Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory
Factor 3 Inhibits Gamma Interferon and Major Histocompatibility
Complex Class II Expression
䌤
†
Katharina Schmidt, Effi Wies, and Frank Neipel*
Virologisches Institut, Universita¨tsklinikum Erlangen, Schlossgarten 4, D-91054 Erlangen, Germany
Received 7 October 2010/Accepted 14 February 2011
Kaposi’s sarcoma-associated herpesvirus (KSHV) carries four genes with homology to human interferon regulatory factors (IRFs). One of these IRFs, the viral interferon regulatory factor 3 (vIRF-3), is expressed in latently infected primary effusion lymphoma (PEL) cells and required for their continuous proliferation. Moreover, vIRF-3 is known to be involved in modulation of the type I interferon (IFN) response. We now show that vIRF-3 also interferes with the type II interferon system and antigen presentation to the adaptive immune system. Starting with an analysis of the transcriptome, we show that vIRF-3 inhibits expression of major histocompatibility complex class II (MHC II) molecules: small interfering RNA (siRNA)-mediated knockdown of vIRF-3 in KSHV-infected PEL cell lines resulted in increased MHC II levels; overexpression of vIRF-3 in KSHV-negative B cells leads to downmodulation of MHC II. This regulation could be traced back to inhibition of class II transactivator (CIITA) transcription by vIRF-3. Reporter assays revealed that the gamma interferon (IFN-␥)-sensitive CIITA promoters PIV and PIII were inhibited by vIRF-3. Consistently, IFN-␥ levels in-creased upon vIRF-3 knockdown in PEL cells. IFN-␥regulation by vIRF-3 was confirmed in reporter assays as well as by upregulation of typical IFN-␥target genes upon knockdown of vIRF-3 in PEL cells. In summary, we conclude that vIRF-3 contributes to the viral immunoevasion by downregulation of IFN-␥and CIITA and thus MHC II expression.
Kaposi’s sarcoma-associated herpesvirus (KSHV), also termed human herpesvirus 8 (HHV-8), belongs to the gamma-herpesvirus-2 subgroup (10). It is associated with all epidemi-ological forms of Kaposi’s sarcoma (KS) and two lymphopro-liferative disorders: primary effusion lymphoma (PEL) (9) and multicentric Castleman disease (52). The genome of KSHV contains a cluster of four genes with homology to cellular interferon regulatory factors (IRFs) (reviewed in reference 25). The viral interferon regulatory factor 3 (vIRF-3), also termed latency-associated nuclear antigen 2 (LANA-2) or K10.5, is among the few viral genes expressed in all latently infected PEL cells (12, 30, 47, 55). Recently,vIRF-3was shown to be required for the continuous proliferation of PEL cells in culture and can therefore be seen as abona fideoncogene of KSHV (55). However, the mechanisms required for the onco-genic activity of vIRF-3 are not sufficiently clear. Possible cel-lular targets of vIRF-3 comprise not only repression of p53 (47) but also the stimulation of c-myc-dependent transcription (31), the stabilization of hypoxia-inducible factor 1␣(HIF-1␣) (51), and inhibition of the proapoptotic cellular IRF-5 (54). Moreover, modulation of the interferon (IFN) system is an important function of vIRF-3 as expected from sequence ho-mology. So far, vIRF-3 has been reported to counteract the interferon class I response by interfering with cellular IRF-3
(30), IRF-7 (21), and IRF-5 (54) as well as by inhibition of protein kinase R (PKR) (15). Until now, vIRF-3 has not been shown to directly modulate the class II interferon response or adaptive immunity. However, a systematic analysis of vIRF-3 functions and effects on the transcriptome has not been pub-lished so far. We thus examined the consequences of vIRF-3 depletion on the transcription of cellular genes. Enhanced transcription of major histocompatibility complex class II (MHC II) genes was the most prominent effect of vIRF-3 knockdown in PEL cells. MHC II expression is normally re-stricted to antigen-presenting cells (B cells, macrophages, and dendritic cells); however, in humans MHC II expression is inducible by gamma interferon (IFN-␥) in almost every cell type (44). The class II transactivator (CIITA) is the key regu-lator of MHC II transcription. Four distinct promoters (PI to PIV) control the transcription of CIITA in a cell-type-specific manner: PI acts in dendritic cells and macrophages, and PIII acts in B lymphocytes. PIV is inducible by IFN-␥in almost every cell type (36).
We show here that the downregulation of MHC II expres-sion by vIRF-3 is essentially due to reduced activity of the IFN-␥-responsive promoters of the main regulator of MHC II transcription, the class II transactivator (CIITA).
MATERIALS AND METHODS
Cell culture and transfection.KSHV-positive PEL cell lines BC-3 (4), JSC-1 (8), and BCBL-1 (45) and KSHV-negative B cell lines (Akata and BJAB) were obtained from the ATCC (Manassas, VA) and cultured as described previously (55). HEK293T cells were obtained from the ATCC and grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS). Jurkat T cells (E6.1; ATCC; TIB-152) were maintained in RPMI 1640 medium (Invit-rogen) supplemented with 10% fetal bovine serum (Invit(Invit-rogen), glutamine, and gentamicin. Cells from the multiple myeloma-derived cell line INA-6 (7) were
* Corresponding author. Mailing address: Virologisches Institut, Universita¨tsklinikum Erlangen, Schlossgarten 4, D-91054 Erlangen, Germany. Phone: 49-9131-8523786. Fax: 49-9131-8526493. E-mail: Frank.neipel@viro.med.uni-erlangen.de.
† Supplemental material for this article may be found at http://jvi .asm.org/.
䌤Published ahead of print on 23 February 2011.
4530
on November 7, 2019 by guest
http://jvi.asm.org/
grown in the presence of 500 U/ml human interleukin-6 (IL-6; Strathmann Biotech, Hannover, Germany).
HEK293T cells were transfected at 70% density using Lipofectamine and Plus reagent (Invitrogen) according to the manufacturer’s instructions in 12-well plates. An 0.2-g amount of a luciferase reporter construct was cotransfected with indicated amounts of expression construct. DNA concentration was ad-justed using empty vector. Jurkat T cells (107cells per sample) were transfected
by electroporation with an Easyject Plus apparatus (Equibio, Boughton, United Kingdom) at 250 V and 1,500F in medium without antibiotics. Eight micro-grams of luciferase reporter plasmid was cotransfected with indicated amounts of expression plasmids. Total amount of DNA was adjusted by the addition of empty vector DNA. BJAB cells (2⫻106
) were electroporated with 10g plasmid DNA by using an Amaxa electroporation apparatus and Nucleofector kit V (Lonza) with program Q01 according to the manufacturer’s instructions. Prior to transfection, BJAB cells were maintained at a density of 200,000 cells/ml for 3 days. Electroporation efficiency was determined by transfection of a green fluo-rescent protein (GFP) expression plasmid and fluorescence-activated cell sorting (FACS) analysis. Approximately 60% of the viable BJAB cells were found to be GFP positive. For small interfering RNA (siRNA) transfection, PEL cells were first maintained at a density of 350,000 cells/ml for 3 days. A total of 106PEL
cells were then transfected with 2.6g siRNA using the HiPerFect (Qiagen) transfection reagent according to the manufacturer’s instructions. siRNAs tar-geted at vIRF-3 (si463 and si976) and the negative-control siRNA (siN) have been described before (55). si716 targets the sequence 5⬘-AAGAUGACCUCA CACUGCUUGAUAA-3⬘of the vIRF-3 transcript.
