JOURNAL OFVIROLOGY, Sept. 2007, p. 9778–9789 Vol. 81, No. 18 0022-538X/07/$08.00⫹0 doi:10.1128/JVI.00360-07
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Type I Interferon Inhibition and Dendritic Cell Activation during
Gammaherpesvirus Respiratory Infection
䌤
Janet L. Weslow-Schmidt,
1Nancy A. Jewell,
1Sara E. Mertz,
1J. Pedro Simas,
3Joan E. Durbin,
1,2and Emilio Flan
˜o
1,2*
Center for Vaccines and Immunity, Columbus Children’s Research Institute, Columbus Children’s Hospital, Columbus,
Ohio 432051; The Ohio State University College of Medicine, Columbus, Ohio2; and Instituto de Medicina Molecular,
Universidade de Lisboa, Lisboa, and Instituto Gulbenkian de Cieˆncia, Oeiras, Portugal3
Received 19 February 2007/Accepted 22 June 2007
The respiratory tract is a major mucosal site for microorganism entry into the body, and type I interferon (IFN) and dendritic cells constitute a first line of defense against viral infections. We have analyzed the interaction between a model DNA virus, plasmacytoid dendritic cells, and type I IFN during lung infection of mice. Our data show that murine gammaherpesvirus 68 (␥HV68) inhibits type I IFN secretion by dendritic cells and that plasmacytoid dendritic cells are necessary for conventional dendritic cell maturation in response to␥HV68. Following␥HV68 intranasal inoculation, the local and systemic IFN-␣/response is below detect-able levels, and plasmacytoid dendritic cells are activated and recruited into the lung with a tissue distribution that differs from that of conventional dendritic cells. Our results suggest that plasmacytoid dendritic cells and type I IFN have important but independent roles during the early response to a respiratory␥HV68 infection.
␥HV68 infection inhibits type I IFN production by dendritic cells and is a poor inducer of IFN-␣/in vivo, which may serve as an immune evasion strategy.
Respiratory viral infections are the leading cause of acute illnesses worldwide, and several members of the herpesvirus family are responsible for severe pneumonia in neonates and immunocompromised patients (52). Herpes simplex virus, cy-tomegalovirus, varicella-zoster virus, Epstein-Barr virus, hu-man herpesvirus 6, and Kaposi’s sarcoma-associated herpesvi-rus (KSHV) have all been associated with respiratory diseases (7, 10, 30, 33, 42, 46). Much of our understanding of immune responses to viral infection of the respiratory tract comes from experimental animal models. Murine gammaherpesvirus 68 (␥HV68) is structurally and biologically similar to the human gammaherpesviruses Epstein-Barr virus and KSHV (16, 49, 50), and it has become a useful in vivo model of herpesvirus infection. Intranasal infection of mice with␥HV68 causes an acute respiratory infection that is rapidly resolved and followed by the establishment of splenic latency mainly in the B-cell compartment (16, 34). Analogous to KSHV (35, 38, 41),
␥HV68 also infects dendritic cells, a process that may act as a mechanism of immune evasion (18, 20).
Plasmacytoid dendritic cells are professional type I inter-feron (IFN)-producing cells that quickly respond to most vi-ruses by secreting large amounts of type I IFNs (28). Type I IFN signaling is important for the control of acute ␥HV68 infection (17, 51). In addition, plasmacytoid dendritic cells secrete cytokines and interact with conventional dendritic cells and T cells (28) and are critical for the defense against paren-teral and mucosal infections (1, 13, 25, 29). Although plasma-cytoid dendritic cells have been detected in the lungs (12, 14) and have been shown to prevent the development of allergic
asthma, we have limited information regarding their role in the antiviral response of the respiratory tract. This is of special importance because the lung is the largest epithelial surface in the body and constitutes a major portal of entry for microor-ganisms (32). The regulation of immune responses in the res-piratory tract must be tightly controlled to elicit an adequate defense against invading agents while maintaining tolerance to innocuous antigens. Dendritic cells have a central role in main-taining homeostasis by discriminating pathogens from harm-less antigens and eliciting the right response to induce immu-nity or tolerance, respectively (44). Not surprisingly, many viruses have developed strategies for disrupting dendritic cell function (36, 37), and the immune system has developed sys-tems such as plasmacytoid dendritic cells to quickly detect and respond to viruses (28).
In this study, we have looked at the interplay between a model double-stranded DNA (dsDNA) virus, plasmacytoid dendritic cells, and type I IFNs during lung infection. The data show that plasmacytoid dendritic cells are necessary for the maturation of conventional dendritic cells and that ␥HV68 inhibits type I IFN secretion by dendritic cells. Following
␥HV68 infection, plasmacytoid dendritic cells are activated and recruited into the lung with a tissue distribution that differs from that of conventional dendritic cells. No local or systemic IFN-␣/activity was detected following intranasal␥HV68 in-stillation, and the production of IFN-␣mRNA was limited to scattered epithelial cells within the respiratory tract. Our re-sults indicate that plasmacytoid dendritic cells and type I IFN have important but independent roles during the early re-sponse to a respiratory DNA virus infection.
MATERIALS AND METHODS
Virus stocks.␥HV68 clone WUMS was propagated and titers were deter-mined on monolayers of NIH 3T3 fibroblasts. Respiratory syncytial virus (RSV) * Corresponding author. Mailing address: Columbus Children’s
Re-search Institute, 700 Children’s Drive WA4015, Columbus, OH 43205. Phone: (614) 722-2735. Fax: (614) 722-3680. E-mail: flanoe@ccri.net.
䌤Published ahead of print on 11 July 2007.
9778
on November 8, 2019 by guest
http://jvi.asm.org/
strain A2 was grown in HEp-2 cells. Newcastle disease virus (NDV) was grown in 10-day-old embryonated chicken eggs, and titers were determined by immu-nofluorescence. Influenza virus A/WSN/33 (H1N1) was grown in Madin-Darby bovine kidney cells, and titers were determined by immunofluorescence.
Animal procedures and virus infection.C57BL/6J mice were purchased from Taconic Farms or Harlan Sprague Dawley Inc. and housed under specific-pathogen-free conditions in biosafety level 2 containment. IFN-␣/ receptor-deficient (IFN-␣/R⫺/⫺) and control (129SvEv strain) mice were bred at the Columbus Children’s Research Institute (CCRI). The Institutional Animal Care and Use Committee at CCRI approved all studies described here. Mice were anesthetized with 2,2,2-tribromoethanol and inoculated with 103
PFU of␥HV68, 102PFU influenza virus A/WSN/33 (H1N1), or 5⫻105PFU NDV in Hanks
balanced salt solution.
Dendritic cell cultures.Dendritic cells were generated from bone marrow cultures in complete tissue culture medium supplemented with 20 ng/ml murine recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) or 100 ng/ml human recombinant Flt3-L (Peprotech). On days 6 to 8, the dendritic cells were infected with 1⫻108to 3.3⫻108PFU of␥HV68,
RSV, or NDV and stimulated with 10g/ml lipopolysaccharide (LPS) (Sigma) or 2g/ml CpG ODN1826 (InvivoGen) if needed.
