0022-538X/08/$08.00⫹0 doi:10.1128/JVI.01141-08
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Evolution of CCR5 Use before and during Coreceptor Switching
䌤
†
Mia Coetzer,
1Rebecca Nedellec,
1Janelle Salkowitz,
1‡ Sherry McLaughlin,
2Yi Liu,
2Laura Heath,
2James I. Mullins,
2and Donald E. Mosier
1*
Department of Immunology, The Scripps Research Institute, La Jolla, California 92037,1and Department of Microbiology,
The University of Washington School of Medicine, Seattle, Washington 981952
Received 30 May 2008/Accepted 15 September 2008
The envelope gene (env) of human immunodeficiency virus type 1 (HIV-1) undergoes rapid divergence from the transmitted sequence and increasing diversification during the prolonged course of chronic infection in humans. In about half of infected individuals or more,envevolution leads to expansion of the use of entry coreceptor from CCR5 alone to CCR5 and CXCR4. The stochastic nature of this coreceptor switch is not well explained by host selective forces that should be relatively constant between infected individuals. Moreover, differences in the incidence of coreceptor switching among different HIV-1 subtypes suggest that properties of the evolving virus population drive the switch. We evaluated the functional properties of sequentialenvclones from a patient with evidence of coreceptor switching at 5.67 years of infection. We found an abrupt decline in the ability of viruses to use CCR5 for entry at this time, manifested by a 1- to 2-log increase in susceptibility to CCR5 inhibitors and a reduced ability to infect cell lines with low CCR5 expression. There was an abnormally rapid 5.4% divergence inenvsequences from 4.10 to 5.76 years of infection, with the V3 and V4/V5 regions showing the greatest divergence and evidence of positive selection. These observations suggest that a decline in the fitness of R5 virus populations may be one driving force that permits the emergence of R5X4 variants.
Human immunodeficiency virus type 1 (HIV-1) infection is usually initiated by outgrowth of one or a few genotypic vari-ants (42, 44, 59, 72, 75, 77) followed by a prolonged period of asymptomatic infection during which virus populations diver-sify and after a period (26) diverge from the founder sequence (61). This process is driven by the high rate of virus genetic mutation and recombination (46, 62) and the selective forces on the diverse virus population imposed by the host (48). Evolution of HIV-1 envelope gene (env) sequences leads to expansion of coreceptor use/cell tropism from CCR5 (macro-phage tropism) to CXCR4 (T-cell line tropism) inⱖ50% of subtype B-infected individuals at late stages of disease (3, 47, 60, 61, 65, 71). Infection with subtype C HIV-1 is associated with a lower incidence of coreceptor switching (8, 54, 56), and infection with subtype D may be associated with more-frequent switching (29). This switch in coreceptor use correlates with more-rapid clinical progression of disease (2, 12, 35), although this correlation does not establish causation.
In vitro studies on coreceptor switching, as well as genetic analysis of viralenvsequences, suggest that viruses at interme-diate stages between CCR5 use and CXCR4 use (the transition from R5 to R5X4 or X4 virus) are less fit than parental R5 populations (49, 50, 52, 31, 73). Intermediate stages in core-ceptor switching may also differ in preference for CCR5 versus CXCR4, and these stages have recently been proposed to be
designated dual-R (CCR5 preference) or dual-X (CXCR4 preference) based on cell line entry assays (29). These presum-ably less fit intermediates are often difficult to detect in patient samples (65, 68), probably because they are at a competitive disadvantage compared to other viruses in the diverse popu-lation. One predictable consequence is that newly emerging R5X4 variants recovered from patients are only distantly re-lated to earlier R5 populations by sequence homology (61, 65), despite the small number of mutations that are usually re-quired for coreceptor switching in vitro (5–7, 22, 34, 52). In vitro models of coreceptor switching fail to reproduce the interactions between members of a large viral quasispecies, and they also may add selective pressures to adapt to levels of CD4 and coreceptors expressed on artificial cell lines and re-move selective pressures imposed by the host, such as immune responses, chemokine levels, and coreceptor expression. It is therefore important to validate that coreceptor switching in-volves transition through a stage of reduced fitness by assess-ment of Env function by using sequences directly isolated from patients.