Plasmids and cloning. Construction of expression plasmids pcK8.1 and pcK10.5 for the expression of gpK8.1 and vIRF-3, respectively, has been de-scribed before (49, 54). CIITA promoter luciferase constructs PIII-luc and PIV-luc were kind gifts of Janet Piskurich (41, 42). The 3.6-kb IFN-␥luciferase construct was provided by Howard Young (16). A GFP expression plasmid was supplied with the Amaxa Nucleofector kit (Lonza).
Western blotting and antibodies. Western blot analysis was performed as described elsewhere (6). Primary antibodies and antisera were obtained from Sigma (anti--tubulin: mouse, T7941; anti--actin: mouse AC-15, A5441), Santa Cruz (anti-STAT-1: rabbit, 9172; anti-phospho-STAT-1 [Tyr701]: rabbit, 9176S antibody) and Abcam (anti-human-MHC II: mouse, ab55152). Rat monoclonal anti-vIRF-3 antibody 3G7 was produced in cooperation with E. Kremmer (Helmholtz Center, Munich, Germany) as described previously (55). Monoclonal antibody BS555 against K8.1 was described previously (24). Secondary antibodies conjugated with horseradish peroxidase were obtained from Dako (Dako Deutschland GmbH, Hamburg, Germany). Peroxidase activity was detected by electrochemiluminescence using an ImageReader LAS-1000 (Fujifilm, Tokyo, Japan) charge-coupled device (CCD) camera and software. Protein expression was quantified using AIDA image analyzer v4.22 (Raytest GmbH, Strauben-hardt, Germany).
Affymetrix GeneChip hybridization and analysis.Sample preparation and microarray hybridization were performed by the Affymetrix Service Provider and Core Facility, KFB-Center of Excellence for Fluorescent Bioanalytics (Regens-burg, Germany; www.kfb-regensburg.de), as described in the Affymetrix GeneChip expression analysis technical manual (Affymetrix, Santa Clara, CA). Briefly, 106 BC-3 cells were transfected with either one siRNA targeted at
vIRF-3 or a negative-control siRNA (targeted at vIRF-3, si476 or si716; control, siN). Cells were harvested 2 days after transfection, and total cellular RNA was purified with RNeasy minicolumns (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Double-stranded cDNA was synthesized from total RNA followed by anin vitrotranscription (IVT) reaction to produce biotin-labeled cRNA. After fragmentation, cRNA was hybridized to Affymetrix Human Genome U133 Plus 2.0 arrays. Hybridized arrays were washed and stained, and the fluorescent signals were measured with an Affymetrix GeneChip 3000-7G scanner. Expression signals for each of the 54,675 probe sets and comparisons between different samples were calculated with the Affymetrix GeneChip oper-ating software (GCOS) using the MAS5 algorithm.
FACS analysis.PEL and BJAB cells were harvested 2 days after transfection with siRNA or expression plasmid, respectively. The cells were washed once with phosphate-buffered saline (PBS) and once with FACS buffer (10% fetal calf serum [FCS] and 0.05% sodium azide in PBS) and then incubated with phyco-erythrin (PE)-coupled primary antibody against HLA-DR (1:100 in FACS buf-fer) for 1 h. After 3 washing steps with FACS buffer, the cells were analyzed on a FACSCalibur system (BD Biosciences, San Jose, CA).
Semiquantitative reverse transcription-PCR (RT-PCR).RNA isolation from 1 million cells was carried out using the Nucleospin RNAII RNA isolation kit (Machery & Nagel, Du¨ren, Germany) according to the manufacturer’s instruc-tions. cDNA was synthesized by using Superscript II reverse transcriptase
(In-vitrogen) and random hexamers (Bioline GmBH) as primers. After an initial activation step (95°C, 2 min), the PCR was performed using denaturation at 95°C for 30 s, annealing at 58°C for 30 s, and elongation at 72°C for 30 s. For each primer pair, the number of cycles required for optimal amplification in the linear range was determined experimentally. The oligonucleotides used for transcript amplification are shown in Table S1 in the supplemental material.
Real-time PCR.Quantitative real-time PCR (qPCR) was performed in an ABI Prism 7500 Thermocycler (Applied Biosystems, Foster City, CA) using the oli-gonucleotides given in Table S1 in the supplemental material and SYBR green dye. Briefly, 45 cycles of amplification were done with a reaction mixture con-taining cDNA and 0.2 mM Rox, 1.5 mM MgCl2, 0.2 mM deoxynucleoside
triphosphates (dNTPs), 0.1⫻SYBR green (Invitrogen), 10 pmol of each primer, 0.6-foldTaqPCR buffer, and 0.25l AmpliTaq Gold DNA polymerase (Applied Biosystems) in a final volume of 50l. MHC II transcripts were normalized to
-actin using the Pfaffl method (40).
Luciferase reporter assay.Two days after transfection, HEK293T cells were harvested by centrifugation, washed with PBS, and resuspended in 150l lucif-erase lysis buffer (50 mM Tris, pH 7.8, 0.5 M dithiothreitol, 1% Triton X-100, 20% glycerol) for 30 min. Lysates were cleared by centrifugation for 5 min at 13,000 revolutions/min. Luciferase activity was determined in duplicates by add-ing 100l assay buffer (100 mM K3PO4, 15 mM MgSO4, 5 mM ATP) and 100l
assay buffer supplemented with 1 mMd-luciferin (Roche Applied Science) using a microplate luminometer (Berthold Detection Systems, Pforzheim, Germany). Total protein content was measured with a Bio-Rad assay, and luciferase counts were normalized to protein concentration.
ELISA.Supernatants of siRNA-transfected PEL cells and controls were har-vested at the indicated time points. Cytokine secretion was measured in dupli-cates with IFN-␥and IL-6 enzyme-linked immunosorbent assays (ELISAs; Bec-ton Dickinson) according to the manufacturer’s instructions.
Microarray data accession number.The microarray data have been deposited in NCBI’s Gene Expression Omnibus (14) and are accessible through GEO series accession number GSE26661.