Fluorescence-activated cell sorter (FACS) analysis.Single-cell suspensions were obtained from tissues after collagenase D (5 mg/ml; Roche) treatment for 45 min. Cells were next incubated with 5 mM phosphate-buffered saline (PBS)– EDTA for 10 min at room temperature to disrupt multicellular complexes. The cells were Fc blocked and stained with combinations of the following antibodies: CD11c, CD11b, B220, CD8a, CD19, NK1.1, CD3, CD80, CD86, Kb, CD54, I-A, and CD40. Samples were washed and resuspended in 1% paraformaldehyde diluted in PBS before analysis. Flow cytometry data were acquired on a FACSCalibur or LSR (Becton-Dickinson) apparatus and analyzed using FlowJo software (TreeStar, Inc.).
Plaque assay.To determine the titer of infectious virus, lungs were stored frozen and mechanically homogenized. The lytic virus concentration of the lung homogenates or of dendritic cell culture supernatants was determined in a standard plaque assay on NIH 3T3 fibroblasts.
IFN-␣/bioassay.An IFN-␣/bioassay was performed as described previ-ously (31). Briefly, supernatants were acid treated to inactivate any input virus as well as other cytokines. Samples were then neutralized, and twofold dilutions of each sample were added to murine fibroblast monolayers. The next day, 1.25⫻
105
PFU of vesicular stomatitis virus (VSV) were added to each well. Controls included untreated monolayers plus and minus VSV infection and IFN-␣/
standards. After 2 days of incubation, wells were fixed and stained. IFN-␣/ concentrations were determined by a comparison of protection from VSV-in-duced cell killing with that seen with known amounts of IFN-␣/.
Immunohistochemistry.Frozen tissue sections were fixed in cold acetone for 10 min. Endogenous peroxidase was neutralized using PBS–0.3% H2O2–0.1%
sodium azide. The sections were stained with CD11c (eBioscience), anti-mPDCA-1 (Miltenyi), or anti-M3 (23) antiserum followed by peroxidase-conju-gated anti-immunoglobulin G antibodies (Jackson ImmunoResearch), and staining was visualized with 3-amino-9-ethylcarbazole. The sections were counterstained with hematoxylin and viewed on an Axioscop 2plus apparatus. Images were captured using a Axiocam HRc digital camera with Axiocam software.
In situ hybridization.Tissues were harvested on days 2, 3, and 7 after infection with influenza virus or␥HV68 or mock infection. To detect type I IFN transcripts in the lung and mediastinal lymph node (MLN) sections, we synthesized digoxi-genin-labeled riboprobes of murine IFN-␣4 and IFN-genes using a digoxigenin (DIG) RNA labeling kit (Roche) according to the manufacturer’s instructions. After deparaffinization and prehybridization of the tissue sections, DIG-labeled riboprobes at 60 ng/sample were diluted into hybridization buffer and incubated with the sections overnight at 42°C. Next, the sections were stained with anti-DIG alkaline phosphatase (Roche), and the signal was detected with nitroblue tetrazolium-BCIP (5-bromo-4-chloro-3-indolylphosphate). The sections were washed in water and counterstained with nuclear fast green. Adjacent serial tissue sections were stained with hematoxylin and eosin.
RESULTS
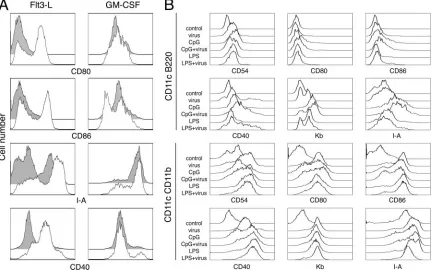
Plasmacytoid dendritic cells are necessary for in vitro acti-vation of conventional dendritic cells in response to␥HV68.
Previous studies of the interaction between␥HV68 and den-dritic cells have shown that␥HV68 infection of bone
marrow-derived dendritic cells cultured with GM-CSF does not induce dendritic cell maturation, although ␥HV68 does not prevent the activation of infected dendritic cells by other stimuli (19). Despite the impact of␥HV68 on dendritic cell function, in-fected mice eventually mount an immune response that con-trols infectious␥HV68 but never clears latent infection (16). It is unclear whether plasmacytoid dendritic cells or other den-dritic cells mediate the recognition of ␥HV68 and how the immune response to this virus is initiated at the site of infec-tion. It is thus possible that cells other than conventional den-dritic cells first detect the presence of ␥HV68 infection and initiate the adaptive immune response. Recognition of dsDNA viruses is mediated by Toll-like receptor 9 (TLR-9) and plas-macytoid dendritic cells (25, 26). However, in the mouse, both conventional and plasmacytoid dendritic cells can be activated in vivo by TLR-4, TLR-7, or TLR-9, but they differ in their requirements for type I IFNs for activation and migration (2). To initially characterize the response of plasmacytoid den-dritic cells to␥HV68, we generated bone marrow-derived den-dritic cells in the presence of GM-CSF or Flt3-L, infected the cultures with ␥HV68, and monitored cell activation by cell surface analysis of the expression of several costimulatory and major histocompatibility complex molecules. As previously re-ported (19),␥HV68 infection of dendritic cells grown in GM-CSF did not up-regulate the surface expression of CD80, CD86, CD40, or I-A molecules compared with mock-infected controls (Fig. 1A). However, the data show that␥HV68 infec-tion of dendritic cells generated in the presence of Flt3-L induced robust cell surface up-regulation of all the activation markers analyzed. Dendritic cells generated in the presence of GM-CSF constitute a homogeneous population with 95% CD11c⫹ CD11b⫹ conventional dendritic cells (5; data not shown). Dendritic cells generated with Flt3-L are heteroge-neous and contain a mixture of 20 to 30% CD11c⫹ B220⫹ plasmacytoid dendritic cells and 70 to 80% CD11c⫹CD11b⫹ conventional dendritic cells (5; data not shown). Next, we ques-tioned whether the activation of Flt3-L-derived dendritic cells by ␥HV68 was homogeneous or whether conventional and plasmacytoid dendritic cell subpopulations had distinct re-sponses to the virus. We analyzed the activation status of each subpopulation of dendritic cells in Flt3-L cultures under dif-ferent stimulatory conditions (Fig. 1B). ␥HV68 induced the up-regulation of surface molecules on plasmacytoid dendritic cells to the same extent as LPS, a TLR-4 agonist. In addition, stimulation with the TLR-9 agonist CpG induced a more ro-bust response by plasmacytoid dendritic cells, and␥HV68 in-fection did not prevent the changes induced by LPS or CpG. Analysis of the conventional dendritic cell subset showed equivalent up-regulation of CD54, CD80, CD86. CD40, Kb, and I-A in response to␥HV68, LPS, or CpG. Taken together, our results indicate that plasmacytoid dendritic cells are nec-essary for conventional dendritic cell activation in response to
␥HV68 infection.
