Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Strain-Dependent Structural Variants of Herpes Simplex Virus Type 1
ICP34.5 Determine Viral Plaque Size, Efficiency of Glycoprotein
Processing, and Viral Release and Neuroinvasive
Disease Potential
Hanwen Mao† and Ken S. Rosenthal*
Northeastern Ohio Universities College of Medicine, Rootstown, Ohio 44272
Received 7 October 2002/Accepted 23 December 2002
The ability of certain strains of herpes simplex virus type 1 (HSV-1) to cause encephalitis or neuroinvasive disease in the mouse upon peripheral infection is dependent on a combination of activities of specific forms of viral proteins. The importance of specific variants of ICP34.5 to neuroinvasive disease potential and its correlation with small-plaque production, inefficient glycoprotein processing, and virus release were suggested by comparison of ICP34.5 from the SP7 virus, originally obtained from the brain of a neonate with dissemi-nated disease, and the tissue culture-passaged progeny of SP7 (SLP5 and SLP10) and the KOS321 virus. SLP5, SLP10, and KOS321 are attenuated and exhibit a large-plaque phenotype, including efficient glycoprotein processing and viral release. We show that expression of the KOS321 ICP34.5 protein in cells infected with SP7 or ICP34.5 deletion mutants promotes large plaque formation and efficient viral glycoprotein processing, while expression of the SP7 ICP34.5 protein decreases efficiency of viral glycoprotein processing. In addition, a recombinant virus, 4hS1, with the SP7 ICP34.5 gene replacing the KOS321-like ICP34.5 gene in the SLP10a background, rescues the small-plaque phenotype and neuroinvasive disease. The major difference in the ICP34.5 gene product is the number of Pro-Ala-Thr repeats in the middle region of the protein, with 18 for SP7 and 3 for KOS321. Strain-dependent differences in the ICP34.5 protein can therefore alter the tissue culture behavior and the virulence of HSV-1.
Herpes simplex virus (HSV) encephalitis is probably the most serious outcome of an HSV type 1 (HSV-1) infection. The ability of the virus to cause such disease is a function of viral properties, the inoculum size, the route of entry, and the immune and health state of the patient. In order to cause encephalitis, neuroinvasive HSV-1 must be able to initiate infection at a local site, travel by retrograde neuronal transport to the brain, and initiate replication prior to control by host protective responses. Different viral strains have different po-tentials to cause disease based on the contributions of specific viral proteins to the ability of the virus to replicate and spread within the host and to escape host protective responses (innate and immune). Attenuation of disease potential can occur fol-lowing passage of virus in tissue culture in the absence of the selective pressures of the host cell machinery, tissue architec-ture, and host protective responses or by selection for rapid dissemination in tissue culture (7).
Several viral genes influence the neuroinvasive disease po-tential of HSV-1. Viral genes important for promoting growth in neurons or specific viral functions can be identified by study-ing the properties of deletion mutants. Comparison of virulent and attenuated strains of HSV can identify important genes and mutations in these genes which may subtly alter, but not
inactivate, the function of an important viral protein. In this manner, specific variants of glycoproteins B and D (gB and gD) were shown to be important for neuroinvasive disease production by comparing the highly passaged KOS and atten-uated ANG viruses to the virulent ANG-PATH virus (26, 34, 45). Studies with virus with a deletion of the ICP34.5 gene but without a compensating mutation (6, 8, 14, 15) identified this gene as important for growth in neuronal cells and for efficient HSV virion maturation and egress (8). Comparison of the SP7 neuroinvasive clinical isolate to the attenuated SLP5 and SLP10 tissue culture-passaged progeny of SP7 and KOS indi-cated a correlation between neuroinvasive disease potential and the following tissue culture behaviors: small-plaque pro-duction and limited glycoprotein processing and virion release (small-plaque phenotype) and importantly, specific gene and protein sequences of ICP34.5 (7).
ICP34.5 is encoded in the inverted repeats of the unique long sequence of HSV, and two copies of the gene are present in the genome (1, 13, 33). The HSV-1 ICP34.5 gene is rich in GC, and there is extensive homology between strains except for major sequence differences in the middle region of the gene in the number of repeats of CCC GCG ACC, encoding pro-line-alanine-threonine (PAT), and the CGC repeats at the N terminus encoding a string of Arg (e.g., RRRRHRGPRRPR, for SP7) (7, 13). The numbers of PAT repeats range from 3 (KOS, SLP5, and SLP10) to 18 (SP7) and 22 (LP5) (7, 13, 32). The C-terminal portion is highly conserved and has sequence homology to GADD34/MYD116 (7, 13). This portion of ICP34.5 binds to protein phosphatase 1 (PP1) and proliferat-ing cell nuclear antigen (9). ICP34.5 bindproliferat-ing to PP1 activates
* Corresponding author. Mailing address: Northeastern Ohio Uni-versities College of Medicine, 4209 State Route 44, Rootstown, OH 44272. Phone: (330) 325-6134. Fax: (330) 325-5914. E-mail: [email protected].
† Present address: Laboratory of Molecular Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Md.
3409
on November 8, 2019 by guest
http://jvi.asm.org/
the enzyme to dephosphorylate and reactivate the eIF2-alpha component of the ribosome, which is phosphorylated by PKR as a host protection against infection, and as a result antago-nizes host shut-off of protein synthesis induced by viral infec-tion or as a consequence of interferon alpha and beta acinfec-tion (11, 22, 23, 24).
Specific regions of the ICP34.5 protein direct the protein to different parts of the cell. Cheng et al. (10) showed that ICP34.5 has three nuclear localization signals, including a nu-cleolar targeting sequence from the Arg-rich cluster within amino acids 1 to 16, a bipartite basic amino acid cluster within amino acids 208 to 236, and also a leucine-rich motif in the center of the protein which facilitates cytoplasmic export of the protein from the nucleus. We showed (32) that the N-terminal arginine-rich region targets the protein to the nucleolus, nu-cleus, and specific regions of the cytoplasm while the strain-dependent differences in the length of the PAT repeats in the center of the protein determine whether the protein is re-stricted to the cytoplasm or can distribute to the nucleus. Most importantly, ICP34.5 also determines the cellular localization of its ligand, PP1.
Our previous study (7) indicated the importance of ICP34.5 for neuroinvasiveness and showed that the KOS-like (also SLP5 and SLP10) form of ICP34.5 is associated with attenuation of neuroinvasive disease potential and the large-plaque phenotype. The purpose of this study was to identify a form of ICP34.5 that can confer the small-plaque phenotype and neuroinvasive disease potential. This was achieved by examination of viral behaviors following infec-tion of cells that transiently expressed different variants of ICP34.5 and by generation of new viruses in which the variant of the ICP34.5 gene had been replaced with an alternate form of ICP34.5, either following passage or by development of a recombinant virus. These studies identify a major determinant of the tissue culture behavior and neu-roinvasive potential or attenuation of HSV-1.