MATERIALS AND METHODS
PCR and cloning.The first-round PCRs used for nested amplification of C2-V5 in the study of Shankarappa et al. (61) were used for the nested ampli-fication of full-length gp160. Briefly, pelleted peripheral blood mononuclear cells were lysed using detergent and proteinase K and treated with GeneReleaser (BioVentures, Murfreesboro, TN), and genomic proviral DNA was amplified with primers that lie outside of envelope (ED3 [5⬘-TTAGGCATCTCCTATGG CAGGAAGAAGCGG-3⬘] and Nef3 [5⬘-TAAGTCATTGGTCTTAAAGGTAC C-3⬘]).
The second-round primers used here were designed to encompass the entire gp160 reading frame and included NheI (5⬘) and BamHI (3⬘) restriction sites to facilitate directional cloning (5⬘primer MP1 [5⬘-AAA TAT GGC TAG CAA GGG GAT CAG GAA GAA TTA TCA G-3⬘] and 3⬘primer JAPCR506 [5⬘ -CGA CGG ATC CTT TGA CCA CTT GCC ACC CAT-3⬘]). PCRs were carried out in a 100-l volume containing 1.25⫻Bio-X-Act PCR buffer (Bioline, Taun-* Corresponding author. Mailing address: Department of
Immunol-ogy, The Scripps Research Institute, La Jolla, CA 92037. Phone: (858) 784-9121. Fax: (858) 784-9190. E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi .asm.org/.
‡ Present address: Department of Biological Sciences, University of Texas at El Paso, 500 W. University Ave., El Paso, TX 79968.
䌤Published ahead of print on 24 September 2008.
11758
on November 8, 2019 by guest
http://jvi.asm.org/
ton, MA), 2.5 mM MgCl2, 200M deoxynucleoside triphosphates, 25 pmol of
each primer (purified by high-performance liquid chromatography), 2.5 U of Bio-X-ActTaqpolymerase, and 2l of first-round PCR product. Paraffin wax was used to achieve a “hot start” to prevent mispriming and primer-dimer formation during the initial temperature ramp-up (25). “Touchdown” PCR (14) was initiated at an annealing temperature of 60°C and decreased by 0.2°C per cycle for 20 cycles, followed by 10 additional cycles at an annealing temperature of 58°C.
PCR products were cloned into mammalian expression vector pWR508 (67). Ligations were transformed into theEscherichia colistrain DH10B. To improve the stability of cloned DNA, bacteria were grown at 30°C in standard LB medium with 50g/ml of ampicillin. To test for transient expression of full-length and posttranscriptionally processed proteins, purified plasmid DNA samples were transfected into COS7 cells and extracts were subjected to analysis by Western blotting and probed with pooled HIV-positive human sera.
Pseudovirions.Infectious pseudovirus was constructed by coexpressing Env with the NL4-3 Env protein-negative, Luc-positive reporter plasmid (11), and infectivity was assayed for U87.CD4.CCR5 cells, U87.CD4.CXCR4 cells (13), MT-2 cells, MT-2.CCR5locells, or MT-2.CCR5hicells (51). The CCR5 inhibitors
PSC-RANTES (23) and TAK779 (1) and the CXCR4 inhibitor AMD3100 (15) were used to confirm coreceptor use and preference as well as sensitivity to inhibition. Fifty percent inhibitory concentrations (IC50) were determined using
full titrations of either PSC-RANTES or TAK779 to inhibit infection of U87.CD4.CCR5 cells, with IC50calculated by using curve fitting programs in
Prism 4 (GraphPad, San Diego, CA) as previously described (50). Coreceptor preference was determined by blocking infection of the MT-2.CCR5hi
cell line that expresses both CXCR4 and CCR5 with one or both coreceptor inhibitors. Viruses that can utilize CCR5 and CXCR4 equally well require both coreceptor inhibitors to block infection, whereas viruses that prefer either CCR5 or CXCR4 are sensitive to the single inhibitor targeting that coreceptor.