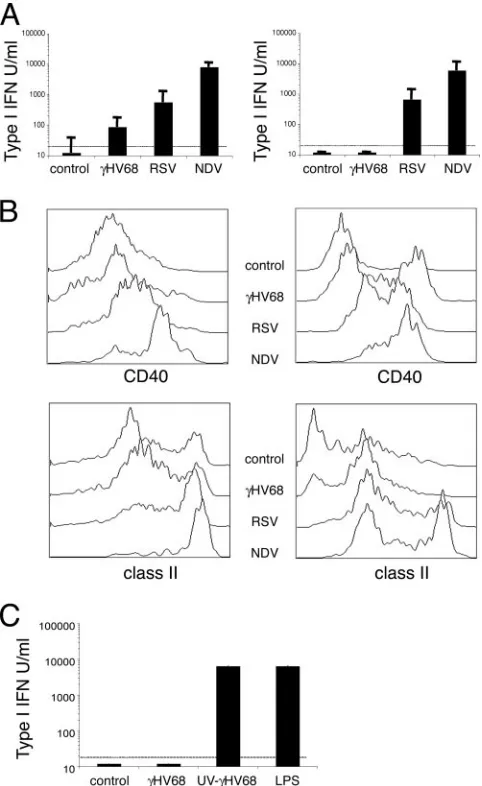
␥HV68 inhibits type I IFN production by dendritic cells.To further investigate the consequences of the interaction of
␥HV68 with dendritic cells for the induction of immunity, we compared dendritic cell activation and type I IFN production during␥HV68 infection with that of two model viruses, RSV and NDV. RSV is a poorly immunogenic virus with reinfec-tions occurring throughout life and a model of inhibition of
on November 8, 2019 by guest
http://jvi.asm.org/
type I IFNs (39, 43). NDV is a potent inducer of both type I IFN production and dendritic cell maturation (6, 22). First, we analyzed levels of IFN-␣/bioactivity in response to viral stim-ulation in dendritic cell cultures. The data reported in Fig. 2A show that NDV induced strong type I IFN production with a response 10-fold greater than that of RSV, which correlates with previous observations (31). ␥HV68 did not induce any significant amounts of type I IFN bioactivity in dendritic cell cultures. A similar pattern of type I IFN bioactivity for all three viruses was obtained using dendritic cells cultured with GM-CSF or with Flt3-L. Second, we analyzed dendritic cell activa-tion after␥HV68, RSV, or NDV infection. As shown in Fig. 2B (left column), both RSV and NDV induced robust activation of GM-CSF dendritic cells, as measured by the up-regulation of surface expression of CD40 and I-A molecules. As expected,
␥HV68 did not induce the up-regulation of the activation markers analyzed. All three viruses induced the activation of Flt3-L-derived dendritic cells although to a different extent (Fig. 2B, right column). Taken together, these results indicate that ␥HV68 is a poor inducer of type I IFN production by dendritic cells. In addition, the data suggest that the lack of IFN-␣/bioactivity is independent of the maturation state of the dendritic cells. To test whether the lack of IFN-␣/ bioac-tivity in culture supernatants is due to an active viral process that requires␥HV68 replication or is due to the poor immu-nogenicity of␥HV68 particles, we used UV-inactivated␥HV68
to stimulate dendritic cell cultures. The data show that UV-inactivated␥HV68 induced 1,000-fold more IFN-␣/synthesis by dendritic cells than did live␥HV68 (Fig. 2C). These data indicate that the inhibition of type I IFN production by den-dritic cells is an active process that requires␥HV68 replication.
Anatomical distribution of dendritic cell subsets within the lung.In vivo dendritic cell activation by microbial products and viruses has been shown to induce dendritic cell redistribution in the spleen (2, 9). However, we have limited information regarding the distribution of different subsets of dendritic cells in the respiratory tract. Until recently, the existence of plas-macytoid dendritic cells in the lung and their immunoregula-tory role in response to inhaled antigens were unknown (12). We analyzed the distribution of conventional dendritic cells and plasmacytoid dendritic cells within the lung and the spleen at early time points after intranasal infection with ␥HV68. Because of the low level of expression of CD11c on plasmacy-toid dendritic cells in vivo, immunohistochemical staining dis-tinguishes between plasmacytoid dendritic cells and conven-tional dendritic cells (2). In naı¨ve control mice (Fig. 3A), numerous CD11c⫹ cells were found interdigitating between respiratory epithelial cells and within the submucosa of con-ducting airways. In addition, scattered CD11c⫹ cells were present in the alveolar spaces. After␥HV68 infection (Fig. 3B and C), CD11c⫹cells were more numerous, forming a contin-uous layer beneath the bronchiolar epithelium and concentrat-FIG. 1. Dendritic cell activation in response to␥HV68 infection. (A) Bone marrow-derived dendritic cells grown in the presence of Flt3-L (left column) or GM-CSF (right column) were infected (empty histograms) or not infected (gray histograms) with␥HV68 as described in Materials and Methods. Forty hours later, the cells were surface stained with antibodies against CD11c and the indicated activation markers to analyze their fluorescence intensity on a flow cytometer. (B) Bone marrow-derived dendritic cells grown in the presence of Flt3-L were infected or not infected with␥HV68 and stimulated with CpG-ODN or LPS as described in Materials and Methods. Forty-eight hours after treatment, the cells were harvested and stained with antibodies against the indicated cell surface markers to analyze their fluorescence intensity. Dendritic cells were previously gated as plasmacytoid dendritic cells (CD11c⫹B220⫹) or conventional dendritic cells (CD11c⫹CD11b⫹). The data are representative of three independent experiments. The data shown in A and B are from two independent experiments.
9780 WESLOW-SCHMIDT ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
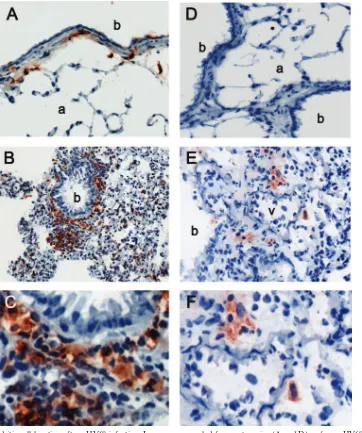
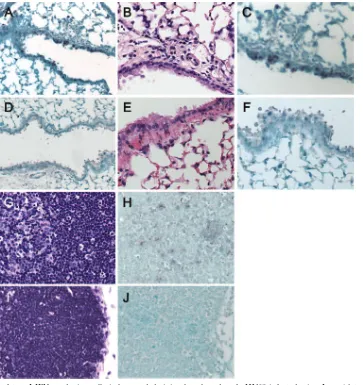
[image:3.585.77.511.71.341.2]ing in areas of inflammation. Cells positive for mPDCA-1, a marker specific for plasmacytoid dendritic cells, could not be visualized in uninfected control mice (Fig. 3D). However, after
␥HV68 infection, mPDCA-1⫹cells were found scattered or in small clusters in areas of inflammation of the lung parenchyma adjacent to blood vessels (Fig. 3D). These results show that conventional and plasmacytoid dendritic cells are distributed differently within the lung.
To determine whether plasmacytoid dendritic cells were re-cruited toward infection sites or to the lung in general, we
analyzed the distribution of the ␥HV68 antigen M3 in the lung of infected mice. As shown in Fig. 4, M3 expression is detected on cells of the airway epithelium, mononuclear cells in the airways, and individual cells in areas of inflam-mation of the lung parenchyma. Altogether, these data sug-gest that mPDCA-1⫹ plasmacytoid dendritic cells and
␥HV68 antigens localize in inflamed areas of the lung parenchyma of infected mice.