MATERIALS AND METHODS
Cells and virus.Vero cells (African green monkey kidney cells; ATCC CCL-81) were grown in medium 199 supplemented with 5% fetal calf serum, penicillin (100 IU/ml), streptomycin (100g/ml), and 2.25 mM NaHCO3at 37°C.
SK-N-SH cells (neuroblastoma; ATCC HTB-11) were grown in Eagle’s minimal essential medium supplemented with 10% fetal calf serum and 1.0 mM sodium pyruvate. U373 cells (glioblastoma, kindly provided by I. Mohr, New York University) were grown in Dulbecco’s minimal essential medium with 5% fetal bovine serum and 5% calf serum.
KOS321 is a plaque-purified and highly passaged strain (provided by Hol-land et al.) (25). SP7 and LP5 were obtained from the brain and lungs, respectively, of a neonate who died of disseminated HSV-1 infection and then were plaque purified (7). SLP10a was picked from a large plaque obtained after 10 passages of SP7 in Vero cells at a low multiplicity of infection (MOI)(7). The SP7 virus is more neuroinvasive than LP5, and SLP10a is attenuated. The ICP34.5 gene from SP7 has 18 nonomer repeats encoding PAT in the middle region and 8 arginines at the N terminus, while SLP10a and KOS321 have only three nonomer repeats and seven arginines at the N terminus. The sequence of the SP7 ICP34.5 used in this and subsequent studies differs from that reported by Bower et al. (7) in that Gly-Glu-Gly-Ala is present at position 153 to 156. Single-amino-acid polymorphisms also distinguish SP7 from SLP10a and KOS321 at positions 140, 158, and 215 with respect to SP7 (7). The only difference between SP7 and LP5 is the number of PAT repeats (for LP5, 22 PAT repeats). The C-terminal GADD34-like sequence is 100% conserved for these and most other HSV-1 strains (13).
HSV-1 strain McKrae and the ICP34.5 null mutantd34.5 were provided by
Perng et al. (36).d34.5 virus has a⬃1.0-kb DNA deletion which deletes the sequence of the ICP34.5 gene in both genetic loci, and the ICP34.5 protein is not produced in cells infected withd34.5 (36). The ICP34.5 null mutant 17termA and its rescued strain, 17termAR, were obtained from Richard Thompson (6). TermA has an insertion of 20 oligonucleotides containing stop codons in all three reading frames at 90 nucleotides after the start codon of the ICP34.5 gene.
Antibodies.Polyclonal anti-gC was kindly provided by G. Cohen and R. Eisen-berg (University of Pennsylvania). Monoclonal anti-myc and anti-myc-horserad-ish peroxidase were purchased from Invitrogen.
Western blot analysis of HSV gC.Whole-cell detergent extracts of HSV-1-infected Vero cells were examined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis using polyclonal anti-gC. The image was developed by chemiluminescence (ECL) (Amersham) and digitally scanned or recorded on film. The ratio of the amount of the mature gC versus precursor gC was determined from densitometry readings from films or scans that were below saturation of the medium.
Viral DNA purification.Viral DNA was purified by the method of Robbins et al. (39).
Cloning of the ICP34.5 gene. (i) Cloning from PCR products.Cloning and insertion of the ICP34.5 coding sequences from SP7 and KOS321 into the mammalian expression vectors, including pcDNA3.1 myc-his B(⫺) and Vitality hrGFP-C (Stratagene) were described earlier (7, 32).
(ii) Cloning from restriction fragments containing the ICP34.5 gene.SP7 DNA was cut completely withBamHI. The products were ligated into the
BamHI site of the pBluescript SK(⫹) II vector (Stratagene). Ligation products were transformed into DH5␣Escherichia coli. Colony PCR (46) was performed on individual colonies using UL34 and 34UL primers covering the ends of the ICP34.5 gene (7) to screen colonies for plasmids containing inserts of the ICP34.5 gene. Briefly, tiny amounts of bacteria were scraped with a sterile 100-l micropipette tip and dissolved into Tris-EDTA buffer containing 0.1% Tween-20. After boiling for 5 min, the lysates were centrifuged at 12,000⫻gfor 5 min and the supernatants were carefully collected. Using 2l of the supernatants as templates, PCR was performed as described above in a total volume of 10l. Plasmid DNA from positive colonies was purified by mini-prep (40) and then amplified following transformation and growth in DH5␣E. coli.
Cotransfection (marker transfer study).Plasmid DNA containing the cloned ICP34.5 gene was linearized withPvuI, which cleaves outside the gene, and then cotransfected with viral DNA into Vero cells using the Lipofectamine Plus reagent (Invitrogen). The next day the medium was changed, and transfected cells were incubated in methylcellulose-M199 medium until viral plaques were well developed.
In vivo selection of progeny viruses generated in the cotransfection study (mouse footpad inoculation model).Male BALB/c mice (3 to 4 weeks old) were purchased from Charles River Laboratories (Wilmington, Mass.). Viral stocks (40 to 50l) were inoculated subcutaneously into the ventral surface of the right rear footpad following pretreatment with 10% saline for 6 h (43). Animals were examined daily for the progression of neurological symptoms. One animal per group was euthanized on day 6 postchallenge, the spine was opened with a rongeur forcep from the dorsal surface, and the spinal cord was exposed. Lum-brosacral ganglia from both sides of the spinal column were removed. Tissues were homogenized in 1 ml of complete M199 medium. The samples were equally split: half was stored at⫺80°C for later use, and the other half was added to confluent Vero cells. Cells were examined up to 2 weeks postinfection (p.i.) for virus production.
Transient expression of ICP34.5 fusion genes.The recombinant DNAs were transfected into Vero cells or SK-N-SH cells (80 to 90% confluent in 6- or 12-well plates) using Lipofectamine PLUS (Invitrogen). After 2 days, cells were either lysed and analyzed by Western blotting or further infected with HSV-1. Some transfected cells were also incubated with G418 (1.0 mg/ml) for 3 days prior to infection and during infection to restrict protein synthesis in untransfected cells.
Selection of cells expressing hrGFP.Vero cells grown to 70 to 80% confluence in 75-cm2flasks were transfected with either of the ICP34.5-hrGFP variants (16 g of DNA/flask) using Lipofectamine PLUS. After 48 h, the cells were trypsinized and washed with Hank’s buffer and Hank’s buffer with 0.5% bovine serum albumin and 1 mM EDTA. The cells with green fluorescent protein (GFP) signals were sorted and isolated using the EPICS ALTRA Flow Cytometer and the HyperSort system (Beckman Coulter) (FACS).
