are required for normal B cell
receptor-mediated signal transduction. Immunity10:399 – 408.http://dx.doi.org
/10.1016/S1074-7613(00)80040-0.
5.Rao S, Matsumura A, Yoon J, Simon MC.1999. SPI-B activates tran-scription via a unique proline, serine, and threonine domain and exhibits
DNA binding affinity differences from PU.1. J. Biol. Chem.274:11115–
11124.http://dx.doi.org/10.1074/jbc.274.16.11115.
6.Mao C, Ray-Gallet D, Tavitian A, Moreau-Gachelin F.1996. Differential
phosphorylations of Spi-B and Spi-1 transcription factors. Oncogene12:
863– 873.
7.Hagemeier C, Bannister AJ, Cook A, Kouzarides T.1993. The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) pro-tein and the transcription factor TFIID in vitro: RB shows sequence
sim-ilarity to TFIID and TFIIB. Proc. Natl. Acad. Sci. U. S. A.90:1580 –1584.
http://dx.doi.org/10.1073/pnas.90.4.1580.
8.Marshall LJ, Dunham L, Major EO. 2010. Transcription factor Spi-B binds unique sequences present in the tandem repeat promoter/enhancer
of JC virus and supports viral activity. J. Gen. Virol.91:3042–3052.http:
//dx.doi.org/10.1099/vir.0.023184-0.
9.Bellizzi A, Anzivino E, Ferrari F, Di Nardo G, Colosimo MT, Fioriti D, Mischitelli M, Chiarini F, Cucchiara S, Pietropaolo V.2011. Polyoma-virus JC reactivation and noncoding control region sequence analysis in pediatric Crohn’s disease patients treated with infliximab. J. Neurovirol.
17:303–313.http://dx.doi.org/10.1007/s13365-011-0036-3.
10. Bellizzi A, Anzivino E, Rodio DM, Cioccolo S, Scrivo R, Morreale M, Pontecorvo S, Ferrari F, Di Nardo G, Nencioni L, Carluccio S, Valesini G, Francia A, Cucchiara S, Palamara AT, Pietropaolo V.2013. Human polyomavirus JC monitoring and noncoding control region analysis in dynamic cohorts of individuals affected by immune-mediated diseases
under treatment with biologics: an observational study. Virol. J.10:298.
http://dx.doi.org/10.1186/1743-422X-10-298.
11. Bellizzi A, Anzivino E, Rodio DM, Palamara AT, Nencioni L, Pi-etropaolo V.2013. New insights on human polyomavirus JC and patho-genesis of progressive multifocal leukoencephalopathy. Clin. Dev. Immu-nol.2013:839719.http://dx.doi.org/10.1155/2013/839719.
12. Marshall LJ, Moore LD, Mirsky MM, Major EO.2012. JC virus promot-er/enhancers contain TATA box-associated Spi-B-binding sites that
sup-port early viral gene expression in primary astrocytes. J. Gen. Virol.93:
651– 661.http://dx.doi.org/10.1099/vir.0.035832-0.
13. Major EO, Frohman E, Douek D. 2013. JC viremia in
natalizumab-treated patients with multiple sclerosis. N. Engl. J. Med.368:2240 –2241.
http://dx.doi.org/10.1056/NEJMc1214233.
14. Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO.1996. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes,
7012.
15. Monaco MC, Shin J, Major EO.1998. JC virus infection in cells from
lymphoid tissue. Dev. Biol. Stand.94:115–122.
16. Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E.2008. Natali-zumab disproportionately increases circulating pre-B and B cells in
mul-tiple sclerosis. Neurology 71:1350 –1354. http://dx.doi.org/10.1212/01
.wnl.0000327671.91357.96.
17. Zohren F, Toutzaris D, Klarner V, Hartung HP, Kieseier B, Haas R. 2008. The monoclonal anti-VLA-4 antibody natalizumab mobilizes
CD34⫹hematopoietic progenitor cells in humans. Blood111:3893–3895.
http://dx.doi.org/10.1182/blood-2007-10-120329.
18. Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO.2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced
demy-elinating disease of the human brain. Clin. Microbiol. Rev.25:471–506.
http://dx.doi.org/10.1128/CMR.05031-11.
19. Jensen PN, Major EO.2001. A classification scheme for human polyo-mavirus JCV variants based on the nucleotide sequence of the noncoding
regulatory region. J. Neurovirol.7:280 –287.http://dx.doi.org/10.1080
/13550280152537102.
20. Kleinschmidt-DeMasters BK, Tyler KL. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and
in-terferon beta-1a for multiple sclerosis. N. Engl. J. Med.353:369 –374.http:
//dx.doi.org/10.1056/NEJMoa051782.
21. Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO.2004. Comparison of PCR-Southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and
cerebrospinal fluid. J. Virol. Methods121:217–221.http://dx.doi.org/10
.1016/j.jviromet.2004.06.021.
22. Rollison DE, Utaipat U, Ryschkewitsch C, Hou J, Goldthwaite P, Daniel R, Helzlsouer KJ, Burger PC, Shah KV, Major EO.2005. Investigation of human brain tumors for the presence of polyomavirus genome
se-quences by two independent laboratories. Int. J. Cancer113:769 –774.
http://dx.doi.org/10.1002/ijc.20641.
23. Frisque RJ.1983. Regulatory sequences and virus-cell interactions of JC
virus. Prog. Clin. Biol. Res.105:41–59.
24. Messam CA, Hou J, Gronostajski RM, Major EO.2003. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann.
Neurol.53:636 – 646.http://dx.doi.org/10.1002/ana.10523.
25. Martin JD, King DM, Slauch JM, Frisque RJ. 1985. Differences in regulatory sequences of naturally occurring JC virus variants. J. Virol.
53:306 –311.
26. Gosert R, Kardas P, Major EO, Hirsch HH.2010. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalop-athy patient samples increase virus early gene expression and replication rate.
J. Virol.84:10448 –10456.http://dx.doi.org/10.1128/JVI.00614-10.
27. Amemiya K, Traub R, Durham L, Major EO.1989. Interaction of a nuclear factor-1-like protein with the regulatory region of the human
polyomavirus JC virus. J. Biol. Chem.264:7025–7032.
28. Amemiya K, Traub R, Durham L, Major EO.1992. Adjacent nuclear factor-1 and activator protein binding sites in the enhancer of the neu-rotropic JC virus. A common characteristic of many brain-specific genes.
J. Biol. Chem.267:14204 –14211.
29. Monaco MC, Sabath BF, Durham LC, Major EO.2001. JC virus multi-plication in human hematopoietic progenitor cells requires the NF-1 class
D transcription factor. J. Virol.75:9687–9695.http://dx.doi.org/10.1128
/JVI.75.20.9687-9695.2001.
30. Ryschkewitsch CF, Jensen PN, Major EO.2013. Multiplex qPCR assay for ultra sensitive detection of JCV DNA with simultaneous identification of genotypes that discriminates non-virulent from virulent variants. J.
Clin. Virol.57:243–248.http://dx.doi.org/10.1016/j.jcv.2013.03.009.
on November 7, 2019 by guest
http://jvi.asm.org/