RESULTS
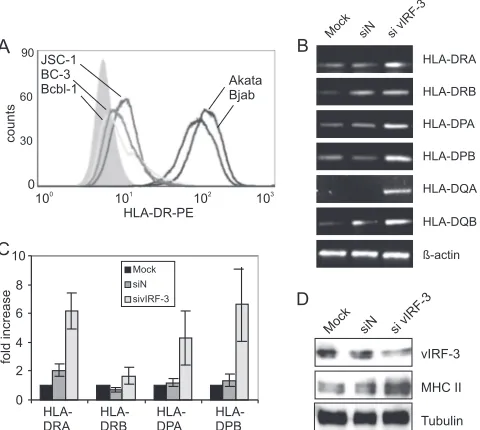
vIRF-3 regulates MHC II levels.vIRF-3 has been shown to interact with several cellular transcription factors. To analyze the overall outcome of these interactions at the level of the transcriptome, vIRF-3 expression was knocked down by RNA interference (RNAi) and genome-wide mRNA expression pro-files were determined by microarray hybridization. Two differ-ent siRNAs (si467 and si716) were used to knock down vIRF-3 expression in the PEL cell line BC-3. An siRNA without ho-mology to any human or viral gene (siN) served as a control. Microarray analysis revealed that all MHC II transcripts— HLA-DR, -DP, and -DQ—were consistently upregulated more than 2-fold upon knocking down vIRF-3 expression (see Table S2 in the supplemental material and GEO database series accession number GSE26661). PEL cells exhibit somatic hy-permutations of the rearranged immunoglobulin genes and are thus likely to be derived from postgerminal B cells (33). With several exceptions, like a lack of antibody production, their gene expression profile resembles that of plasma cells (20). Compared to other B cell lines like BJAB or Akata, they express MHC II at relatively low levels (Fig. 1A). The observed upregulation of MHC II transcripts upon knockdown of vIRF-3 was verified by semiquantitative RT-PCR. All MHC II transcripts (HLA-DRA/B, -DPA/B, and -DQA/B) were found to be upregulated upon knockdown of vIRF-3 expression in the PEL cell line BC-3 compared to untransfected (mock) and siN-transfected cells (Fig. 1B). Results similar to those shown in Fig. 1 for BC-3 were obtained using the dually KSHV- and Epstein-Barr virus (EBV)-infected PEL cell line JSC-1 (data not shown). qPCR with normalization against -actin con-firmed that knockdown of vIRF-3 expression in BC-3 cells resulted in a 2- (DRB) to 6-fold (DRA and
on November 7, 2019 by guest
http://jvi.asm.org/
DBP) increase of MHC II transcription (Fig. 1C). This in-crease in MHC II transcript levels was reflected by a marked increase of MHC II protein levels as shown in Fig. 1D. Den-sitometric analysis with normalization against tubulin revealed that the reduction of vIRF-3 by 60% resulted in an up to 4.4-fold increase of MHC II protein expression. The data shown in Fig. 1 were obtained using si463 targeted at vIRF-3. Comparable results were obtained with another siRNA against vIRF-3 (si976; data not shown). In addition to vIRF-3 deple-tion, experiments with overexpression of vIRF-3 in the
[image:3.585.44.284.71.286.2]KSHV-and EBV-negative B cell line BJAB were performed to cor-roborate the negative regulation of MHC II expression by vIRF-3. BJAB cells were transfected with expression con-structs for either vIRF-3, the KSHV glycoprotein gpK8.1 (6), or empty vector. Expression of vIRF-3 or gpK8.1 protein was verified by Western blotting (Fig. 2A). vIRF-3 overexpression led to a decrease of all MHC II transcripts (HLA-DRA/B, -DPA/B, and -DQA/B) (Fig. 2B, middle lane) compared to control transfections (Fig. 2B, left and right lanes).-Actin expression was essentially unaltered on both protein (Fig. 2A) and transcript (Fig. 2B) levels. Again, downregulation of MHC II upon vIRF-3 overexpression could be verified on the protein level. FACS analysis revealed that HLA-DRA expression on the surface of BJAB cells was reduced in cells expressing vIRF-3 (Fig. 2C) compared to that on BJAB cells transfected with empty vector (Fig. 2C). These results were consistent with the data obtained via vIRF-3 knockdown (Fig. 1). Thus, we FIG. 1. Knockdown of vIRF-3 leads to an upregulation of MHC
class II. (A) Low surface expression of MHC II in PEL cells compared to KSHV-negative B cell lines. Three KSHV-positive (BC-3, JSC-1, and BCBL-1) and two KSHV-negative (BJAB and Akata) B cell lines were stained with PE-labeled monoclonal antibody against HLA-DRA. The surface expression of HLA-DRA was determined by FACS analysis. Filled histogram, unstained cells; open histograms, left, light gray line, BCBL-1; dark gray line, BC-3; black line, JSC-1; right, dark gray line, BJAB; black line, Akata. (B) Upregulation of MHC II transcripts upon vIRF-3 knockdown. BC-3 cells were either mock transfected or transfected with a nonsense siRNA (siN) or an siRNA targeted at vIRF-3 (si vIRF-3) and maintained in culture for 2 days. Cells were harvested, and total RNA was isolated. Expression of MHC II (HLA-DRA/B, -DPA/B, and -DQA/B) mRNA was analyzed by semiquantitative RT-PCR (HLA-DRA/B and -DPA/B, 30 PCR cycles; HLA-DQA/B, 35 PCR cycles;-actin, 25 PCR cycles). Amplification of-actin was used to normalize MHC II expression. One represen-tative gel out of three independent experiments is shown. (C) Upregu-lation of MHC II transcripts upon vIRF-3 knockdown. BC-3 cells were either mock transfected or transfected with a nonsense siRNA (siN, dark gray) or an siRNA targeted at vIRF-3 (sivIRF-3, light gray) and maintained in culture for 2 days. Cells were harvested, and total RNA was isolated. Expression of MHC II transcripts HLA-DRA/B and HLA-DPA/B was quantified by real-time PCR analysis. Amplification of-actin was used for normalization. Error bars indicate the standard deviations of three independent experiments. (D) Upregulation of MHC II upon vIRF-3 knockdown in BC-3 PEL cells. BC-3 cells were either mock transfected or transfected with a nonsense siRNA (siN) or an siRNA targeted at vIRF-3 (si vIRF-3) and maintained in culture for 2 days. Cells were then harvested, and lysates were separated by SDS-PAGE and analyzed by Western blotting using monoclonal antibodies against vIRF-3 or MHC II. Detection of tubulin was used as a loading control. One representative blot out of three independent experiments is shown.