Statistical analysis.The significant differences in plaque size of viruses were examined using thettest included with Microsoft Excel software.
on November 8, 2019 by guest
http://jvi.asm.org/
RESULTS
Passage of SP7 yields a large-plaque-producing virus with a KOS321-like ICP34.5 gene. Passage of SP7 at a low MOI in Vero cells resulted in selection of the SLP5 (small to large plaque) and SLP10a viruses, which have a large-plaque phe-notype and are attenuated for neuroinvasive disease (7). Virus obtained from each of the passages leading to the generation of SLP10a was reexamined for plaque size, efficiency of virus release, glycoprotein processing, and the PCR products of the ICP34.5 gene in order to determine if the phenotype and genotype conversions were concurrent.
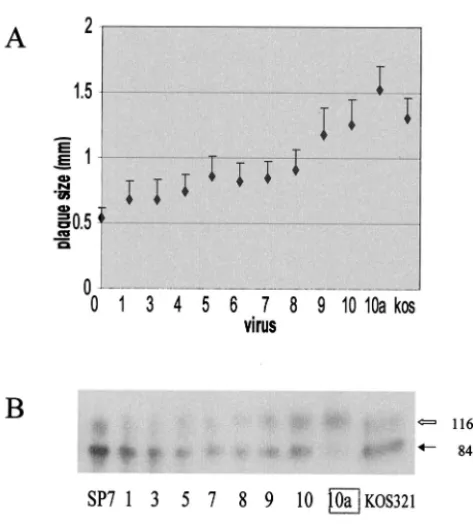
The PCR-amplified ICP34.5 gene for virus obtained from each passage up to passage 8 resembled the parental SP7 isoform (7). The change in the ICP34.5 gene was noted within one passage such that virus from passages 9 and 10 had an ICP34.5 gene that resembled the ICP34.5 gene from KOS321.
The average plaque size (⬃20 plaques [chosen at random])
gradually increased until passage 8, with a large increase in plaque size occurring between passages 8 and 9 (Fig. 1A). Statistical analysis indicated that the only significant difference
in plaque size for virus from any two consecutive passages was
between passage 8 (P8) and passage 9 (SLP9) (P⬍0.0001,t
test). The plaque sizes for virus from the initial passages (⬍5)
were similar to SP7, whereas SLP9 and SLP10 were similar to KOS321. SLP10 replication efficiency in Vero and U373 cells resembled SP7 and LP5 and was more efficient than that with KOS (Table 1). Virus release for SLP10a from Vero cells was also efficient, with a ratio of extracellular/intracellular virus of 1.59, similar to that with KOS321 (1.3) and larger than that with LP5 (0.33) and SP7 (0.037) (Table 1). Similarly, the effi-ciency of glycoprotein processing increased, as indicated by an increase in the ratio of the mature to precursor forms of gC (gC/pgC) (Fig. 1B). The efficiency of gC processing is repre-sentative of that for other HSV glycoproteins but is easier to distinguish (7). For SP7, the gC/pgC ranged from 0.14 to 0.37, for SP7 and virus from passages 1 to 8 and increased to 0.69 for passage 9 and 0.84 for passage 10, and for SLP10a, it was 4.3. The pattern for the last passages resembled the glycoprotein profile for KOS321 (gC/pgC ratio 0.52 in this experiment).
The selection for SLP10a virus during passage of SP7 virus suggests that virus with the large-plaque-producing phenotype (more efficient spread) was dominant in tissue culture. The increases in efficiency of glycoprotein processing for SLP9 and SLP10, plaque size, and efficiency of virus release occurred concurrently with the deletion of 135 nucleotides encoding 15 PAT repeats. The gradual increase in the plaque size during passage indicates that other possible gene mutations accumu-lated within the viral genome during passage and would be present in SLP10a.
Expression of the different ICP34.5 protein variants modu-lates the properties of an infecting virus.Vero cells transfected with plasmids capable of expressing the ICP34.5 gene as a fusion protein with the 6-histidine, c-myc epitope peptide or hrGFP were infected with HSV-1 strains lacking ICP34.5 or expressing a different variant of ICP34.5 in order to determine whether a specific variant of the ICP34.5 protein is responsible for the differences in viral behavior between SP7, KOS321, and SLP10a. A change in phenotype can be attributed to the ICP34.5 protein, because only the coding portion of the ICP34.5 gene amplified by PCR was cloned and expressed from a strong promoter, and its activity dominated the infect-ing virus. In addition, it is unlikely that a mutation in the ICP34.5 sequences would occur, unlike what may happen
[image:3.603.48.285.66.328.2]dur-FIG. 1. Change in the tissue culture behavior upon passage of SP7 leads to the generation of SLP10a. (A) Average plaque size (n⫽ ⬃20) of virus from SP7 passages 1 to 10 in Vero cells. A large plaque was picked from passage 10 from which virus stocks for SLP10a were prepared. SLP10a has a KOS321-like ICP34.5 gene (see reference 7). (B) HSV-1 gC from Vero cells infected with SP7, KOS321, or virus from each passage of SP7. Whole-cell detergent extracts from Vero cells infected with each virus were examined by SDS-PAGE and West-ern blot analysis using polyclonal anti-gC. The image was developed by chemiluminescence and digitally scanned. The black arrow indicates the precursor gC with an apparent mass of 84 kDa, while the open arrow represents the mature gC, which is 116 kDa in mass. The ratio of the amount of the processed gC versus precursor gC is indicated below each lane. PCR analysis indicated a deletion within the ICP34.5 gene for passages 9 and 10.
TABLE 1. Replication of variants of HSV-1 in various cell lines
Virus Viral replication
a
Extra/intra ratiob
Vero U373
SP7 4.3⫻108 4.3⫻108 0.037
LP5 3.0⫻108 NDc 0.330
KOS321 4.0⫻107 3.0⫻106 1.30
SLP10a 7.2⫻108 4.2⫻108 1.59
4hS1 1.9⫻108 1.5⫻108 0.89
4ds 4.1⫻108 2.9⫻108 2.14
aConfluent cells were infected with HSV-1 at a MOI of 5. The production of
virus at 24 h p.i. was determined by plaque assay on Vero cells.
bConfluent Vero cells were infected with HSV-1 at a MOI of 0.001. The
supernatant and cell fraction were separately collected at 72 h p.i. The progeny viruses in both fractions were quantitated on Vero cells. A ratio of extracellular and intracellular viral titers was calculated.
cND, not done.
on November 8, 2019 by guest
http://jvi.asm.org/
ing passage of a virus, selection of a recombinant, or develop-ment of an ICP34.5-expressing cell line. Although the ideal approach for studying the effects of different ICP34.5 protein variants on the tissue culture behavior of HSV-1 would be to develop stable cell lines that express a specific variant of the ICP34.5 protein, efforts to establish these stable cell lines failed due to a growth-limiting or toxic influence of the expressed ICP34.5.
gC processing efficiency was analyzed as an indicator of the influence of ICP34.5 on tissue culture behavior. Examination of plaque size was not possible due to the presence of untrans-fected cells. Cells were transuntrans-fected with the appropriate ICP34.5 plasmid or control plasmid and then incubated for 2 days to allow expression of ICP34.5 (32) prior to treatment with 1 mg of G418/ml and infection with an HSV-1 strain lacking or encoding a different ICP34.5 gene. G418 inhibited viral protein (including gC) synthesis in untransfected cells (data not shown), allowing selective observation of gC produc-tion and processing in only those Vero cells which had been transfected with the empty or ICP34.5-containing pcDNA 3.1 plasmid expressing the neomycin resistance gene.