Sequence analysis.Sequences of the gp160 region from 45 Env genes used to make pseudoviruses were determined by Sanger sequencing and aligned with ClustalX (41). Manual editing and predicted protein translation were performed using BioEdit (version 7.0.5.3). Phylogenetic analysis (using neighbor-joining algorithms with bootstrap testing of 200 replicates), determination of genetic distance, and estimation of the proportions of synonymous (dS) and nonsynony-mous (dN) sites (with Jukes-Cantor correction and complete deletion) were determined using MEGA (version 3.1) software. Initial phylogenetic analysis included reference sequences obtained from the Los Alamos HIV sequence database to investigate sequence quality (including contamination and intersub-type recombination). Viral divergence from the founder strain and viral diversity were estimated for each time point. A clone with a sequence most homologous to the consensus B sequence was used as the founder strain, but similar results were obtained using a consensus sequence from the first time point. The posi-tion-specific scoring matrix (PSSM) for each sample was obtained with a Web-based program (http://indra.mullins.microbiol.washington.edu/pssm/), and the
⫻4r5 matrix was used. Potential N glycosylation sites (PNGS) were identified using N-GlycoSite (76). Positive selection of specific amino acids at each position of gp120 was analyzed using the methods described by Liu et al. (43). Potential cytotoxic-T-lymphocyte (CTL) epitopes (PCTLep) were identified based on alignment with the gp160 CTL/CD8⫹Epitope Map (36) with the HLA genotype of Multicenter AIDS Cohort Study (MACS) patient 5 taken into account (HLA A.0201 homozygous, B.0702/2705, C.0202/0.0702).
Nucleotide sequence accession numbers.All nucleotide sequences used in this study were submitted to GenBank and can be found under the accession num-bers FJ222409 to FJ222440 (see Fig. 1).
RESULTS
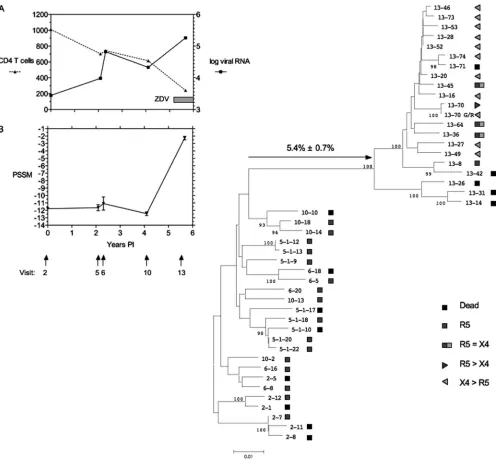
We have characterized 30 full-length functional envclones from a total of 82 clones collected longitudinally from patient 5 described in the study of Shankarappa et al. (57, 61). This patient from the MACS cohort (33) showed the appearance of
envsequences consistent with R5 to R5X4 switching at 5.67 years postinfection (p.i.), at the time of maximum divergence from the founderenvC2-V5 sequence (61). Only clones de-rived from 5.67 years p.i. (visit 13; clones numbered 13-n, wheren is a given number) showed expansion of coreceptor use from CCR5 to CXCR4, and routine typing of all functional clones on U87 target cells assigned the R5X4 phenotype
pre-dicted by the original V3 sequence (31, 61) and by the PSSM score (Fig. 1B). Data on the evolution and function of full-length env sequences (as opposed to the C2-V5 sequences analyzed by Shankarappa et al. [61]) in patient 5 are presented in Fig. 1.
Env sequence evolution.The sequences of the full-lengthenv
clones from the last time point sampled, 5.67 years p.i., were closely related to each other and substantially diverged from
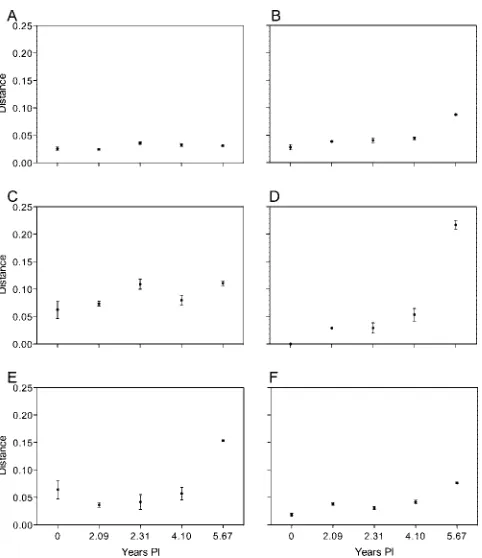
envsequences from each earlier time point (Fig. 1). Envelope sequences continued to diverge from the founder sequence at the expected rate (⬃1%/year) until visit 10 at 4.10 years p.i., but a further 5.4% divergence was observed over the next 18 months. There was no evidence, however, to indicate that this subject was superinfected with another subtype B strain, as the level of divergence is low relative to that of unlinked strains and the emerging sequences were phylogenetically closely linked to the preexisting sequences (61). To determine if this divergence was uniform across Env proteins, we analyzed amino acid diversity from the founder sequence by region (Fig. 2). The largest contributions to the increased diversity in the year 5.67 p.i. samples were made by the V3 and V4/V5 regions (Fig. 2D and E). We then analyzed the nucleotide sequences for evidence of positive selection by comparing the numbers of dS and dN substitutions (Fig. 3). The dN/dS ratio for the entire gp160 coding sequence showed no evidence of positive selec-tion (Fig. 3A) and only weak evidence of positive selecselec-tion when only the V3 region was analyzed (Fig. 3B). However, the V4/V5 region (Fig. 3C) showed evidence of strong positive selection (dN/dS⬎1.0). By contrast, there was no evidence of positive selection of the V1/V2 or gp41 region (data not shown) by this method. Because the dN/dS ratio does not take into account selection for specific amino acid substitutions, we performed a second analysis of positive selection using a more powerful method.