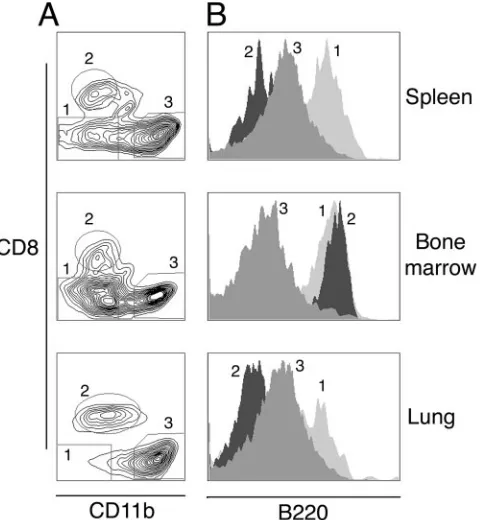
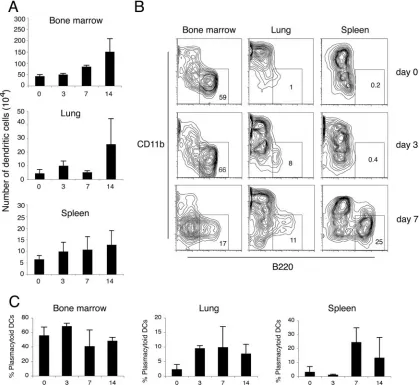
Plasmacytoid dendritic cell recruitment to the lung follow-ing␥HV68 infection.We have used enzymatic tissue digestion and flow cytometry to analyze the migration of dendritic cells in the respiratory tract in response to␥HV68 infection. We have compared this information with that for the spleen, where dendritic cell subsets are well described, and bone marrow, where dendritic cell precursors are generated. During the study, lineage-positive cells (CD19⫹, CD3⫹, and NK1.1⫹) and macrophages (CD11b⫹CD11cdim and/or large forward-scat-ter/side-scatter autofluorescent cells) were excluded from the analysis. Dendritic cells were gated using forward scatter/side scatter and low autofluorescence as plasmacytoid dendritic cells (CD11cdim B220⫹) and conventional dendritic cells (CD11chighCD8␣⫹or CD11chighCD11b⫹). Figure 5 shows a representative plot of the CD11c⫹population in lung, bone marrow, and spleen 5 days after infection and the B220 ex-pression profile of three different subsets of dendritic cells. The different subsets of respiratory tract dendritic cells presented the same B220 staining profile as their splenic counterparts.
We next did a temporal kinetic analysis of the numbers of dendritic cells in various tissues following ␥HV68 infection. The data in Fig. 6A show an increase in the absolute numbers of dendritic cells after␥HV68 infection in all the tissues ana-lyzed. This increase starts at day 3 after infection in lung and spleen, although at that time, viral replication is restricted to the respiratory tract (8, 45). By the time that lytic virus is cleared from the respiratory tract and viral latency peaks in the spleen at day 14 (8), the dendritic cell numbers have increased two- to threefold in lung, spleen, and bone marrow. To analyze the composition of the different subsets of dendritic cells and the frequency of plasmacytoid dendritic cells present in the lung at different times after ␥HV68 infection, we digested whole lungs and compared them with spleen and bone marrow. As observed in the representative plots shown in Fig. 6B, plasmacytoid dendritic cells migrate into the lung after intra-nasal instillation with␥HV68, and by days 3 to 7, a distinct population can be observed in the FACS contour plots. Simul-taneously, the frequency of plasmacytoid dendritic cells in the spleen also increased, with a distinct population becoming evident by day 7. An analysis of the frequency of plasmacytoid dendritic cells in lung showed a twofold increase in the per-centage of plasmacytoid dendritic cells from days 3 to 14 after infection and up to a sixfold increase in the spleen at day 7 after infection (Fig. 6C). Altogether, these data indicate that (i) plasmacytoid dendritic cells are differentially recruited into the lung after gammaherpesvirus infection and (ii) plasmacy-toid dendritic cells are also being recruited into the spleen, although viral replication is restricted to the lung.
[image:4.585.43.284.69.462.2]Kinetics of dendritic cell activation and type I IFN secretion in response to␥HV68 infection.Dendritic cells play a central role in the induction of adaptive and innate immune responses to respiratory tract infections. Although “myeloid” dendritic FIG. 2.␥HV68 inhibits type I IFN production by dendritic cells.
(A) Type I IFN induction by␥HV68, RSV, or NDV was measured in cell culture supernatants 40 h after infection using an IFN-␣/ bioas-say. The dendritic cells were grown in the presence of GM-CSF (left column) or Flt3-L (right column). (B) The relative abilities of␥HV68, RSV, or NDV to induce maturation of dendritic cells were tested using bone marrow-derived dendritic cells grown in the presence of GM-CSF (left column) or Flt3-L (right column). The cell cultures were infected as described in Materials and Methods, and 40 h later, the cells were surface stained with antibodies against CD11c, CD40, and I-A. The histograms shown have been previously gated as CD11c⫹ cells. (C) Type I IFN induction by UV-inactivated␥HV68 in dendritic cell cultures supplemented with GM-CSF. The data presented are the means and standard deviations of triplicate dendritic cell cultures.
on November 8, 2019 by guest
http://jvi.asm.org/
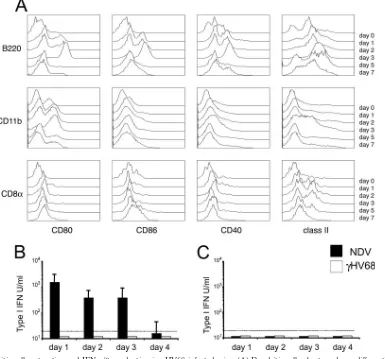
cells constitute the predominant population of pulmonary den-dritic cells in humans (47) and mice (11), other subsets also play essential roles in the response to inhaled antigens (12, 48). Our previous in vitro results suggest that plasmacytoid den-dritic cells are essential for host detection of␥HV68 infection. Thus, we questioned which subset(s) of respiratory dendritic cells was activated in response to infection in vivo. To investi-gate the kinetics of plasmacytoid and conventional dendritic cell activation in the lung, we monitored the surface expression of several costimulatory (CD80, CD86, and CD40) and major histocompatibility complex (I-A) molecules on B220⫹ plasma-cytoid dendritic cells, conventional CD11b⫹ dendritic cells, and conventional CD8␣⫹ dendritic cells. Plasmacytoid den-dritic cells up-regulated all activation markers analyzed by 24 h
after infection, and this state of activation was maintained until day 5 after infection (Fig. 7A). Conventional dendritic cells did not show any phenotypic changes until days 2 to 3 and then only partially up-regulated some of the markers analyzed: CD80 (CD11b⫹ dendritic cells) and class II (CD11b⫹ and CD8␣⫹dendritic cells). Thus, plasmacytoid dendritic cells are the first dendritic cell subpopulation at the site of infection that up-regulates activation markers in response to␥HV68. In ad-dition, plasmacytoid dendritic cells display a more robust state of activation than their conventional dendritic cell counter-parts in the lung. These data suggest that plasmacytoid den-dritic cells are the first lung denden-dritic cell population to detect
␥HV68 infection after intranasal inoculation.