The efficiency of gC processing decreased in cells expressing the SP7 form of ICP34.5. For cells infected with either of two
ICP34.5 null viruses,d34.5 virus (36) or 17termA virus (6), the
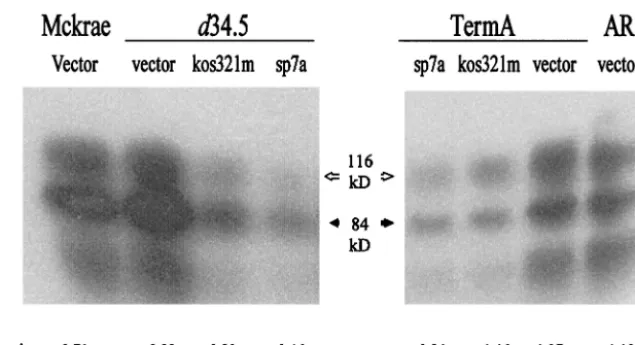
gC/pgC ratio decreased from 0.89 and 1.37 in cells transfected with the empty vector to 0.46 and 0.84 in cells transfected with the SP7 ICP34.5-myc, respectively. In contrast, gC was pro-cessed efficiently to levels similar to that for the parental
Mc-Krae ford34.5 and to the rescued 17termAR for 17termA virus
(Fig. 2) upon infection of Vero cells transfected with vector (pcDNA3.1) or the kos321m plasmid containing the ICP34.5 gene from KOS321. The gC/pgC ratio in cells transfected with
the empty vector was 0.89 and 1.37 for d34.5 and 17termA,
respectively, and for the KOS321 ICP34.5-myc, gC/pgC was
0.80 and 1.46 ford34.5 and 17termA, respectively.
The influence of the ICP34.5 variant on gC processing was more pronounced in cells infected after the cells were selected
for expression of the appropriate ICP34.5-hrGFP variant using the fluorescence-activated cell sorter (Fig. 3). Expression of the SP7 ICP34.5 variant had a large inhibitory effect on the processing of gC in SLP10a-infected cells, reducing the gC/pgC ratio to 0.34, compared to 2.8 for the SLP10a-infected cells expressing only hrGFP. In contrast, the ratio of mature gC to precursor gC increased from 0.13 for SP7 virus in the cells expressing only hrGFP to 0.75 in the cells expressing the KOS321 ICP34.5-hrGFP, an enhancement of gC processing.
This approach clearly demonstrates the importance and in-fluence of specific forms of the ICP34.5 protein on the tissue culture behavior of HSV-1 because there is no other viral function expressed in the target cell other than the infecting virus. The ectopically expressed ICP34.5 variant protein from SP7 appears to dominate over ICP34.5 from the infecting virus and acts on the cell to limit the processes important for viral glycoprotein processing, while the ectopically expressed KOS variant of the ICP34.5 protein promotes the efficiency of viral glycoprotein processing.
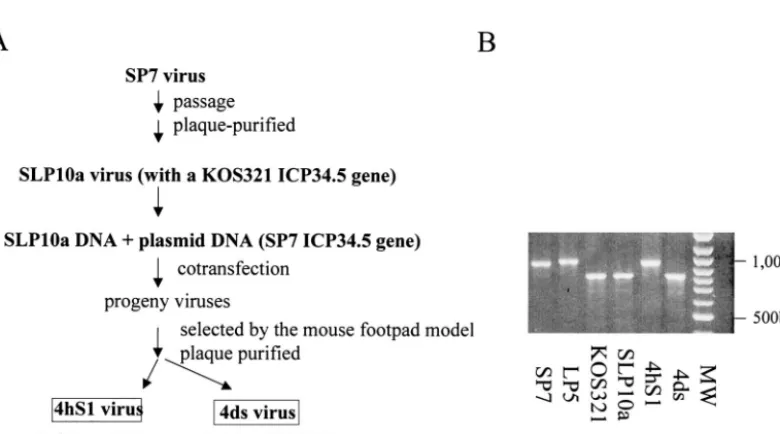
Tissue culture behavior of a recombinant SLP10a virus with an SP7 ICP34.5 gene. A recombinant virus was constructed from SLP10a in which its KOS321-like ICP34.5 gene was re-placed with the SP7 ICP34.5 gene. SLP10a virus was derived from SP7 and shares a very similar genetic background with its parent. The KOS321 form of the ICP34.5 protein is the same as the SLP10 isoform. Attempts to isolate a small-plaque-producing recombinant in tissue culture were unsuccessful, requiring that an in vivo selection be used to isolate the re-combinant virus (Fig. 4) based on the transfer of the neuroin-vasive property of the SP7 virus which is lacking for the SLP10a or KOS321 virus. Similar procedures were used to isolate neu-roinvasive recombinant viruses with KOS as the genomic back-ground (43). Cell lysates prepared from monolayers of Vero
cells transfected with the SP7BamHI SP fragment and SLP10a
[image:4.603.135.453.67.240.2]viral DNA were injected into the footpads of 3- to 4-week-old BALB/c mice. After 5 to 7 days, the lumbrosacral dorsal root ganglia (DRG) were collected and tested for the presence of
FIG. 2. Ectopic expression of ICP34.5-myc variants determines the tissue culture behaviors of an infecting virus. Vero cells were transfected, and 2 days later they were treated with 1 mg of G418/ml for 3 days to inhibit protein synthesis in untransfected cells and then infected with ICP34.5 deletion mutants (d34.5 or TermA) or wild-type viruses (McKrae or AR, a rescue virus from TermA) and harvested 2 days later. Detergent extracts were analyzed by Western blotting. The solid arrows indicate the precursor gC, with an apparent mass of 84 kDa, while the open arrows represent the mature gC, which is 116 kDa. The ratio of the amount of the processed gC to that of precursor gC is indicated below each lane.