FIG. 2. Downregulation of MHC II expression upon overexpres-sion of vIRF-3. (A) Control of vIRF-3 expresoverexpres-sion in transfected BJAB cells. BJAB cells were transfected by Amaxa electroporation with either empty vector, vIRF-3 expression plasmid, or an expression plas-mid for KSHV glycoprotein K8.1 (gpK8.1). Electroporation of a GFP expression plasmid followed by FACS analysis revealed that about 60% of the viable cells were successfully transfected. Cells were har-vested after maintenance in culture for 2 days, and cell lysates were separated by SDS-PAGE and analyzed by Western blotting using monoclonal antibodies against vIRF-3 (top) and K8.1 (middle). De-tection of-actin was used as a loading control (bottom). (B) Down-regulation of MHC II transcripts upon vIRF-3 overexpression. BJAB cells were transfected by Amaxa electroporation with either empty vector, vIRF-3 expression plasmid, or an expression plasmid for KSHV gpK8.1 as a control. Total cellular RNA was isolated after mainte-nance in culture for 2 days. Expression of transfected vIRF-3 and gpK8.1 and expression of MHC II (HLA-DRA/B, -DPA/B, and -DQA/B) mRNA were analyzed by semiquantitative RT-PCR (HLA-DRA/B and -DPA/B, 30 PCR cycles; HLA-DQA/B, 35 PCR cycles; -actin, 25 PCR cycles). Amplification of-actin was used to normal-ize HLA expression. One representative gel out of three independent experiments is shown. (C) Downregulation of MHC II surface expres-sion upon vIRF-3 overexpresexpres-sion. BJAB cells were transfected by Amaxa electroporation with either empty vector, vIRF-3 expression plasmid, or an expression plasmid for KSHV gpK8.1. After mainte-nance in culture for 3 days, surface expression of HLA-DRA proteins was determined by FACS analysis using PE-labeled monoclonal anti-body. Filled histogram, unstained cells; open histogram, gray line, BJAB cells transfected with empty vector; black line, vIRF-3-trans-fected BJAB cells. One representative graph out of three independent experiments is shown.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.320.515.71.220.2]conclude that vIRF-3 downregulates the expression of MHC II molecules in PEL cells.
MHC II regulation by vIRF-3 is mediated by regulation of CIITA.MHC II expression is regulated by one master switch protein, the class II transactivator (CIITA). CIITA expression is essentially driven from the three cell-type-specific promot-ers, PI, PIII, and PIV, that give rise to distinct transcripts (reviewed in reference 44). In order to determine whether vIRF-3 inhibits MHC II transcription directly or via suppres-sion of CIITA, the influence of vIRF-3 on CIITA expressuppres-sion was examined. Semiquantitative RT-PCR was performed using oligonucleotide pairs specific for transcripts from either PI, PIII, or PIV. The macrophage-specific isoform of CIITA is transcribed from promoter PI. Transcripts generated from pro-moter PI were not detectable in BC-3 or BJAB cells (Fig. 3A and B, first row). However, both the B cell-specific transcript originating from the CIITA promoter PIII and the IFN-␥ -inducible isoform transcribed from promoter PIV were detect-able in untreated PEL cells (Fig. 3A, second and third rows, respectively). Knockdown of vIRF-3 expression in BC-3 cells by RNAi resulted in enhanced transcription from both PIII and PIV (Fig. 3A, second and third rows, respectively; lane si vIRF-3 versus lane Mock or siN). In addition, regulation of CIITA by vIRF-3 was verified by overexpression of vIRF-3 in KSHV-negative BJAB cells. Both the B cell-specific PIII tran-script and the IFN-␥-inducible PIV transcript were decreased upon overexpression of vIRF-3 in BJAB cells (Fig. 3B, middle lane) compared to control transfections with either empty vec-tor (Fig. 3B, left lane) or the KSHV lytic cycle protein gpK8.1 (Fig. 3B, right lane). The observed repression of CIITA pro-moters PIII and PIV by vIRF-3 was further confirmed by reporter assays. HEK293T cells were transfected with lucifer-ase constructs controlled by either CIITA promoter PIII or CIITA promoter PIV (Fig. 3C and D, respectively). Cotrans-fection of a vIRF-3 expression plasmid repressed the activity of CIITA promoter PIII dose dependently up to 80% compared to empty vector (Fig. 3C). A similar reduction of PIII activity by vIRF-3 was observed upon cotransfection of vIRF-3 into lymphoid Jurkat T cells, which exhibit a higher basal activity of the PIII promoter (data not shown). In contrast to PIII, the basal activity of the IFN-␥-inducible PIV in HEK293T cells was not affected by cotransfection of vIRF-3 (Fig. 3D, left columns). However, induction of the CIITA promoter PIV by IFN-␥was completely reverted by expression of vIRF-3 (Fig. 3D, right columns). In summary, these observations demon-FIG. 3. vIRF-3 regulation of MHC II is mediated by regulation of
CIITA. (A) Upregulation of CIITA upon vIRF-3 knockdown. BC-3 cells were either mock transfected or transfected with a nonsense siRNA (siN) or an siRNA targeted at vIRF-3 (si vIRF-3) and main-tained in culture for 2 days. Cells were harvested, and total RNA was isolated. Expression of CIITA mRNA from promoters PI, PIII, and PIV was analyzed by semiquantitative RT-PCR (CIITA PI, 40 cycles; CIITA PIII/PIV, 30 cycles;-actin, 25 cycles). Amplification of-actin was used to normalize CIITA expression. One representative gel out of three independent experiments is shown. (B) Downregulation of CIITA upon vIRF-3 overexpression. BJAB cells were transfected by Amaxa electroporation with either empty vector, vIRF-3 expression plasmid, or an expression plasmid for KSHV glycoprotein K8.1. After maintenance in culture for 2 days, total RNA was isolated. Expression of CIITA mRNA from the cell-type-specific promoters PI, PIII, and PIV was analyzed by semiquantitative RT-PCR (CIITA PI, 40 cycles; CIITA PIII/PIV, 30 cycles;-actin, 25 cycles). Amplification of-actin was used to normalize CIITA transcripts. One representative gel out of three independent experiments is shown. (C) Inhibition of CIITA promoter PIII by vIRF-3. HEK293T cells were transfected with PIII luciferase reporter construct and either empty vector or increasing amounts of vIRF-3 expression construct. Luciferase assays were per-formed on day 2 after transfection. Values are given relative to the activity measured in cells transfected with reporter construct and empty expression plasmid only (ø). Bars represent arithmetic means
and standard deviations from three independent experiments. Aster-isks indicate a significant reduction (P⬍0.05,ttest). (D) Inhibition of CIITA promoter PIV by vIRF-3. HEK293T cells were transfected with PIV luciferase reporter construct and either empty vector or vIRF-3 expression construct. Cells were either left unstimulated (left) or stim-ulated 1 day after transfection with 1,000 U/ml IFN-␥for 24 h (right). Luciferase assays were performed on day 2 after transfection. Values are given relative to the luciferase activity measured in cells transfected with reporter construct and empty expression plasmid only (ø). Bars represent arithmetic means and standard deviations from three inde-pendent experiments. The asterisk indicates a significant reduction (P⬍0.05,ttest). n.s., not significant.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.43.281.66.495.2]strate that the inhibition of MHC II expression by vIRF-3 is mediated by the repression of CIITA expression.