Accordingly, we analyzed the sequence changes from 4.10 years p.i. to 5.67 years p.i. for evidence of positive selection by using methods described previously by Liu et al. (43). These results are presented graphically in Fig. S1 in the supplemental material and summarized in Table 1. The positive selection evident between 4.10 and 5.67 years p.i. was associated with 31 amino acid substitutions in gp120, with the majority of substi-tutions occurring in variable regions. There were seven changes in the V3 region, including an S11R substitution and increased positive charge, changes previously associated with CXCR4 use (6, 30, 39). There were 10 changes in the V4/V5 region, resulting in an increase in the net charge of the V4 region (Table 1). The PNGS within this patient were also investigated. The number of PNGS within the gp160 region varied from 26 to 30 with no net increase or decrease over time. Most of the shifting PNGS were located in insertions within the variable regions. We also determined PCTLep in gp120 (Table 1) and gp41 to determine if the robust CTL response to Env observed in this patient (57) might account for the high level of positive selection (43). Of the 31 sites in gp120 showing evidence of positive selection, 8 were in PCTLep, predicted based on the patient HLA genotype. These included several substitutions in V3 that are also associated with core-ceptor switching. We also observed 15 sites in gp41 with evi-dence of positive selection, and 7 of these sites were within predicted CTL epitopes.
on November 8, 2019 by guest
http://jvi.asm.org/
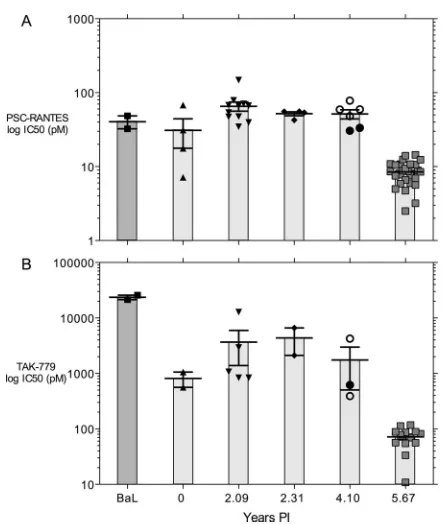
Functional evolution of Env.Thirty of 82envclones tested yielded functional Env proteins as evidenced by mediating entry of pseudotyped virus into U87.CD4.CCR5 and (for year 5.67 p.i.envclones only) U87.CD4.CXCR4 target cells. The CCR5 use of each functionalenvclone was evaluated by one of two assays. The first assay measured the sensitivity of single-cycle infection of U87.CD4.CCR5 cells to the CCR5 inhibitor PSC-RANTES (23) or TAK779 (55), and the
sec-ond measured ability to infect MT-2.CD4.CCR5 cells ex-pressing high or low levels of CCR5 (51). Figure 4 shows the changes in IC50of PSC-RANTES (Fig. 4A) or TAK779 (Fig.