on November 8, 2019 by guest
http://jvi.asm.org/
virus by incubation with Vero cells. The collected virus was then plaque purified, and the ICP34.5 gene was examined by PCR. The in vivo selection procedure enriched the population of recombinant viruses for viruses with the SP7 ICP34.5 gene, and as a result, the chances for their isolation increased, bring-ing them from undetectable to a detectable level. Although most of the plaque-purified viruses obtained from the DRG produced large plaques, e.g., 4ds, small-plaque DRG isolates were detectable. The progression of the parental SLP10a to
[image:5.603.102.492.447.664.2]the DRG was predicted based on previous findings in a similar in vivo selection experiment in which KOS was blocked at the level of the spinal ganglia following footpad inoculation (43). One of the small-plaque isolates, 4hS1, was picked, cloned, and analyzed. The PCR product of the 4hS1 ICP34.5 gene had the same agarose gel mobility as that of the SP7 gene (Fig. 4B) and has the SP7 ICP34.5 gene sequence with 18 (CCC GCG ACC) repeats encoding PAT in the bridge region and 8 CGC repeats encoding Arg at the N terminus. The success of the in
FIG. 3. Ectopic expression of ICP34.5-GFP variants determines the tissue culture behaviors of an infecting virus. Vero cells expressing GFP from the vector or vector containing one of the variants of ICP34.5-GFP were selected based on the expression of GFP using fluorescence-activated cell sorter analysis. The cells were infected with the indicated strain of HSV-1, and gC was analyzed as described in the legend to Fig. 1. The transfecting ICP34.5 variant DNA and the infecting HSV-1 strains are indicated below the image. The solid arrow indicates the precursor gC, with an apparent mass of 84 kDa, while the open arrow represents the mature gC, of 116 kDa. The ratio of the amount of the processed gC to that of precursor gC is indicated below the image.
FIG. 4. Construction of recombinant virus 4hS1 with an SP7 ICP34.5 gene in an SLP10a background. (A) SLP10a genomic DNA was cotransfected with the ICP34.5-containingBamHI fragment of SP7. The progeny viruses were inoculated into footpads of mice. The lumbrosacral DRGs were collected, and two types of viruses were identified, one with an SP7-like ICP34.5 gene (4hS1) and the other with a KOS321-like gene (4ds). (B) PCR products of the ICP34.5 gene from the different strains were analyzed by electrophoresis. Positions of size standards are indicated in base pairs. LP5 is a clinical strain obtained from the lung of the same neonate as SP7. The LP5 ICP34.5 gene has 22 nucleotide repeats encoding PAT (7).
on November 8, 2019 by guest
http://jvi.asm.org/
vivo selection procedure supports the association of the neu-roinvasive property with the SP7 form of ICP34.5.
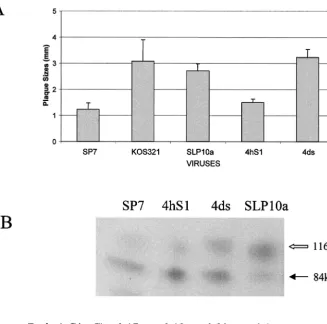
The 4hS1 virus, which has the SP7-like ICP34.5 gene, pro-duced plaques that were similar in size to SP7, significantly smaller than those of the parental SLP10a strain (Fig. 5A). The 4ds virus, with the SLP10a-like ICP34.5 gene, generated large plaques with sizes similar to those of SLP10a. The small plaque size is not due to a decrease in replication ability, because the replication competence of the 4hS1 virus in Vero and U373 (glioblastoma) cells was comparable to that of SP7 and SLP10a (Table 1). The efficiency of gC processing for 4hS1 infections was reduced to levels similar to that for SP7, for which the gC/pgC ratio was less than 0.2. This was different from the 4ds virus with a KOS321-like ICP34.5 gene, for which gC was
processed with higher efficiency (gC/pgC ⫽ 0.80) (Fig. 5B).
The 4hS1 virus was also more cell associated than the parental SLP10a or 4ds viruses, with a ratio of extracellular to intracel-lular virus at 72 h p.i. of 0.89, lower than for SLP10a (1.59) and 4ds (2.14) (Table 1). Analysis of the properties of 4hS1 indi-cates that the replacement of the SLP10a (KOS321-like) form of ICP34.5 with the SP7 ICP34.5 gene rescued the small-plaque phenotype (small small-plaque size, limited glycoprotein pro-cessing, and limited virus release) from SLP10a virus.
Neuroinvasiveness of a recombinant SLP10a virus with an SP7 ICP34.5 gene. The neuroinvasive potential of the 4hS1 recombinant virus was compared with those of the parental and 4ds virus strains. BALB/c mice were challenged with sim-ilar amounts of virus in the right hind footpad, and each mouse was monitored for signs of neurological disease. The extent of disease was compared on the sixth day p.i. (Table 2). Mice inoculated with SLP10a or 4ds did not exhibit any neurological signs, similar to previous results for KOS321 (7). Consistent with earlier findings (7), SP7 was the most neurovirulent virus, and all the challenged mice presented with neurological signs ranging from a crippled foot (paresis) to monoplegia, paraple-gia, or death. Importantly, the 4hS1 virus induced neurological disease, although mild, such that two out of the five mice exhibited paresis in the challenged leg on day 6.
The lumbosacral DRG was extracted from one mouse from each group on day 6 and tested for the presence of virus by cocultivation with Vero cells. Virus was recovered from the DRG of mice challenged with SP7 and 4hS1, but no virus was isolated from mice inoculated with SLP10a or 4ds (Table 2).
These results show that the 4hS1 virus is neuroinvasive and has acquired the potential to cause neuroinvasive disease, un-like its attenuated parent, SLP10a. These in vivo results
dem-FIG. 5. Tissue culture behavior of recombinant virus 4hS1 with an SP7 ICP34.5 gene in an SLP10a background. (A) Average plaque size (n
⫽ ⬃20) of virus grown in Vero cells. (B) HSV-1 gC from infected Vero cells. Whole-cell detergent extracts from Vero cells infected with each virus were examined by SDS-PAGE and Western blot analysis using polyclonal anti-gC. The image was developed by chemiluminescence and digitally scanned. The black arrow indicates the precursor gC, with an apparent mass of 84 kDa, while the open arrow represents the mature gC, which is 116 kDa. The ratio of the amount of the processed gC to that of precursor gC is indicated below each lane.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.603.135.462.73.397.2]onstrate that the SP7 ICP34.5 gene product can confer neuro-invasive potential to an attenuated virus. The neuroneuro-invasive potential, like the plaque size of 4hS1, is not equivalent to that of SP7, but this may be due to other mutations in the virus suggested by the observation of small changes in plaque size during passage of SP7 during the generation of SLP10a. The properties of 4hS1, 4ds, and their parental viruses are summa-rized in Table 3.