vIRF-3 downregulates IFN-␥.IFN-␥is a major player in the regulation of the CIITA expression. First, it is the main regu-lator of CIITA promoter PIV. There are also reports that the lymphocyte-specific promoter PIII is induced by IFN-␥(41). As we showed above that the transcription of CIITA in PEL cells is inhibited by vIRF-3, we hypothesized that vIRF-3 in-hibits the production of IFN-␥. As shown in Fig. 4A, all PEL cell lines examined expressed IFN-␥, albeit to a variable extent. BC-3 cells showed the highest IFN-␥transcript level (Fig. 4A). In contrast to PEL cells, IFN-␥transcripts were not detectable in the KSHV-negative B cell lines Akata and BJAB (Fig. 4A). Upon knockdown of vIRF-3 expression by si467 (si vIRF-3), IFN-␥transcription was markedly induced in BC-3 cells com-pared to mock and siN controls (Fig. 4B). To further verify that vIRF-3 regulates IFN-␥, the concentration of IFN-␥ in the supernatant of PEL cell lines BC-3 (Fig. 4C) and JSC-1 (Fig. 4E) or the KSHV-negative B cell line BJAB (Fig. 4G) was determined by ELISA. The concentration of human IL-6 was determined as a control (Fig. 4D and F). Cells were either left untreated or transfected with a nonsense siRNA or with an siRNA targeted at vIRF-3 (sivIRF-3). Knockdown of vIRF-3 expression resulted in an up to 4-fold increase of IFN-␥in the cell culture supernatant of both BC-3 (Fig. 4C) and JSC-1 (Fig. 4E) cells. Transfection with the siRNA targeted at vIRF-3 did not induce IFN-␥secretion in KSHV-negative BJAB cells (Fig. 4G). This shows that the increased IFN-␥production observed in KSHV-positive PEL cells upon transfection of sivIRF-3 was not caused by an unspecific or off-target mechanism. The con-centration of IL-6 was not altered by knocking down vIRF-3 expression in PEL cells (Fig. 4D and F). Similar data were obtained with a second siRNA targeted at vIRF-3 (si976; data not shown).
Next, we used vIRF-3 overexpression in order to comple-ment the results obtained by vIRF-3 knockdown. A 3.6-kb IFN-␥promoter construct driving luciferase expression served as reporter. As unstimulated HEK293T cells do not produce IFN-␥, transcription from the promoter construct was induced by stimulation with phorbol myristate acetate (PMA)-ionomy-cin for 20 h in HEK293T cells. vIRF-3 was able to downregu-late the activity of the IFN-␥promoter dose dependently (Fig. 5A). To show that the vIRF-3-mediated repression of IFN-␥ promoter activity is not dependent on PMA-ionomycin stimu-lation, we performed the same experiment in Jurkat T cells. These cells secrete IFN-␥ and exhibit activity of the IFN-␥ promoter construct without stimulation. In Jurkat T cells, both the basal and the PMA-ionomycin-stimulated IFN-␥promoter activity could be downregulated dose dependently by vIRF-3 (Fig. 5B). We therefore conclude that vIRF-3 represses the IFN-␥promoter.
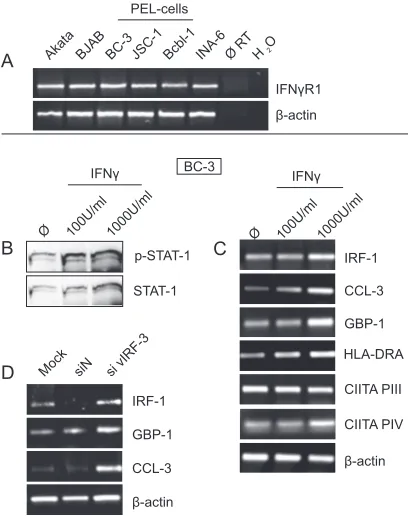
PEL cells respond to IFN-␥.It is a well-established notion that CIITA transcription is regulated by IFN-␥(reviewed in reference 26). However, our finding that vIRF-3 inhibits IFN-␥ expression at the level of transcription does not necessarily imply that the reduced expression of IFN-␥results in reduced CIITA (and thus MHC II) transcription. To test whether the secreted IFN-␥is functional in PEL cells, we first determined IFN-␥ receptor (IFN-␥R1) expression in different PEL cell lines by RT-PCR. IFN-␥R1 transcripts were present at
com-parable amounts in all PEL cell lines examined (Fig. 6A). Treatment of BC-3 cells with different amounts of recombinant IFN-␥induced the typical IFN-␥signaling cascade as indicated by phosphorylation of STAT-1 (Fig. 6B). Consequently, IFN-␥ signaling resulted in induction of typical IFN-␥target genes FIG. 4. Effects of vIRF-3 knockdown on cytokine secretion. (A) IFN-␥expression levels in different PEL cell lines. Total RNA was isolated from the indicated cell lines. Expression of IFN-␥mRNA was analyzed by semiquantitative RT-PCR (IFN-␥, 35 PCR cycles;-actin, 25 PCR cycles). Amplification of-actin was used to normalize IFN-␥ expression. (B) Upregulation of IFN-␥upon vIRF-3 knockdown. BC-3 cells were either mock transfected or transfected with a nonsense siRNA (siN) or an siRNA targeted at vIRF-3 (si vIRF-3) and main-tained in culture for 2 days. Cells were harvested, and total RNA was isolated. Expression of IFN-␥mRNA was analyzed by semiquantita-tive RT-PCR (IFN-␥, 30 PCR cycles;-actin, 25 PCR cycles). Ampli-fication of-actin was used to normalize IFN-␥expression. One rep-resentative gel out of three independent experiments is shown. (C to G) Influence of vIRF-3 knockdown in B cells on cytokine secretion. positive BC-3 cells (C and D), JSC-1 cells (E and F), or KSHV-negative BJAB cells (G) (106of each line) were either mock
trans-fected (light gray lines and diamonds) or transtrans-fected with a nonsense siRNA (dark gray lines and squares) or an siRNA targeted at vIRF-3 (black lines and triangles) and maintained in culture for 3 days. Su-pernatants were collected 24 h, 48 h, and 72 h after transfection. Amounts of secreted IFN-␥(C, E, and G) or IL-6 (D and F) were measured by ELISA. Arithmetical mean values from 3 independent experiments are shown. Error bars indicate standard deviations. The data shown in panels C and D and panels E and F are from the same set of experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
like IRF-1, GBP-1, CCL-3, and HLA-DRA in BC-3 PEL cells (Fig. 6C). Increased expression of HLA-DRA in BC-3 cells upon IFN-␥treatment could be traced back to an increase of CIITA transcription from promoter PIV, whereas the activity of CIITA PIII remained unchanged (Fig. 6C).