[image:3.585.45.546.66.526.2]4B). These data show a remarkable and highly significant increase in susceptibility to both CCR5 inhibitors that co-incides with the appearance of R5X4 variants. Moreover, clone 10-18 was the most sensitive to inhibition by PSC-RANTES of the clones from 4.10 years p.i. (individual data
FIG. 1. Genetic distance between sequentialenvclones from MACS patient 5.envclone numbers correspond to patient visit number: 2-1, 2-5, and 2-7 are from just after seroconversion; 5.1-nclones are from 2.09 years p.i.; 6-nclones are from 2.31 years p.i.; 10-nclones are from 4.10 years p.i.; and 13-nclones are from 5.67 years p.i. Panels A and B indicate the clinical course of the patient with respect to CD4 T-cell counts/mm3and viral RNA copy number (log10scale; panel A) and the PSSM score for V3 sequences at each time point. The right side of the figure shows a phylogenetic tree depicting the relationship betweenenvgp160 sequences of the biological clones studied here. Symbols indicate coreceptor use and preference of each clone. Only a subset of nonfunctionalenvclones is depicted. The coreceptor preference of eachenvclone with evidence of R5X4 tropism is indicated by the symbols for R5 or X4. R5⫽X4 indicates that both PSC-RANTES and AMD3100 were required to inhibit infection of MT-2.CCR5hicells. R5⬎X4 indicates that PSC-RANTES alone could partially inhibit infection but AMD3100 alone could not. X4⬎ R5 indicates that AMD3100 alone was more inhibitory than PSC-RANTES alone. ZDV, zidovudine.
on November 8, 2019 by guest
http://jvi.asm.org/
points in Fig. 4A) and was the closest ancestor to clones from the last visit (Fig. 1).
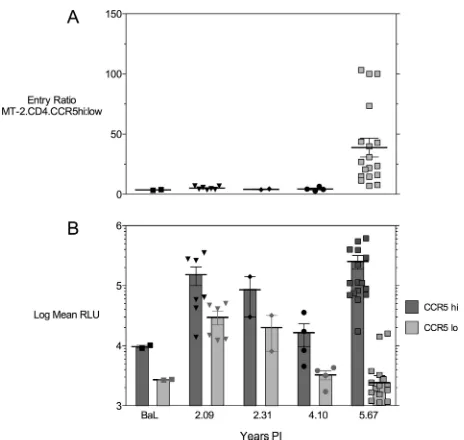
Figure 5 shows the results of various CCR5 densities on target cells on infection mediated by Env proteins from patient 5. Figure 5A shows the ratio of infected CCR5hicells to
in-fected CCR5locells, which is less than 5 until 5.67 years p.i.,
when the ratio increases to a mean of 39.79⫾7.75. Figure 5B shows that the change in ratio at year 5.67 p.i. is due both to a loss of infection efficiency with low CCR5 levels and to a gain of infection efficiency, with high CCR5 levels indicated by increased mean relative light unit signals on MT-2.CD4.CCR5hicells. Also note that there were significant
de-creases in the use of CCR5 for both cells with low CCR5 levels and cells with high CCR5 levels at year 4.10 p.i., a time before any R5X4 Env clones were detected.
A coreceptor preference assay provided additional evidence of functional differences between year 5.67 p.i. Env proteins. Coreceptor preference was assessed by determining the sensi-tivity of infection to either CCR5 or CXCR4 inhibition on target cells expressing both coreceptors (see Materials and Methods for details). The results are depicted schematically in Fig. 1, and the inhibition data are presented in Fig. 6. The majority of Env proteins from 5.67 years p.i. were more sen-sitive to the CXCR4 inhibitor AMD3100 than the CCR5 in-hibitor PSC-RANTES and were thus scored as having a pref-erence of X4⬎R5. A smaller number of Env proteins were
equally inhibited by both agents and were scored as R5⫽X4. Only two Env proteins, 13-70 and the poorly infectious clone 13-8, showed a strong preference for CCR5 in this assay. By contrast, all Env clones from earlier time points were fully sensitive to PSC-RANTES inhibition and were not inhibited by AMD3100. The coreceptor preference assay used with a single target cell and coreceptor-specific inhibitors was more sensi-tive in assigning use of CCR5 and CXCR4 than in determining relative entry into different target cells expressing either CCR5 or CXCR4 (Fig. 6B), although cells with strong CXCR4 pref-erence were identified in both assays. Env 13-70 had the un-usual V3 crown sequence GPGG, and changing this to the consensus GPGR sequence (13-70 G/R) changed the corecep-tor preference from CCR5 to CXCR4 (Fig. 1 and 6). However, Env clone 13-70 and the V3 consensus clone 13-70 G/R had identical high sensitivities to CCR5 inhibitors and poor abili-ties to infect target cells with low CCR5 levels (data not shown), suggesting that loss of CCR5 binding was influenced by sequence changes elsewhere in the Env protein.