DISCUSSION
Many different viral functions contribute to the encephalitic and neuroinvasive disease potential of HSV-1, including activ-ities which promote growth in neuronal and other cells, effi-cient spread to and through the central nervous system, and escape from host innate and immune responses. Comparison of the properties of the SP7 virus, isolated from the brain of a neonate who died of disseminated HSV infection, and its tissue culture-passaged and attenuated progeny, SLP10a, suggested that the ICP34.5 gene is important for neuroinvasive disease, in addition to its known importance for neurovirulence (12, 14, 31). These studies were facilitated by the observation that the neuroinvasive isolates SP7 (7, 21) and 490 (18) share a com-mon tissue culture behavior, the small-plaque phenotype, which includes a restriction in viral glycoprotein processing and virion release, and an ICP34.5 protein with 18 PAT re-peats in the center of the protein. In contrast, many attenuated viruses, such as KOS321, SLP5, and SLP10, produce large plaques and have only three PAT repeats (7, 18). The ability of the pL/ST plasmid from KOS to genetically transfer the large-plaque phenotype into SP7 and 490 indicated that sequences containing ICP34.5 could eliminate the small-plaque pheno-type (7; also data not shown), but the present studies demon-strate that specific isoforms of the ICP34.5 protein confer both
the plaque phenotype and neuroinvasive disease potential onto the virus.
Although viruses with different numbers of PAT repeats in their ICP34.5 protein replicate efficiently and PAT repeats are not required for HSV-2 replication (33), the major structural difference between the ICP34.5 proteins of the neuroinvasive, small-plaque-producing SP7 virus and the attenuated, large-plaque-producing SLP10a and KOS321 viruses is the number
of PAT repeats. The association of the (PAT)3 variant of
ICP34.5 with large-plaque production and attenuation was strengthened by the observation that multiple passaging of SP7 reproducibly results in the concurrent change in tissue culture behavior to favor a virus capable of rapid dissemination (large-plaque production), truncation of the number of PAT repeats in ICP34.5 from 18 for SP7 to 3 repeats, and also attenuation of neuroinvasive disease production. The SLP5 and SLP10a (7) viruses were the products of this tissue culture selection pro-cess. Interestingly, the evolution of KOS321 probably occurred in a similar manner, since the low-passage, related KOS79 (19) (obtained from the same site and person as KOS321) produces small plaques, is neuroinvasive (19), and has 11 PAT repeats (32).
The importance of the ICP34.5 protein and specific struc-tural features of the protein to the tissue culture behavior of HSV-1 is shown by the ability of the specific ICP34.5 protein variants expressed in transfected cells to confer their parental phenotype onto an infecting virus. This approach clearly dem-onstrates the importance and influence of specific forms of the ICP34.5 protein, because there is no other viral function ex-pressed in the target cell other than that of the specific ICP34.5 and the infecting virus. Expression of the SP7 variant of ICP34.5 limited the processing of gC during infection by the deletion mutants, d34.5 and TermA, and for the viruses with
the (PAT)3ICP34.5 variant, SLP10a and KOS321.
Unexpect-TABLE 2. Neurological symptoms (day 6) of mice following HSV infectionaof the footpad
SP7 (6.7⫻106) 4hS1 (7.6⫻106) 4ds (6.7⫻106) SLP10A (1.1⫻107)
Mouse no. Symptom Mouse no. Symptom Mouse no. Symptom Mouse no. Symptom
Ib Paralysis, both legs I —c I — I —
IId(⫹) Paralysis, both legs IId(⫹) Paresis, right leg IId(⫺) — IId(⫺) —
III Paresis, right leg III Paresis, right leg III — III —
IVb Paralysis, both legs IV — IV — IV —
V Paresis, right leg V — V —
aThe numbers in parentheses are the viral doses used for the inoculation. bThese mice died within 2 days after 6th day postchallenge.
cNo neurological symptoms.
dMice were sacrificed on day 6, and the lumbosacral DRG were extracted. The presence of virus in the DRG was examined by coincubation with Vero cells.⫹or
[image:7.603.44.542.83.161.2]⫺indicates the presence or absence of virus in the DRG tissue, respectively.
TABLE 3. Summary of genetic and phenotypic properties
Virus Parentalvirus ICP34.5 geneproduct No. of PATrepeats N-terminalNo. of arginines
Plaque
size Glycoproteinprocessing Viral release Neuroinvasivepotential
SP7 18 8 Small Limited Very limited High
KOS321 3 7 Large Efficient Efficient None
SLP10a SP7 KOS321-like 3 7 Large Very efficient Very efficient None
4HS1 SLP10a SP7-like 18 8 Small Limited Limited Some
4ds SLP10a KOS321-like 3 7 Large Efficient Efficient None
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.603.44.547.643.725.2]edly, glycoprotein processing of the deletion mutants in Vero cells appeared normal, in contrast with observations from other studies (8). Expression of the KOS321-like ICP34.5 vari-ant (structurally similar to the SLP10a ICP34.5 protein) pro-moted efficient processing of the viral glycoprotein for the
(PAT)18-containing SP7 virus, a characteristic of the
large-plaque phenotype.
The connection and contribution of the SP7 form of ICP34.5 to the small-plaque phenotype and neuroinvasive disease po-tential was demonstrated by the ability of the SP7 variant of the ICP34.5 gene to rescue both of these properties upon creation of the 4hS1 recombinant virus. These properties were lost during the creation of the parental SLP10a virus during pas-sage of SP7. The in vivo selection procedure required for isolation of 4hS1 demonstrated that insertion of the SP7 ICP34.5 gene product into SLP10a enhanced the ability of the virus to reach the DRG following peripheral inoculation. The incorporation of SP7 ICP34.5 sequences into SLP10a to gen-erate 4hS1 resulted in a virus which exhibits the small-plaque phenotype and now can cause limited neurological disease following inoculation of mouse footpads. In addition, the 4hS1 virus could be isolated from the DRG on the sixth day after inoculation of the footpad, whereas the SLP10a could not. The inability of the SP7 ICP34.5 gene to promote an equivalent level of neuroinvasiveness upon insertion into the SLP10a background is probably due to the presence of spontaneous mutations elsewhere in the genome. This was suggested by the gradual increase in plaque size that was observed with each passage of SP7 to generate SLP10a. Similar attempts to genet-ically transfer neuroinvasive activity into the ANG virus with sequences from ANG-PATH demonstrated the importance of gD but similarly could not rescue ANG to the virulence level demonstrated by ANG-PATH (26). In a similar experiment, insertion of the SP7 ICP34.5 gene into the closely related LP5 viral genome yielded a virus producing smaller plaques than LP5 (data not shown). Neuroinvasiveness was not tested, since LP5 causes neuroinvasive disease (7).