[image:5.585.113.475.68.501.2]The recognition of dsDNA viruses by plasmacytoid dendritic FIG. 3. Lung dendritic cell location after␥HV68 infection. Lungs were sampled from naı¨ve mice (A and D) or from␥HV68-infected mice at 7 days postinfection (B and E). Serial sections were stained with anti-CD11c (left panels) or anti-mPDCA-1 (right panels) as described in Materials and Methods. Objective magnification,⫻20. C and F show a detail of the tissue area adjacent to the bronchi (b) or blood vessel (v) from B and E, respectively. One representative staining out of three mice per group is shown from three independent experiments. a, arteriole.
9782 WESLOW-SCHMIDT ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
cells triggers IFN-␣secretion (28), and the activation and mi-gration of plasmacytoid dendritic cells is thought to be depen-dent on type I IFN (2). To determine the role of IFN-␣/ during␥HV68 respiratory infection, we measured the amount of type I IFN present in bronchoalveolar lavage (BAL) sam-ples and sera of␥HV68-infected mice. Mice intranasally in-fected with NDV were used as positive controls. The data in Fig. 7B show that no IFN-␣/ activity could be detected in BAL samples or sera of ␥HV68-infected mice. In addition, although intranasal infection with NDV induces potent
IFN-␣/bioactivity in BAL samples, the amount of systemic type I IFNs in serum was below the level of detection of our bioassay.
Type I IFN production and dendritic cell activation during
␥HV68 infection of the lung.Type I IFNs are critical in the defense against viral infections by direct inhibition of viral replication in infected cells as well as through immunoregula-tory effects (4). IFN-␣/has been shown to be important for plasmacytoid dendritic cell activation and for conventional dendritic cell activation and migration (2, 22). Our previous results suggested that plasmacytoid dendritic cell activation may be critical for the induction of a conventional dendritic cell response to␥HV68 in mice and demonstrated that␥HV68 does not induce a detectable type I IFN response on dendritic cell cultures or in the lung. Next, we analyzed lung virus titers and dendritic cell activation and recruitment into the lung of
␥HV68-infected mice using IFN-␣/R⫺/⫺and wild-type mice (Fig. 8). As expected, IFN-␣/R⫺/⫺mice showed a 100-fold increase in infectious virus compared with normal mice. These data corroborate the important role of type I IFN signaling in the control of␥HV68 lytic infection (17, 51). In addition, and despite the increased virus production, IFN-␣/R⫺/⫺ mice showed a two- to threefold decrease in the recruitment of dendritic cells into the lung after␥HV68 infection (Fig. 8B). The frequency of activated dendritic cells as measured by class II expression was also lower in IFN-␣/R⫺/⫺mice (40%) than in wild-type mice (80%) after␥HV68 infection (Fig. 8C and D). These data suggest that type I IFN signaling enhances, but is not an absolute requirement, for dendritic cell recruitment into and activation in the lung after␥HV68 infection.
There is a discrepancy between the important role of
IFN-␣/in controlling␥HV68 respiratory infection and our inabil-ity to detect IFN-␣/bioactivity in BAL and serum samples of infected mice. To resolve this apparent contradiction, we used in situ hybridization to analyze the production of type I IFN in lung and draining lymph nodes and to determine which cells are responsible for its production in vivo. We analyzed infected tissues on days 2, 3, and 7 after intranasal viral inoculation using probes for IFN-␣4 and for IFN-. Mice infected with 102 PFU of influenza virus were used as positive controls. The data show that in influenza virus-infected mice, a strong IFN-␣/ signal is detected in the epithelium of the bronchi and in a few scattered cells in the airways (Fig. 9A to C). On the contrary,
[image:6.585.45.285.390.650.2]␥HV68-infected mice showed a very weak IFN-␣/-positive FIG. 4. Location of viral antigens in the lung of␥HV68-infected mice. Lungs were sampled from␥HV68-infected mice at 7 days postinfection, and sections were stained with anti-␥HV68 M3 antiserum. (A) Area of inflammation in the lung parenchyma. Magnification,⫻20. (B) Detail of the bronchi (b) from A. Magnification,⫻40.
FIG. 5. Identification of dendritic cell subsets in respiratory tract mPDCA-1⫹cells.(A) Expression of CD8␣and CD11b in previously gated CD11c⫹cells defines different dendritic cell subsets in the lung, bone marrow, and spleen of mice 5 days after␥HV68 infection: 1, plasmacytoid dendritic cells; 2, CD8 conventional dendritic cells; 3, conventional CD11b dendritic cells. (B) B220 cell surface expression on mouse dendritic cell subsets as previously gated in A. Data are representative of three independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
signal in lung sections exclusively in scattered bronchiolar epi-thelial cells (Fig. 9D to F). The analysis of tissue sections from MLN of influenza virus-infected mice revealed IFN-␣/ -posi-tive cells distributed throughout the lymph node (Fig. 9G and H). Mice infected with␥HV68 showed no IFN-␣/signal in the draining lymph node (Fig. 9I and J). Similar results were obtained using the␣ and  probes and at all different time points analyzed. Altogether, the data indicate that intranasal
␥HV68 infection does not induce a potent type I IFN response in either the lung or its draining lymph nodes.
DISCUSSION
In this paper, we show that␥HV68 infection inhibits type I IFN production in dendritic cells and is a poor inducer of IFN-␣/in vivo. In addition, our data show that dendritic cell
activation and recruitment to the lung after␥HV68 respiratory infection occur in spite of an IFN-␣/response that is below the limit of detection or in mice that lack IFN-␣/ signaling. Our data also indicate that plasmacytoid dendritic cells play an important role in the response to gammaherpesviruses by de-tecting␥HV68 and promoting the activation of conventional dendritic cells regardless of the weak IFN-␣/ response to infection.
Type I IFNs are essential for the defense against viral infec-tions (4), and␥HV68 is not an exception. IFN-␣/is important for the control of acute␥HV68 infection (17, 51) and also for the control of latency (3). Thus, it is not surprising that␥HV68 has evolved strategies to subvert type I IFN responses. Our data showing a lack of type I IFN production in response to
[image:7.585.83.502.70.455.2]␥HV68 infection by cultured dendritic cells generated in the presence of GM-CSF or Flt3-L suggest the existence of specific FIG. 6. Dendritic cell migration into the lung and spleen in response to␥HV68 infection. (A) Time course analysis of the absolute dendritic cell numbers in bone marrow, lung, and spleen after␥HV68 infection. The data presented are the means and standard deviations of three independent experiments, each containing three mice. (B) Plasmacytoid dendritic cells migrate into the lung and spleen after␥HV68 infection. Numbers indicate the percentages of cells inside the gate. One representative of three experiments is shown. (C) Time course analysis of the frequency of plasmacytoid dendritic cells (DC) in bone marrow, lung, and spleen after␥HV68 infection. The data presented are the means and standard deviations of three independent experiments, each containing three mice. In all the panels shown, dendritic cells were analyzed as CD11c⫹and lineage-negative (CD3, CD19, and NK1.1) nonautofluorescent cells.