DISCUSSION
[image:4.585.44.285.65.344.2]The results presented here demonstrate two striking fea-tures of HIV-1 envelope evolution associated with coreceptor expansion from CCR5 alone to CCR5 and CXCR4. First, there was a major divergence in envsequence from the last pure R5 virus to the first R5X4 variant, with the greatest divergence observed in the V3 and V4/V5 domains and the
[image:4.585.345.494.71.360.2]FIG. 2. Diversity of different regions of Env during the course of infection. (A) Pairwise amino acid diversity across gp160 at each time point. (B) Amino acid divergence from the founder Env clone 2-12 for gp160. (C) Amino acid divergence from the founder for V1/V2 region. (D) Amino acid divergence from the founder for the V3 loop. (E) Amino acid divergence from the founder for the V4/V5 region. (F) Amino acid divergence from the founder for the gp41 region.
FIG. 3. Analysis of dS and dN nucleotide substitutions during the course of infection. (A) dN/dS ratios from the founder for gp160. (B) dN/dS ratios from founder for the V3 region. (C) dN/dS ratios from founder for the V4/V5 region.
on November 8, 2019 by guest
http://jvi.asm.org/
greatest evidence of positive selection in the same variable domains (Fig. 1 to 3; Table 1). Second, there was a major change in the efficiency with which Env engaged CCR5, dem-onstrated by increased sensitivity to CCR5 inhibitors (Fig. 4) and a diminished ability to infect target cells with low CCR5 expression (Fig. 5). A detailed functional analysis ofenvclones from the time of first appearance of R5X4 variants showed a spectrum of CCR5 versus CXCR4 preference as coreceptor (Fig. 6). These results confirm and expand upon prior work (19, 40, 61, 65) indicating that coreceptor switching in patients involves sequence changes both in V3 and in other regions of Env. They are also consistent with our earlier observations in cell culture models (49, 50, 52) that V3 mutations, while nec-essary for coreceptor switching, must be accompanied by com-pensatory mutations elsewhere in env to produce virus with reasonable competitive fitness. The rapid evolution of the V4/V5 domain has been observed in at least one other patient described in a recent publication (21), although the association of the sequence changes with coreceptor switching is unknown. Extensive sequence changes in V4/V5 following coreceptor switching have been noted in other studies (28, 63), but the kinetics of these changes were not presented. Our results con-firm that rapid and extensive changes in the V4/V5 sequence can accompany coreceptor switching.
The results presented in Fig. 5 are potentially very important
[image:5.585.46.282.90.406.2]because they show a decrease in the ability of Env clones to mediate infection of MT-2 target cells with either high or low CCR5 expression at 4.10 years after infection, a time point that precedes the detection of any R5X4 variants by about 18 months. This observation suggests that it may be possible to develop phenotypic assays that predict the impending onset of coreceptor switching, if the data from this single patient can be extended to a larger number of HIV-1-infected patients. The gradual increase in PSSM score observed to occur prior to the appearance of X4 viruses, including R5X4 viruses with inter-mediate values (31), also indicated that a predictive measure is possible prior to the occurrence of a phenotypic switch. The same data show that the ability to infect target cells with high CCR5 expression is either sustained or improved, while the ability to infect target cells with low CCR5 levels declines (Fig. 5B). This result suggests that the lower avidity for CCR5 pre-dicted by increased sensitivity to CCR5 inhibitors has a major impact only when CCR5 levels are limiting. Very high CCR5 levels may allow increased avidity of Env binding by engaging multiple CCR5 molecules in dense local aggregates, as pre-dicted by prior studies of infection (37, 38) and cryoelectron tomography of virus-target cell interactions (64). It is possible that entry via high CCR5 levels could be mediated by binding to only the CCR5 N-terminal domain (74), whereas entry via low CCR5 levels would require the more-efficient binding of
[image:5.585.310.531.362.624.2]FIG. 4. Sensitivity to CCR5 coreceptor inhibitors. Env-pseudo-typed virus was used to infect U87.CD4.CCR5 cells in the presence of increasing concentrations of PSC-RANTES (A) or TAK779 (B). BaL, reference R5 Env. Data are the means⫾standard errors of the IC50 for each Env clone in two or three replicate experiments (as illustrated in Fig. S2 in the supplemental material), with the results for individual Env clones represented by filled symbols. The data in panels A and B at 4.10 years p.i. shown as filled circles are results forenvclone 10-18, theenvclone closest to the ancestor of those obtained at 5.67 years p.i. (Fig. 1), while the unfilled circles represent otherenvclones from this time point.