The number of PAT repeats is a major determinant of the intracellular distribution of ICP34.5 within the cell and also the distribution of its ligand, PP1 (32). The KOS321 variants of ICP34.5 and PP1 are concentrated in the nucleolus, whereas the SP7 variants of ICP34.5 and PP1 are concentrated in the cytoplasm to the exclusion of the nucleus in Vero cells con-taining the ectopically expressed ICP34.5 variants. As such, ICP34.5 appears to act like one of several cell-encoded PP1 binding proteins, including PNUT (3), neurofilament L (41), and spinophilin (4), that bind to PP1 and localize it to the nucleus, neuronal plasma membrane, or dendritic spines, re-spectively. These are proteins which direct PP1 to different cellular locations, influence its substrate specificity, and mod-ulate its enzymatic activity. Targeting of the PP1 to the cyto-plasm by the SP7 variant of ICP34.5, but not the KOS variant,
could promote greater interaction with eIF2-␣on the ribosome
and to cellular processes involved in virion egress and viral glycoprotein processing. ICP34.5 activation of PP1 promotes
the dephosphorylation of eIF2-␣, to reverse the inhibition of
protein synthesis induced by HSV replication or by the alpha interferon-induced antiviral state (22, 23, 27, 35). PP1 has also been reported to be involved in membrane fusion, proper function of the endoplasmic reticulum networks, and vesicular
transport in yeast (37),Xenopusegg (2), and mammalian cells
(17). Inhibition of vesicular transport would restrict HSV gly-coprotein processing and virion release (20), resulting in small-plaque production. In contrast, binding of PP1 to the KOS321 variant of ICP34.5 and sequestration of PP1 in the nucleus would limit exposure of PP1 to these processes and thus could prevent an inhibitory activity.
The ability of ICP34.5 to facilitate HSV replication in neu-rons is an important feature for promoting neurovirulence but is not sufficient for promoting neuroinvasion. A neuroinvasive virus is also able to avoid host immune control and travel efficiently through the central nervous system. The presence of
⬎11 PAT repeats (as for KOS79) in the protein appears to be
important for neuroinvasive disease potential. Although the lack of PAT repeats in the HSV-2 protein (38) might seem to contradict this statement, HSV-2 is less likely to cause enceph-alitis than HSV-1 and much more likely to cause meningitis (5, 16) in an immunocompetent individual and is less likely to induce brain lesion formation in a mouse model (42). HSV-2 is also more sensitive to interferon alpha and beta (29, 30). The ability of ICP34.5 to counteract interferon action is important for the virulence of the virus (11, 14, 27, 28, 35), and strain-dependent differences in the cellular location of ICP34.5 and its ligand, PP1, may influence the efficiency of this activity. In addition, limiting the release of virion particles and cell surface expression of viral glycoproteins for the small-plaque pheno-type would limit the display and availability of viral antigen to reduce exposure and hence reduce the induction of immune responses. A recent study indicated that glioblastoma cells infected with an HSV-1 strain expressing an ICP34.5 variant with 10 PAT repeats expressed fewer cell surface major histo-compatibility complex class II molecules than an ICP34.5 de-letion mutant (44), suggesting another means of immune es-cape. The combination of these effects would enhance virus replication in neuronal cells, promote escape from the innate interferon alpha response, and reduce exposure to antigen-induced immunity to allow the virus to progress more exten-sively into the central nervous system, beyond the site of in-fection, to promote neuroinvasive disease progression.
ACKNOWLEDGMENTS
This research was supported by Public Health Service research grant R15 NS40324-01 from the National Institute for Neurological Diseases and Stroke to K.S.R.
We thank William Lynch for advice and help with the FACS and Tom Kim and Scott Shors for helpful scientific discussions and reviews of our manuscript.
REFERENCES
1. Ackermann, M., J. Chou, M. Sarmiento, R. A. Lerner, and B. Roizman.1986. Identification by antibody to a synthetic peptide of a protein specified by a diploid gene located in the terminal repeats of the L component of herpes simplex virus genome. J. Virol.58:843–850.
2. Allan, V.1995. Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J. Cell Biol.
128:879–891.
3. Allen, P. B., Y. G. Kwon, A. C. Nairn, and P. Greengard.1998. Isolation and characterization of PNUTS, a putative protein phosphatase 1 nuclear tar-geting subunit. J. Biol. Chem.273:4089–4095.
4. Allen, P. B., C. C. Ouimet, and P. Greengard.1997. Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. USA94:9956–9961.
5. Bergstrom, T., A. Vahlne, K. Alestig, S. Jeansson, M. Forsgren, and E. Lycke.1990. Primary and recurrent herpes simplex virus type 2-induced meningitis. J. Infect. Dis.162:322–330.
on November 8, 2019 by guest
http://jvi.asm.org/
6. Bolovan, C. A., N. M. Sawtell, and R. L. Thompson.1994. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn⫹are attenuated for neuroviru-lence in mice and for replication in confluent primary mouse embryo cell cultures. J. Virol.68:48–55.
7. Bower, J. R., H. Mao, C. Durishin, E. Rozenbom, M. Detwiler, D. Rempinski, T. L. Karban, and K. S. Rosenthal.1999. Intrastrain variants of HSV-1 isolated from a neonate with fatal disseminated infection differ in the ICP34.5 gene, glycoprotein processing, and neuroinvasive disease. J. Virol.
73:3843–3853.
8. Brown, S. M., A. R. MacLean, J. D. Aitken, and J. Harland.1994. ICP 34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J. Gen. Virol.75:3679–3686.
9. Brown, S. M., A. R. MacLean, E. A. McKie, and J. Harland.1997. The herpes simplex virus virulence factor ICP34.5 and the cellular protein MyD116 complex with proliferating cell nuclear antigen through the 63-amino-acid domain conserved in ICP34.5, MyD116, and GADD34. J. Virol.
71:9442–9449.
10. Cheng, G., M.-E. Brett, and B. He.2002. Signals that dictate nuclear, nucle-olar, and cytoplasmic shuttling of the␥134.5 protein of herpes simplex virus
type 1. J. Virol.76:9434–9445.
11. Chou, J., J.-J. Chen, M. Gross, and B. Roizman.1995. Association of a novel M190,000 phosphoprotein with PKR kinase in cells exhibiting enhanced
phosphorylation of eIF-2␣and premature shutoff of protein synthesis after infection with␥134.5⫺mutants of herpes simplex virus 1. Proc. Natl. Acad.
Sci. USA92:10516–10520.
12. Chou, J., E. R. Kern, R. J. Whitley, and B. Roizman.1990. Mapping of herpes simplex virus 1 neurovirulence to␥134.5, a gene nonessential for
growth in cell culture. Science252:1262–1266.
13. Chou, J., and B. Roizman.1990. The herpes simplex virus 1 gene for ICP34.5, which maps in inverted repeats, is conserved in several limited-passage isolates but not in strain 17syn⫹. J. Virol.64:1014–1020. 14. Chou, J., and B. Roizman.1992. The gamma134.5 gene of herpes simplex
virus precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programmed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA9:3266–3270.
15. Chou, J., and B. Roizman.1994. Herpes simplex virus gamma-1 34.5 gene function, which blocks the host response to infection, maps in the homolo-gous domain of the genes expressed during growth arrest and DNA damage. Proc. Natl. Acad. Sci. USA91:5247–5251.
16. Craig, C. P., and A. J. Nahmias.1973. Different patterns of neurologic involvement with herpes simplex virus types 1 and 2: isolation of herpes simplex virus type 2 from the buffy coat of two adults with meningitis. J. Infect. Dis.127:365–372.