9784 WESLOW-SCHMIDT ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
IFN-inhibitory mechanisms. The activation of Flt3-L-derived dendritic cells by infectious␥HV68 in the absence of type I IFN production also supports this conclusion. In addition, the ability of UV-inactivated, but not live,␥HV68 to induce
IFN-␣/production in dendritic cell cultures gives strong support to the hypothesis that␥HV68 inhibition of type I IFN production is an active process. Several mechanisms common to gamma-herpesviruses may account for the following findings: (i) M2 gene expression inhibits IFN-mediated transcriptional activa-tion by down-regulating STAT1 and STAT2 (27), and (ii) ORF45, a gene conserved among the gammaherpesviruses that is essential for␥HV68 replication (24), blocks interferon reg-ulatory factor 7 (IRF-7) phosphorylation and nuclear accumu-lation (54). Regardless of the mechanism inhibiting IFN-␣/ production, the data show that ␥HV68 inhibits type I IFN synthesis by cultured dendritic cells, a process that may help the virus to evade immune control in vivo. This idea is also supported by (i) in vivo data showing the lack of detectable IFN-␣/ bioactivity in BAL and serum samples of ␥
HV68-infected mice, (ii) in situ hybridization data showing weak IFN-␣and IFN-signals only from respiratory epithelial cells, and (ii) previous studies showing that␥HV68 infects dendritic cells (18–20).
[image:8.585.101.486.71.430.2]Our results indicate that plasmacytoid dendritic cells are the first dendritic cell population in the lung to show signs of activation in response to ␥HV68 intranasal inoculation and that␥HV68 induces the activation of conventional dendritic cells in vitro only when external “help” in the form of plasma-cytoid dendritic cells is present. These data correlate with a requirement for plasmacytoid dendritic cell “help” in conven-tional dendritic cell function during cutaneous herpes simplex virus infection (53). However, in a model of genital herpes simplex virus type 2 infection, plasmacytoid dendritic cells were not required to mediate Th1 immunity (29). Thus, it seems that the type of herpesvirus and/or the route of infection is likely to contribute to fundamental differences in the re-sponse. In addition, our data demonstrate that in response to a gammaherpesvirus, (i) plasmacytoid dendritic cell activation FIG. 7. Dendritic cell maturation and IFN-␣/production in␥HV68-infected mice. (A) Dendritic cell subsets undergo differential maturation in the lung in response to␥HV68 infection. Lung dendritic cells were analyzed at different time points after␥HV68 infection (days 0 to 7) for the level of cell surface expression of several activation markers (CD80, CD86, CD40, and I-A). Histograms are previously gated as CD11c⫹B220⫹ (plasmacytoid dendritic cells) (first row), CD11c⫹CD11b⫹(conventional dendritic cells) (second row), or CD11c⫹CD8a⫹(conventional dendritic cells) (third row). (B) Type I IFN bioactivity in BAL fluid of␥HV68- and NDV-infected mice at different time points after infection. (C) Type I IFN bioactivity in serum of␥HV68- and NDV-infected mice at different time points after infection. The data presented are the means and standard deviations for three to four mice.
on November 8, 2019 by guest
http://jvi.asm.org/
in culture is independent of IFN-␣/, (ii) plasmacytoid den-dritic cell recruitment and activation in the lung occur in the presence of a local and systemic IFN-␣/ response that is below the limits of detection by bioassay, and (iii) dendritic cell activation and recruitment into the lung, albeit reduced, still occur in IFN-␣/R⫺/⫺mice. Altogether, our findings suggest that type I IFN signaling is important but not an absolute requirement for dendritic cells to respond to␥HV68 infection. These findings contrast with the previously observed require-ment for IFN-␣/ signaling during the activation of conven-tional dendritic cells in culture (22) as well as the need for type I IFN signaling for plasmacytoid dendritic cell migration and activation in response to TLR-7 and TLR-9 ligands (2). It is possible that low levels of IFN-␣/below the limit of detection of our assays may contribute to the recruitment and activation of dendritic cells and that dendritic cell activation can also be induced by alternative mechanisms such as a CD40/CD40L interaction or membrane-bound interleukin-15. In addition, it is likely that the response to a DNA virus infection in vivo is more complex and may account for the differences observed between experimental systems.
The ability of plasmacytoid dendritic cells to recognize and respond to viruses is critical for providing a first line of defense at mucosal surfaces. The importance of plasmacy-toid dendritic cells during the immune response to␥HV68 is supported by our analysis of infected mice. The immunohis-tochemistry and flow cytometry data show that plasmacytoid dendritic cells are rapidly recruited to the lung after
intra-nasal instillation of␥HV68. This increase in the number of respiratory plasmacytoid dendritic cells is accompanied by the early induction of an activation phenotype. The tissue distribution and migration of plasmacytoid dendritic cells into the respiratory tract are less well defined in comparison to those of conventional dendritic cells. Our data are con-sistent with human and mouse data indicating that conven-tional dendritic cells in the lung are mainly CD11b⫹ or “myeloid” dendritic cells and that they form a contiguous subepithelial network (40, 47). Lung plasmacytoid dendritic cells have recently been described as BDCA2⫹/CD123⫹ cells in humans (14) and as Gr-1⫹/B220⫹ cells scattered throughout the lung interstitium in mice (12). Our analysis indicates that plasmacytoid dendritic cells constitute a small fraction (2%) of pulmonary dendritic cells and that they were not visualized during steady-state conditions by immu-nohistochemistry using mPDCA-1. However, after respira-tory ␥HV68 inoculation, plasmacytoid dendritic cells (CD11c⫹B220⫹) are recruited into the lung, constitute 10% of lung dendritic cells, and can be visualized as mPDCA-1⫹ cells in areas of inflammation. Thus, the distribution of plasmacytoid dendritic cells in the lung is different from that of conventional CD11c⫹dendritic cells. While conventional dendritic cells mostly form a dense network underneath the epithelium and in areas of inflammation, plasmacytoid den-dritic cells appear to migrate into the lung in response to an inflammatory stimulus.
It is becoming increasingly evident that plasmacytoid den-FIG. 8. Role of type I IFN signaling in virus control and dendritic cell recruitment and activation in the lung. (A) Infectious virus titers in the lung of 129SvEv or IFN-␣/R⫺/⫺mice were determined by plaque assay on day 5 after virus inoculation. (B) Frequency of CD11c⫹
dendritic cells in the lung of␥HV68-infected mice. (C) Representative histograms of the level of expression of class II molecules on CD11c⫹ cells in the lung. Numbers indicate the percentage of cells inside the gate. (D) Frequency of class II expression on CD11c⫹cells in the lung. FACS analysis was performed on naı¨ve and␥HV68-infected mice on day 7. The data presented are the means and standard deviations for three individual mice.
9786 WESLOW-SCHMIDT ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
dritic cells play an essential dual role in the initiation of anti-viral responses: as professional type I IFN producer cells and in regulating the function of conventional dendritic cells by IFN-independent pathways (9, 28). The data presented here support this hypothesis using the␥HV68 model of infection. Due to the key role of type I IFNs in antiviral defense, it is not surprising that many viruses have developed immune evasion mechanisms to block their production (21). Plasmacytoid den-dritic cells are resistant to many of these strategies either because they cannot be infected by the virus or because the virus targets IRF-3-dependent pathways, which are not essen-tial for type I IFN induction by TLR-7 and TLR-9 (15). The exceptions to this rule include some highly successful
patho-gens including measles virus, RSV, KSHV (21, 54), and
␥HV68.