TABLE 1. Site-by-site analysis of positive selection in gp120afrom
year 4.10 p.i. to year 5.67 p.i.
Position
Residue
Region Change(s) Original Mutation
15 R K C1
16 W G C1
25 M C1 Insertion
33 N D C1 No PNGS, PCTLep
65 V A C1
85 V I C1
138 T D V1 PNGS conserved
153 G V1 Insertion
154 I V1 Insertion
158 T K V1 Charge added
173 R K V2 Charge conserved
195 N V2 No PNGS, insertion
261 R K C2 Charge conserved, PCTLep
308 P L V3 PCTLep
315 S R V3 Charge added
317 H T V3 PCTLep
318 I M V3 PCTLep
323 A V V3 PCTLep
324 F Y V3 PCTLep
336 Q K V3 Charge added, PCTLep
361 F Y C3
396 T A V4
397 Q K V4 Charge added
404 M I V4
405 F N V4
406 N V4 PNGS shifted, deletion
407 S G V4
414 E G V4
472 T V5 PNGS shifted, deletion
475 T N V5 PNGS added
477 V T V5
a
Fifteen additional sites in gp41 showed evidence of positive selection. Seven of 15 sites in gp41 under positive selection were in PCTLep.
on November 8, 2019 by guest
http://jvi.asm.org/
both the N-terminal domain and other extracellular domains. This explanation is consistent with a two-site binding model wherein the base of the V3 loop contacts the CCR5 N terminus and residues near the tip of the loop contact CCR5 EC2 (16, 24). Env 13-70 from the last visit may represent the capture of an intermediate in this process, since it has poor entry function on CCR5 and even worse entry function via CXCR4. The functionality of clone 13-70 was largely repaired by changing the V3 crown sequence from GPGG to GPGR, suggesting that this conserved sequence may be important for binding to CXCR4. Env 13-70 shares the divergent V4/V5 region with other clones from this time point, so it appears to have accu-mulated the compensatory mutations necessary for efficient CXCR4 use, and it is likely that the GPGR to GPGG mutation was a recent event, since it should reduce competitive fitness. The loss of apparent CCR5 binding avidity noted in our results is consistent with prior observations suggesting that R5X4 viruses are more sensitive to a CCR5 inhibitor than are R5 viruses (73). Moreover, a recent publication (45) reported entry data consistent with our observations. Entry efficiency in the Trofile assay (69) continued to increase in patients who progressed without any detection of R5X4 variants, but the signal on R5 targets declined in those patients who developed R5X4 (or dual/mixed) variants (45). These entry effects were not seen in the sample of 30 individual Env clones from a single subject studied in our experiments but were apparent in the population of the 785 patient samples used in the Trofile assay (45). The increase in entry efficiency via CCR5 was neg-atively correlated with CD4 T-cell counts in those patients who failed to develop R5X4 or dual/mixed viruses (45), suggesting that such a change may accelerate disease progression.
The stochastic nature of coreceptor switching (i.e., it occurs in a subset of infected patients but is correlated with high viral load, low CD4 T-cell numbers, and reduced expression of
CCR5) (3, 66) suggests that target cell availability and core-ceptor density are selective forces. Our results are consistent with a model whereby the bifurcation point is determined by either continuing to improve CCR5 binding (as in patients who do not undergo coreceptor switching) or starting down a path-way of diminished CCR5 binding that is permissive for core-ceptor switching. The continued improvement in CCR5 bind-ing has been inferred from prior studies of RANTES inhibition (32) and is assumed to be driven by a combination of mutation and selection acting on the viral quasispecies in each patient. The accumulation of mutations that improve CCR5 use should be limited by a fitness peak whereby additional mutations are more likely to decrease than increase fitness (58, 70). For our results, it is important to note that every Env from the last visit had lost efficiency of entry via CCR5 and that the closest ancestor at the prior sampling time (clone 10-18) had the poorest use of CCR5 of any Env from that time point (Fig. 4A). We thus propose that an important event in coreceptor switching is anenvmutation(s) that diminishes CCR5 binding and that mutations deleterious for CCR5 use continue to ac-cumulate as mutations that improve CXCR4 occur. Efficient use of CXCR4 appears to require multiple mutations in both V3 and V4/V5 (although the importance of V4/V5 mutations has not been formally demonstrated; see Table 1), suggesting that there may be a population bottleneck at a point where CCR5 entry efficiency has diminished and CXCR4 entry has yet to be optimized. It is not obvious from this proposed ex-planation that every mutational pathway that impairs CCR5 use inevitably leads to CXCR4 use; it is possible that additional mutations can restore CCR5 binding and reverse the trend toward CXCR4 engagement (31). The evidence for strong positive selection for V3 and V4/V5 replacement mutations (Fig. 3 and Table 1; see Fig. S1 in the supplemental material) suggests that changes in these domains are essential for bind-ing to both CCR5 and CXCR4 and that the envelope surface near the conserved bridging sheet must be remodeled to ac-commodate this new functionality. Additional selection pres-sure may have been exerted by Env-specific CTLs, which were readily detectable in this patient (57), and could have helped drive the many substitutions in the V3 region. Note, however, that PCTLep were not identified in the V4/V5 region and that antibody selective pressure on variable regions is expected to diminish with increasing duration of infection (4).