17. Davidson, H. W., C. H. McGowan, and W. E. Balch.1992. Evidence for the regulation of exocytic transport by protein phosphorylation. J. Biol. Chem.
116:1343–1355.
18. Dick, J. W., and K. S. Rosenthal.1995. A block in glycoprotein processing correlates with small plaque morphology and virion targeting to cell-cell junctions for an oral and an anal strain of herpes simplex virus type-1. Arch. Virol.140:2163–2181.
19. Dix, R. D., R. R. McKendall, and J. R. Baringer.1983. Comparative neuro-virulence of herpes simplex virus type 1 strains after peripheral or intrace-rebral inoculation of BALB/c mice. Infect. Immun.40:103–112.
20. Enquist, L. W., P. J. Husak, B. W. Banfield, and G. A. Smith.1999. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virol. Res.
51:237–347.
21. Goel, N., H. Mao, Q. Rong, J. J. Docherty, D. Zimmerman, and K. S. Rosenthal.2002. The ability of an HSV strain to initiate zosteriform spread correlates with its neuroinvasive disease potential. Arch. Virol.147:763–773. 22. He, B., J. Chou, D. A. Liebermann, B. Hoffman, and B. Roizman.1996. The carboxyl terminus of the murine MyD116 gene substitutes for the corre-sponding domain of the gamma-1 ICP34.5 of herpes simplex virus to pre-clude the premature shutoff of total protein synthesis in infected human cells. J. Virol.70:84–90.
23. He, B., M. Gross, and B. Roizman.1997. The␥-1 34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1␣to dephosphorylate the␣subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA94:843–848.
24. He, B., M. Gross, and B. Roizman.1998. The␥-1 34.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem.273:20737–20743. 25. Holland, T. C., S. D. Marlin, M. Levine, and J. Glorioso.1983. Antigenic
variants of herpes simplex virus selected with glycoprotein-specific monoclo-nal antibodies. J. Virol.45:672–682.
26. Izumi, K. M., and J. G. Stevens.1990. Molecular and biological character-ization of an HSV-1 neuroinvasion gene. J. Exp. Med.172:487–496. 27. Leib, D. A., T. E. Harrison, K. M. Laslo, M. A. Machalek, N. J. Moorman,
and H. W. Virgin.1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med.189:663–672. 28. Leib, D. A., M. A. Machalek, B. R. G. Williams, R. H. Silverman, and W. V.
Herbert.2000. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA97:6097– 6101.
29. Lerner, A. M., and E. J. Bailey.1976. Differential sensitivity of SHV types 1 and 2 to human interferon: antiviral effects of interferon plus 9-beta-D -arabinofuranosyladenine. J. Infect. Dis.134:400–404.
30. Leventon-Kriss, S., M. Movshovitz, Z. Smetana, M. Shewach-Millet, T. Doerner, and T. Gotlieb-Stematsky.1987. Sensitivity in vitro of herpes sim-plex virus isolates to human fibroblast interferon. Med. Microbiol. Immunol.
176:151–159.
31. MacLean, A., L. Robertson, E. McKay, and S. M. Brown.1991. The RL neurovirulence locus in herpes simplex virus type 2 strain HG52 plays no role in latency. J. Gen. Virol.72:2305–2310.
32. Mao, H., and K. S. Rosenthal.2002. An N-terminal arginine-rich cluster and a proline-alanine-threonine repeat region determine the cellular localization of the herpes simplex virus type 1 ICP34.5 protein and its ligand protein phosphatase 1. J. Biol. Chem.277:11423–11431.
33. McGeoch, D. J., C. Cunningham, G. McIntyre, and A. Dolan.1991. Com-parative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J. Gen. Virol.72:3057–3075.
34. Mitchell, B. M., and J. G. Stevens.1996. Neuroinvasive properties of herpes simplex virus type I glycoprotein variants are controlled by the immune response. J. Immunol.156:246–255.
35. Mossman, K. L., and J. R. Smiley.2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol.76:1995–1998.
36. Perng, G. C., R. L. Thompson, N. M. Sawtell, W. E. Taylor, S. M. Slanina, H. Ghiasl, R. Kaiwar, A. B. Nesburn, and S. L. Wechsler.1995. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol.69:3033–3041.
37. Peters, C., P. D. Andrews, M. J. Stark, S. Cesaro-Tadic, A. Glatz, A. Pod-telejnikov, M. Mann, and A. Mayer.1999. Control of the terminal step of intracellular membrane fusion by protein phosphatase 1. Science285:1084– 1087.
38. Ravi, V., P. G. Kennedy, and A. R. MacLean.1998. Functional analysis of the herpes simplex virus type 2 strain HG52 RL1 gene: the intron plays no role in virulence. J. Gen. Virol.79:1613–1617.
39. Robbins, A. K., M. E. Whealy, and L. W. Enquist.1988. Centrifugation procedures for studying herpes viruses using the Sorvall RC-28 SUPRAspeed centrifuge. Dupont Biotech. Update3:1–3.
40. Sambrook, J., E. F. Fritsch, and T. Maniatis.1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
41. Terry-Lorenzo, R. T., M. Inoue, J. H. Connor, T. A. J. Haystead, B. N. Armbruster, R. P. Gupta, C. J. Oliver, and S. Shenolikar.2000. Neurofila-ment-L is a protein phosphatase-1-binding protein associated with neuronal plasma membrane and post-synaptic density. J. Biol. Chem.275:2439–2446. 42. Thomas, H. C., R. D. Kapadia, G. I. Wells, A. M. Gresham, D. Sutton, H. A. Solleveld, S. K. Sarkar, S. B. Dillon, and R. Tal-Singer.2001. Differences in pathogenicity of herpes simplex virus serotypes 1 and 2 may be observed by histopathology and high-resolution magnetic resonance imaging in a murine encephalitis model. J. Neurovirol.7:105–116.
43. Thompson, R. L., M. L. Cook, G. B. Devi-Rao, E. K. Wagner, and J. G. Stevens.1986. Functional and molecular analyses of the avirulent wild-type herpes simplex virus type 1 strain KOS. J. Virol.58:203–211.
44. Trgovcich, J., D. Johnson, and B. Roizman.2002. Cell surface major histo-compatibility complex class II proteins are regulated by the products of the gamma-1 34.5 and Ul41 genes of herpes simplex virus 1. J. Virol.76:6974– 6986.
45. Yuhasz, S. A., and J. G. Stevens.1993. Glycoprotein B is a specific deter-minant of herpes simplex virus type 1 neuroinvasiveness. J. Virol.67:5948– 5954.46Zon, L. I., D. M. Dorfman, and S. H. Orkin.1989. The polymerase chain reaction colony minipre. Biotechniques7:696–698.
46. Zon, L. I., D. M. Dorfman, and S. H. Orkin.1989. The polymerase chain reaction colony miniprep. Biotechniques7:696–698.