We further verified that PEL cells respond to the elevated secretion of IFN-␥observed upon knockdown of vIRF-3. For this purpose, expression of IFN-␥target genes IRF-1, GBP-1, and CCL-3 was analyzed upon vIRF-3 knockdown in BC-3 cells by semiquantitative RT-PCR. As shown in Fig. 6D, tran-script levels of these IFN-␥targets were strongly increased upon vIRF-3 knockdown compared to mock and siN controls (Fig. 6D). In summary, the observed induction of the IFN-␥ signaling cascade as demonstrated not only by STAT-1 phos-phorylation but also by upregulation of typical IFN-␥target genes confirmed that PEL cells respond to IFN-␥.
DISCUSSION
Expression of MHC II molecules on the surface of PEL cells is low compared to that on B cell lines established from KSHV-negative lymphomas (Fig. 1A). In this study, we report that vIRF-3 downmodulates the expression of MHC II molecules on the cell surface of latently KSHV-infected PEL cells. In
[image:6.585.318.522.69.327.2]addition, we show that MHC II suppression by vIRF-3 was carried out on the transcriptional level: vIRF-3 expression re-sulted in reduced transcription of the main MHC II regulator CIITA from both the IFN-␥-inducible promoter PIV and the B cell-specific promoter PIII. For CIITA promoter PIV, the lat-ter could be traced back to the reduction of IFN-␥expression in PEL cells by vIRF-3. However, transcription from the CIITA promoter PIII could not be induced by IFN-␥ treat-ment of PEL cells. Inhibition of IFN-␥expression by vIRF-3 was verified both by IFN-␥promoter assays and by analyzing the expression of common target genes upon knockdown of FIG. 5. vIRF-3 represses the IFN-␥ promoter. HEK293T cells
(A) or Jurkat T cells (B) were transfected with a 3.6-kb IFN-␥ pro-moter luciferase reporter construct and increasing amounts of vIRF-3. DNA amounts were adjusted by cotransfection of empty vector. Where indicated, IFN-␥promoter activity was stimulated 24 h after transfec-tion with PMA-ionomycin for 20 h (light gray bars in panel A and right half of panel B). Luciferase assays were performed on day 2 after transfection. Bars represent arithmetic means and standard errors of relative luciferase activities (transfections with reporter construct and empty vector ⫽ 1) from three independent experiments. Asterisks indicate statistically significant reductions (P⬍0.05,ttest).
FIG. 6. PEL cells respond to IFN-␥. (A) Expression of IFN-␥R1 in PEL cell lines. Total RNA was isolated from the indicated cell lines. Expression of IFN-␥R1 and-actin mRNAs was analyzed by semi-quantitative RT-PCR (IFN-␥R1, 30 PCR cycles;-actin, 25 PCR cy-cles). (B) IFN-␥ induces phosphorylation of STAT-1 in PEL cells. BC-3 cells were either left untreated or treated for 24 h with indicated amounts of recombinant human IFN-␥. Cells were harvested, and lysates were separated by SDS-PAGE and analyzed by Western blot-ting with an antibody to phospho-STAT-1 (Tyr701, upper row). Blots were stripped and reprobed with anti-STAT-1 monoclonal antibody (lower row). (C) Upregulation of IFN-␥target genes in PEL cells in response to IFN-␥. BC-3 cells were either left untreated or treated for 24 h with indicated amounts of recombinant human IFN-␥. Cells were harvested, and total RNA was isolated. Transcript levels of the IFN-␥ target genes IRF-1, GBP-1, CCL-3, and HLA-DRA as well as CIITA mRNAs originating from promoter PIII or PIV were analyzed by semiquantitative RT-PCR (IRF-1, 25 PCR cycles; GBP-1, 25 PCR cycles; CCL-3, HLA-DRA, CIITA PIII, and CIITA PIV, 30 PCR cycles;-actin, 25 PCR cycles). Amplification of-actin was used as a control. (D) Upregulation of IFN-␥target genes upon vIRF-3 knock-down. BC-3 cells were either mock transfected or transfected with a nonsense siRNA (siN) or an siRNA targeted at vIRF-3 (si vIRF-3) and maintained in culture for 2 days. Cells were harvested, and total RNA was isolated. mRNA expression levels of the IFN-␥target genes IRF-1, GBP-1, and CCL-3 were analyzed by semiquantitative RT-PCR as described above. Amplification of-actin was used as a control. One representative gel out of three independent experiments is shown (all panels).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.73.254.70.332.2]vIRF-3. Thus, we showed that vIRF-3 suppresses the MHC II synthesis in PEL cells by both IFN-␥-dependent (PIV) and -independent (PIII) pathways.
It has been shown before that vIRF-3 is involved in the regulation of the class I interferon response in many ways. It counteracts the interferon class I response by interfering with cellular IRF-3 (30), IRF-7 (21), and IRF-5 (54). For example, it has been shown that vIRF-3 inhibits IFN class I transcription by acting as a dominant-negative inhibitor of cellular IRF-5 and preventing IRF-5 binding to IFN class I promoters (5, 54). It is also able to block apoptosis by double-stranded RNA (dsRNA)-activated protein kinase R (15). It is therefore seen as a defense protein against class I interferons. Although there is a limited cross talk between signal transduction pathways from interferon class I and II receptors, to our knowledge there are no reports that either vIRF-3 or any of the cellular IRFs inhibited by vIRF-3 is involved in the regulation of IFN-␥ or CIITA promoter PIII. However, it is likely that vIRF-3 inhibits IFN-␥ and CIITA transcription by a similar mecha-nism, i.e., by counteracting cellular transcription factors.
Escape from antigen presentation is a central strategy of evasion from the adaptive immune response by KSHV (re-viewed in reference 3). It is a well-established finding that antigen presentation in the context of MHC class I is inhibited by KSHV proteins vIRF-1 (23) and K3/K5 (19) by either down-regulation or degradation of MHC I molecules, respectively. Inhibition of MHC II presentation by viruses was observed or at least reported far less frequently than was MHC I inhibition. This is not surprising, as intracellular pathogens like viruses are supposed to be presented mainly via MHC I. However, it became apparent in recent years that endogenous peptides can also be presented on MHC class II molecules to CD4⫹T cells via autophagy (reviewed in reference 22). Notably, the EBV nuclear antigen EBNA-1 is presented on MHC II molecules to CD4⫹T cells after autophagic processing (27, 38). CD4⫹T cells are usually thought to govern the adaptive immune sponse and provide help for maintenance of the cytotoxic re-sponse by CD8⫹T cells. However, more recent studies showed that CD4⫹ T cells can eliminate infected cells directly (re-viewed in reference 18). It has been shown for the closely related gammaherpesvirus murine herpesvirus 68 (MHV68) that virus-specific CD4⫹ T cells alone are able to eliminate MHV68-infected cells. Elimination of MHV68-infected cells occurred in the absence of antibodies or CD8⫹ T cells but relied on the presence of IFN-␥(11). Furthermore, CD4⫹T lymphocytes inhibited the outgrowth of MHV68-transformed B lymphocytes in an animal model (48). In addition, IFN-␥ -and granzyme B-secreting CD4⫹ T cells efficiently killed B lymphocytes presenting EBV glycoproteins and inhibited the generation of EBV-transformed B cell linesin vitro(2). This suggests that also KSHV proteins could be presented via MHC II. Consequently, KSHV would benefit from inhibition of the MHC II pathway in infected B lymphocytes.