ACKNOWLEDGMENTS
We thank the Morphology Core at CCRI for technical help. This work was supported by NIH grant AI-59603 and by CCRI.
REFERENCES
1.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O’Garra, C. Biron, F. Briere, and G. Trinchieri.2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol.2:1144–1150.
[image:10.585.111.474.67.452.2]2.Asselin-Paturel, C., G. Brizard, K. Chemin, A. Boonstra, A. O’Garra, A. Vicari, and G. Trinchieri.2005. Type I interferon dependence of plas-macytoid dendritic cell activation and migration. J. Exp. Med.201:1157– 1167.
FIG. 9. Absence of type I IFN-producing cells in lung and draining lymph nodes of␥HV68-infected mice. Lung (A to F) and MLN (G to J) tissue sections from influenza virus-infected (A to C, G, and H) or␥HV68-infected (D to F, I, and J) mice (days 2, 3, and 7) or control mice were labeled with riboprobes of murine IFN-␣4 and IFN- genes. (A) Influenza virus-infected lung, day 2, with an IFN- probe. Magnification,⫻20. A detail is shown is shown in B and C. (B) Influenza virus-infected lung, day 2, with staining with hematoxylin and eosin. Magnification,⫻40. (C) Influenza virus-infected lung, day 2, with an IFN-probe. Magnification,⫻40. (D)␥HV68-infected lung, day 2, with an IFN-probe. Magnification,⫻20. A detail is shown in E and F. (E)␥HV68-infected lung, day 2, with staining with hematoxylin and eosin. Magnification,⫻40. (F)␥HV68-infected lung, day 2, with IFN-probe. Magnification,⫻40. (G) Influenza virus-infected MLN, day 3, with staining with hematoxylin and eosin. Magnification,⫻40. (H) Influenza virus-infected MLN, day 3, with IFN-␣4 probe. Magnification,⫻40. (I)␥HV68-infected MLN, day 3, with staining with hematoxylin and eosin. Magnification,⫻40. (J)␥HV68-infected MLN, day 3, with IFN-␣4 probe. Magnification,⫻40.
on November 8, 2019 by guest
http://jvi.asm.org/
3.Barton, E. S., M. L. Lutzke, R. Rochford, and H. W. Virgin IV.2005. Alpha/beta interferons regulate murine gammaherpesvirus latent gene ex-pression and reactivation from latency. J. Virol.79:14149–14160. 4.Biron, C. A.2001. Interferons alpha and beta as immune regulators—a new
look. Immunity14:661–664.
5.Brawand, P., D. R. Fitzpatrick, B. W. Greenfield, K. Brasel, C. R. Maliszewski, and T. De Smedt.2002. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand supplemented bone marrow cultures are immature APCs. J. Immunol.169:6711–6719.
6.Brehm, G., and H. Kirchner.1986. Analysis of the interferons induced in mice in vivo and in macrophages in vitro by Newcastle disease virus and by polyinosinic-polycytidylic acid. J. Interf. Res.6:21–28.
7.Bruynseels, P., P. G. Jorens, H. E. Demey, H. Goossens, S. R. Pattyn, M. M. Elseviers, J. Weyler, L. L. Bossaert, Y. Mentens, and M. Ieven.2003. Herpes simplex virus in the respiratory tract of critical care patients: a prospective study. Lancet362:1536–1541.
8.Cardin, R. D., J. W. Brooks, S. R. Sarawar, and P. C. Doherty.1996. Progressive loss of CD8⫹T cell-mediated control of a gamma-herpesvirus in the absence of CD4⫹T cells. J. Exp. Med.184:863–871.
9.Colonna, M., G. Trinchieri, and Y. J. Liu.2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol.5:1219–1226.
10.Cool, C. D., P. R. Rai, M. E. Yeager, D. Hernandez-Saavedra, A. E. Serls, T. M. Bull, M. W. Geraci, K. K. Brown, J. M. Routes, R. M. Tuder, and N. F. Voelkel.2003. Expression of human herpesvirus 8 in primary pulmonary hypertension. N. Engl. J. Med.349:1113–1122.
11.de Heer, H. J., H. Hammad, M. Kool, and B. N. Lambrecht.2005. Dendritic cell subsets and immune regulation in the lung. Semin. Immunol.17:295– 303.
12.de Heer, H. J., H. Hammad, T. Soullie, D. Hijdra, N. Vos, M. A. Willart, H. C. Hoogsteden, and B. N. Lambrecht.2004. Essential role of lung plas-macytoid dendritic cells in preventing asthmatic reactions to harmless in-haled antigen. J. Exp. Med.200:89–98.
13.Delale, T., A. Paquin, C. Asselin-Paturel, M. Dalod, G. Brizard, E. E. Bates, P. Kastner, S. Chan, S. Akira, A. Vicari, C. A. Biron, G. Trinchieri, and F. Briere.2005. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J. Immunol.175:6723–6732.
14.Demedts, I. K., G. G. Brusselle, K. Y. Vermaelen, and R. A. Pauwels.2005. Identification and characterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol.32:177–184.
15.Diebold, S. S., M. Montoya, H. Unger, L. Alexopoulou, P. Roy, L. E. Haswell, A. Al-Shamkhani, R. Flavell, P. Borrow, and C. Reis e Sousa.2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon pro-ducers. Nature424:324–328.
16.Doherty, P. C., J. P. Christensen, G. T. Belz, P. G. Stevenson, and M. Y. Sangster.2001. Dissecting the host response to a gamma-herpesvirus. Philos. Trans. R. Soc. Lond. B Biol. Sci.356:581–593.
17.Dutia, B. M., D. J. Allen, H. Dyson, and A. A. Nash.1999. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology261:173–179.
18.Flano, E., S. M. Husain, J. T. Sample, D. L. Woodland, and M. A. Blackman.2000. Latent murine gamma-herpesvirus infection is estab-lished in activated B cells, dendritic cells, and macrophages. J. Immunol.
165:1074–1081.
19.Flano, E., B. Kayhan, D. L. Woodland, and M. A. Blackman.2005. Infection of dendritic cells by a gamma2-herpesvirus induces functional modulation. J. Immunol.175:3225–3234.
20.Flano, E., I. J. Kim, J. Moore, D. L. Woodland, and M. A. Blackman.2003. Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J. Immunol.170:3828– 3834.
21.Hengel, H., U. H. Koszinowski, and K. K. Conzelmann.2005. Viruses know it all: new insights into IFN networks. Trends Immunol.26:396–401. 22.Honda, K., S. Sakaguchi, C. Nakajima, A. Watanabe, H. Yanai, M. Matsumoto,
T. Ohteki, T. Kaisho, A. Takaoka, S. Akira, T. Seya, and T. Taniguchi.2003. Selective contribution of IFN-alpha/beta signaling to the maturation of den-dritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA100:10872–10877.