Several other herpesviruses are known to affect MHC II presentation. Most of them do not act on the transcriptional level but rather by enhancing MHC II degradation or by avoid-ing MHC II surface presentation. For example, human cyto-megalovirus (HCMV) protein US2 (37, 53) leads to protea-somal degradation of MHC II and HCMV US3 prevents assembly and peptide loading of newly synthesized MHC II
molecules (17). EBV gp42 sterically blocks the interaction of T cell receptors with MHC class II/peptide complexes (46). How-ever, the EBV-encoded transcriptional regulator Zta inhibits MHC II expression on the transcriptional level by binding to and repression of the CIITA promoter PIII (28), comparable to the regulation of MHC II by vIRF-3 reported in this study. Like KSHV, several other herpesviruses modulate IFN-␥ signaling. In contrast to the mechanism identified here for KSHV, other herpesviruses usually interfere with the IFN-␥ signaling cascade and are not known to directly inhibit IFN-␥ production. For example, infection with EBV, varicella-zoster virus (VZV), or HCMV renders the cells insensitive for IFN-␥-mediated induction of MHC II (1, 34, 35). The spectrum of cells producing IFN-␥is essentially limited to the T-cell lin-eage, and most of the cells infected by these viruses do not produce IFN-␥. Thus, it is obviously more efficient for most herpesviruses to inhibit signal transduction from the IFN-␥ receptor rather than blocking IFN-␥ transcription. Similarly, the lytically expressed KSHV proteins K3 and K5 affect the IFN-␥response by downregulating the IFN-␥receptor (29). However, B cells produce IFN-␥under certain conditions, too. In the presence of antigen and Th-1 cytokines, naïve B cells differentiate into type 1 effector B cells (Be-1), which produce IFN-␥, IL-12, and tumor necrosis factor alpha (TNF-␣). This results in a positive feedback loop, further driving T cells toward the Th-1 phenotype and thus enhanced activation of cytotoxic T cells (32).
Inhibition of IFN-␥secretion is likely to be advantageous for KSHV in ways other than downregulation of MHC II expres-sion. IFN-␥is an important mediator of tumor immunosurveil-lance. Defects in IFN-␥signal transduction are often associ-ated with enhanced tumor growth (reviewed in reference 13). First, reduced levels of IFN-␥ result in downregulation of MHC I and II expression and thus reduced immunosurveil-lance. Second, IFN-␥also has antiproliferative and proapop-totic effects that directly inhibit tumor growth (reviewed in reference 50). Interestingly, antiproliferative effects of IFN-␥ are mediated by inhibition of c-myc (43) and activation of PKR (39). As vIRF-3 counteracts IFN-␥, this is in line with reports that show that vIRF-3 activates c-myc-dependent transcription (31) and inhibits PKR (15). IFN-␥ not only inhibits tumor growth but also triggers the immune system to recognize tu-mors by polarizing CD4⫹T cells toward a Th-1 phenotype, which is required for tumor rejection. It is therefore likely that inhibition of IFN-␥by vIRF-3 does not only result in immune escape due to reduced MHC II expression. It is also a mech-anism that contributes to the antiapoptotic and proprolifera-tive functions recently identified for vIRF-3 (55).
ACKNOWLEDGMENTS
This work was supported by the Akademie der Wissenschaften und der Literatur (Mainz), the European Community research project TargetHerpes, Deutsche Forschungsgemeinschaft graduate school 1071 “Viruses of the Immune System,” and the Interdisciplinary Cen-ter for Clinical Research (IZKF) at the University Hospital of the University of Erlangen-Nuremberg.
We thank Andrea Kress for helpful discussions.
REFERENCES
1.Abendroth, A., et al.2000. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J. Virol.74:1900–1907.
on November 7, 2019 by guest
http://jvi.asm.org/
2.Adhikary, D., et al.2006. Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J. Exp. Med.203:995–1006. 3.Areste´, C., and D. J. Blackbourn.2009. Modulation of the immune system by
Kaposi’s sarcoma-associated herpesvirus. Trends Microbiol.17:119–129. 4.Arvanitakis, L., et al.1996. Establishment and characterization of a primary
effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood88:2648–2654.
5.Bi, X., L. Yang, M. E. Mancl, and B. J. Barnes.6 December 2010. Modu-lation of interferon regulatory factor 5 activities by the Kaposi sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3 contrib-utes to immune evasion and lytic induction. J. Interferon Cytokine Res. [Epub ahead of print.] doi:10.1089/jir.2010.0084.
6.Birkmann, A., et al.2001. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J. Virol.
75:11583–11593.
7.Burger, R., et al.1998. Human herpesvirus type 8 interleukin-6 homologue is functionally active on human myeloma cells. Blood91:1858–1863. 8.Cannon, J. S., et al.2000. A new primary effusion lymphoma-derived cell line
yields a highly infectious Kaposi’s sarcoma herpesvirus-containing superna-tant. J. Virol.74:10187–10193.
9.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles.1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-re-lated body-cavity-based lymphomas. N. Engl. J. Med.332:1186–1191. 10.Chang, Y., et al.1994. Identification of herpesvirus-like DNA sequences in
AIDS-associated Kaposi’s sarcoma. Science266:1865–1869.
11.Christensen, J. P., R. D. Cardin, K. C. Branum, and P. C. Doherty.1999. CD4(⫹) T cell-mediated control of a gamma-herpesvirus in B cell-deficient mice is mediated by IFN-gamma. Proc. Natl. Acad. Sci. U. S. A.96:5135– 5140.
12.Cunningham, C., S. Barnard, D. J. Blackbourn, and A. J. Davison.2003. Transcription mapping of human herpesvirus 8 genes encoding viral inter-feron regulatory factors. J. Gen. Virol.84:1471–1483.
13.Dunn, G. P., L. J. Old, and R. D. Schreiber.2004. The three Es of cancer immunoediting. Annu. Rev. Immunol.22:329–360.
14.Edgar, R., M. Domrachev, and A. E. Lash.2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res.30:207–210.
15.Esteban, M., et al.2003. The latency protein LANA2 from Kaposi’s sarco-ma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol.84:1463–1470. 16.Gonsky, R., R. L. Deem, J. Bream, H. A. Young, and S. R. Targan.2004.
Enhancer role of STAT5 in CD2 activation of IFN-gamma gene expression. J. Immunol.173:6241–6247.
17.Hegde, N. R., et al.2002. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J. Virol.76:10929–10941.
18.Heller, K. N., C. Gurer, and C. Munz.2006. Virus-specific CD4⫹T cells: ready for direct attack. J. Exp. Med.203:805–808.
19.Ishido, S., C. Wang, B. S. Lee, G. B. Cohen, and J. U. Jung.2000. Down-regulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol.74:5300–5309. 20.Jenner, R. G., et al.2003. Kaposi’s sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA100:10399–10404.
21.Joo, C. H., et al.2007. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol.81:8282–8292.
22.Klein, L., C. Munz, and J. D. Lunemann.2010. Autophagy-mediated antigen processing in CD4(⫹) T cell tolerance and immunity. FEBS Lett.584:1405– 1410.
23.Lagos, D., et al.2007. Kaposi sarcoma herpesvirus-encoded vFLIP and vIRF1 regulate antigen presentation in lymphatic endothelial cells. Blood
109:1550–1558.
24.Lang, D., et al.2000. Generation of monoclonal antibodies directed against the immunogenic glycoprotein K8.1 of human herpesvirus 8. Hybridoma
19:287–295.
25.Lee, H. R., M. H. Kim, J. S. Lee, C. Liang, and J. U. Jung.2009. Viral interferon regulatory factors. J. Interferon Cytokine Res.29:621–628. 26.LeibundGut-Landmann, S., et al.2004. Mini-review: specificity and
expres-sion of CIITA, the master regulator of MHC class II genes. Eur. J. Immunol.
34:1513–1525.
27.Leung, C. S., T. A. Haigh, L. K. Mackay, A. B. Rickinson, and G. S. Taylor.
2010. Nuclear location of an endogenously expressed antigen, EBNA1, re-stricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. U. S. A.107:2165–2170.
28.Li, D., et al.2009. Down-regulation of MHC class II expression through inhibition of CIITA transcription by lytic transactivator Zta during Epstein-Barr virus reactivation. J. Immunol.182:1799–1809.
29.Li, Q., R. Means, S. Lang, and J. U. Jung.2007. Downregulation of gamma
interferon receptor 1 by Kaposi’s sarcoma-associated herpesvirus K3 and K5. J. Virol.81:2117–2127.
30.Lubyova, B., M. J. Kellum, A. J. Frisancho, and P. M. Pitha.2004. Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcrip-tional activity of cellular IRF-3 and IRF-7. J. Biol. Chem.279:7643–7654. 31.Lubyova, B., M. J. Kellum, J. A. Frisancho, and P. M. Pitha.2007.
Stimu-lation of c-Myc transcriptional activity by vIRF-3 of Kaposi sarcoma-associ-ated herpesvirus. J. Biol. Chem.282:31944–31953.
32.Lund, F. E.2008. Cytokine-producing B lymphocytes-key regulators of im-munity. Curr. Opin. Immunol.20:332–338.
33.Matolcsy, A., R. G. Nador, E. Cesarman, and D. M. Knowles.1998. Immu-noglobulin VH gene mutational analysis suggests that primary effusion lym-phomas derive from different stages of B cell maturation. Am. J. Pathol.
153:1609–1614.
34.Miller, D. M., et al.1998. Human cytomegalovirus inhibits major histocom-patibility complex class II expression by disruption of the Jak/Stat pathway. J. Exp. Med.187:675–683.
35.Morrison, T. E., A. Mauser, A. Wong, J. P. Ting, and S. C. Kenney.2001. Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity15:787–799.
36.Muhlethaler-Mottet, A., L. A. Otten, V. Steimle, and B. Mach.1997. Expres-sion of MHC class II molecules in different cellular and functional compart-ments is controlled by differential usage of multiple promoters of the trans-activator CIITA. EMBO J.16:2851–2860.
37.Odeberg, J., B. Plachter, L. Branden, and C. Soderberg-Naucler.2003. Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR alpha-chain. Blood101:4870– 4877.
38.Paludan, C., et al.2005. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science307:593–596.
39.Patterson, J. B., D. C. Thomis, S. L. Hans, and C. E. Samuel.1995. Mech-anism of interferon action: double-stranded RNA-specific adenosine deami-nase from human cells is inducible by alpha and gamma interferons. Virology
210:508–511.
40.Pfaffl, M. W.2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res.29:e45.
41.Piskurich, J. F., M. W. Linhoff, Y. Wang, and J. P. Ting.1999. Two distinct gamma interferon-inducible promoters of the major histocompatibility com-plex class II transactivator gene are differentially regulated by STAT1, in-terferon regulatory factor 1, and transforming growth factor beta. Mol. Cell. Biol.19:431–440.
42.Piskurich, J. F., Y. Wang, M. W. Linhoff, L. C. White, and J. P. Ting.1998. Identification of distinct regions of 5⬘flanking DNA that mediate constitu-tive, IFN-gamma, STAT1, and TGF-beta-regulated expression of the class II transactivator gene. J. Immunol.160:233–240.
43.Ramana, C. V., et al.2000. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J.19:263–272. 44.Reith, W., S. LeibundGut-Landmann, and J. M. Waldburger.2005. Regu-lation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol.5:793–806.
45.Renne, R., et al.1996. Lytic growth of Kaposi’s sarcoma-associated herpes-virus (human herpesherpes-virus 8) in culture. Nat. Med.2:342–346.
46.Ressing, M. E., et al.2003. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. U. S. A.100:11583–11588.
47.Rivas, C., A. E. Thlick, C. Parravicini, P. S. Moore, and Y. Chang.2001. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol.75:429–438.
48.Robertson, K. A., E. J. Usherwood, and A. A. Nash.2001. Regression of a murine gammaherpesvirus 68-positive b-cell lymphoma mediated by CD4 T lymphocytes. J. Virol.75:3480–3482.
49.Sander, G., et al.2008. Intracellular localization map of human herpesvirus 8 proteins. J. Virol.82:1908–1922.
50.Schroder, K., P. J. Hertzog, T. Ravasi, and D. A. Hume.2004. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol.
75:163–189.
51.Shin, Y. C., C. H. Joo, M. U. Gack, H. R. Lee, and J. U. Jung.2008. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hy-poxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res.68:1751–1759.
52.Soulier, J., et al.1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castlemans disease. Blood86:1276–1280. 53.Tomazin, R., et al.1999. Cytomegalovirus US2 destroys two components of
the MHC class II pathway, preventing recognition by CD4⫹T cells. Nat. Med.5:1039–1043.
54.Wies, E., et al.2009. The Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 inhibits cellular IRF-5. J. Biol. Chem.284:8525–8538.
55.Wies, E., et al.2008. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood111:
320–327.