23.Jensen, K. K., S. C. Chen, R. W. Hipkin, M. T. Wiekowski, M. A. Schwarz, C. C. Chou, J. P. Simas, A. Alcami, and S. A. Lira.2003. Disruption of CCL21-induced chemotaxis in vitro and in vivo by M3, a chemokine-binding protein encoded by murine gammaherpesvirus 68. J. Virol.77:
624–630.
24.Jia, Q., V. Chernishof, E. Bortz, I. McHardy, T. T. Wu, H. I. Liao, and R. Sun.2005. Murine gammaherpesvirus 68 open reading frame 45 plays an essential role during the immediate-early phase of viral replication. J. Virol.
79:5129–5141.
25.Krug, A., A. R. French, W. Barchet, J. A. Fischer, A. Dzionek, J. T. Pingel, M. M. Orihuela, S. Akira, W. M. Yokoyama, and M. Colonna.2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity21:107– 119.
26.Krug, A., G. D. Luker, W. Barchet, D. A. Leib, S. Akira, and M. Colonna.
2004. Herpes simplex virus type 1 activates murine natural interferon-pro-ducing cells through Toll-like receptor 9. Blood103:1433–1437.
27.Liang, X., Y. C. Shin, R. E. Means, and J. U. Jung.2004. Inhibition of interferon-mediated antiviral activity by murine gammaherpesvirus 68 latency-associated M2 protein. J. Virol.78:12416–12427.
28.Liu, Y. J.2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol.23:275–306. 29.Lund, J. M., M. M. Linehan, N. Iijima, and A. Iwasaki.2006. Cutting edge: plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J. Immunol.177:7510–7514. 30.Mackie, P. L.2003. The classification of viruses infecting the respiratory
tract. Paediatr. Respir. Rev.4:84–90.
31.Martinez-Sobrido, L., N. Gitiban, A. Fernandez-Sesma, J. Cros, S. E. Mertz, N. A. Jewell, S. Hammond, E. Flano, R. K. Durbin, A. Garcia-Sastre, and J. E. Durbin.2006. Protection against respiratory syncytial virus by a recom-binant Newcastle disease virus vector. J. Virol.80:1130–1139.
32.Masten, B. J.2004. Initiation of lung immunity: the afferent limb and the role of dendritic cells. Semin. Respir. Crit. Care Med.25:11–20.
33.Merk, J., F. X. Schmid, M. Fleck, S. Schwarz, C. Lehane, S. Boehm, B. Salzberger, and D. E. Birnbaum.2005. Fatal pulmonary failure attributable to viral pneumonia with human herpes virus 6 (HHV6) in a young immu-nocompetent woman. J. Intensive Care Med.20:302–306.
34.Nash, A. A., B. M. Dutia, J. P. Stewart, and A. J. Davison.2001. Natural history of murine gammaherpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci.356:569–579.
35.Offermann, M. K.1999. Consideration of host-viral interactions in the patho-genesis of Kaposi’s sarcoma. J. Acquir. Immune Defic. Syndr.21(Suppl. 1):S58–S65.
36.Palucka, K., and J. Banchereau.2002. How dendritic cells and microbes interact to elicit or subvert protective immune responses. Curr. Opin. Im-munol.14:420–431.
37.Rescigno, M., and P. Borrow.2001. The host-pathogen interaction: new themes from dendritic cell biology. Cell106:267–270.
38.Rettig, M. B., H. J. Ma, R. A. Vescio, M. Pold, G. Schiller, D. Belson, A. Savage, C. Nishikubo, C. Wu, J. Fraser, J. W. Said, and J. R. Berenson.1997. Kaposi’s sarcoma-associated herpesvirus infection of bone marrow dendritic cells from multiple myeloma patients. Science276:1851–1854.
39.Schlender, J., V. Hornung, S. Finke, M. Gunthner-Biller, S. Marozin, K. Brzozka, S. Moghim, S. Endres, G. Hartmann, and K. K. Conzelmann.2005. Inhibition of Toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J. Virol.79:5507–5515.
40.Schon-Hegrad, M. A., J. Oliver, P. G. McMenamin, and P. G. Holt.1991. Studies on the density, distribution, and surface phenotype of intraepi-thelial class II major histocompatibility complex antigen (Ia)-bearing dendritic cells (DC) in the conducting airways. J. Exp. Med.173:1345– 1356.
41.Schulz, T. F.1998. Kaposi’s sarcoma-associated herpesvirus (human herpes-virus-8). J. Gen. Virol.79:1573–1591.
42.Simoons-Smit, A. M., E. M. Kraan, A. Beishuizen, R. J. S. van Schijndel, and C. M. Vandenbroucke-Grauls.2006. Herpes simplex virus type 1 and respi-ratory disease in critically-ill patients: real pathogen or innocent bystander? Clin. Microbiol. Infect.12:1050–1059.
43.Spann, K. M., K. C. Tran, B. Chi, R. L. Rabin, and P. L. Collins.2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epi-thelial cells and macrophages. J. Virol.78:4363–4369.
44.Steinman, R. M., D. Hawiger, and M. C. Nussenzweig.2003. Tolerogenic dendritic cells. Annu. Rev. Immunol.21:685–711.
45.Sunil-Chandra, N. P., S. Efstathiou, J. Arno, and A. A. Nash. 1992. Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J. Gen. Virol.73:2347–2356.
46.Verboon-Maciolek, M. A., T. G. Krediet, L. J. Gerards, A. Fleer, and T. M. van Loon.2005. Clinical and epidemiologic characteristics of viral infections in a neonatal intensive care unit during a 12-year period. Pediatr. Infect. Dis. J.24:901–904.
47.Vermaelen, K., and R. Pauwels.2005. Pulmonary dendritic cells. Am. J. Respir. Crit. Care Med.172:530–551.
48.Vermaelen, K. Y., I. Carro-Muino, B. N. Lambrecht, and R. A. Pauwels.
2001. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med.193:51–60.
49.Virgin, H. W., and S. H. Speck.1999. Unraveling immunity to gamma-herpesviruses: a new model for understanding the role of immunity in chronic virus infection. Curr. Opin. Immunol.11:371–379.
50.Virgin, H. W., IV, P. Latreille, P. Wamsley, K. Hallsworth, K. E. Weck, A. J. Dal Canto, and S. H. Speck.1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol.71:5894–5904.
51.Weck, K. E., A. J. Dal Canto, J. D. Gould, A. K. O’Guin, K. A. Roth, J. E. Saffitz, S. H. Speck, and H. W. Virgin.1997. Murine gamma-herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-gamma
respon-9788 WESLOW-SCHMIDT ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
siveness: a new model for virus-induced vascular disease. Nat. Med.3:1346– 1353.
52.World Health Organization. 2004. Changing history. The World Health Report. World Health Organization, Geneva, Switzerland.
53.Yoneyama, H., K. Matsuno, E. Toda, T. Nishiwaki, N. Matsuo, A. Nakano, S. Narumi, B. Lu, C. Gerard, S. Ishikawa, and K. Matsushima.2005.
Plas-macytoid DCs help lymph node DCs to induce anti-HSV CTLs. J. Exp. Med.
202:425–435.
54.Zhu, F. X., S. M. King, E. J. Smith, D. E. Levy, and Y. Yuan.2002. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumula-tion. Proc. Natl. Acad. Sci. USA99:5573–5578.