Coreceptor switching is less frequent in subtype C HIV-1 infections. How do our results explain this observation? There are recent data suggesting that different V3 conformations in subtype C Env limit adaptation to CXCR4 (53). It is also possible that compensatory mutations in regions other than V3 that are necessary for efficient CXCR4 use are more difficult to accomplish. Sequences from subtype C show more variation in the C3 region than in the V3 region (8–10), and the rare R5X4 subtype C isolates show major divergence from earlier R5 isolates (9). These observations are thus consistent with the probability of coreceptor switching being determined by the starting Env sequence and the mutational distance to a fit R5X4 descendant, which would vary with subtype.
[image:6.585.48.280.65.285.2]Our observations and model for coreceptor switching do not entirely rule out other hypotheses, such as those implicating target cell selection (17, 18) or immune pressures (20, 27), that preferentially slow the emergence of R5X4 HIV-1 strains.
FIG. 5. Entry efficiency of Env-pseudotyped virus on target cells expressing high or low levels of CCR5. (A) Ratios of entry on MT-2.CCR5hiversus MT-2.CCR5locells. (B) Relative light units (RLU; in log10scale) used to calculate the ratios in panel A.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 6. Coreceptor (CoR) preference results for Env clones from year 5.67 p.i. (A) Entry of Env-pseudotyped viruses into MT-2.CD4.CCR5hi cells was inhibited by PSC-RANTES to block CCR5-mediated entry, AMD3100 to block CXCR4-mediated entry, or both inhibitors (P⫹A) as indicated in the key. Data are plotted as the means and standard errors for percent inhibition for three replicate experiments. HXB2 Env is the control for CXCR4 use, ADA Env is the control for CCR5 use, and ADA-1M45 is the control for equivalent CCR5 and CXCR4 use. Preference for CCR5 is defined as inhibition by PSC-RANTES that was statistically significantly greater than that by AMD3100. Preference for CXCR4 is defined by greater inhibition by AMD3100 than by PSC-RANTES. If the two inhibitors showed equivalent inhibition levels, then the coreceptor preference is designated R5⫽X4. (B) Relative entry of the same set of Env-pseudotyped viruses into U87.CD4.CCR5 target cells (log CCR5 relative light units [RLU]) versus U87.CD4.CXCR4 target cells (log CXCR4 RLU). Data are displayed as means⫾standard errors for RLU in each assay. Only a subset of Env clones are identified by the clone name; these showed the largest differences between CCR5- and CXCR4-mediated entry.
on November 8, 2019 by guest
http://jvi.asm.org/
However, the collapse of CCR5 use documented in these stud-ies does provide a potential explanation for the stochastic nature of coreceptor switching and its linkage to the subtype of HIV-1, and we propose that it is an important early event that allows other selective forces to become operative.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01AI52778 (D.E.M.), R01AI058894 (J.I.M.), R37AI047734 (J.I.M.), and PO1AI051649 (D.E.M.). M.C. was partially supported by a fellowship from the La Jolla Foundation for Microbicide Research. We appreciate additional support from the James B. Pendleton Trust.
This is publication number 19454 from The Scripps Research Insti-tute.
REFERENCES
1.Baba, M., O. Nishimura, N. Kanzaki, M. Okamoto, H. Sawada, Y. Iizawa, M. Shiraishi, Y. Aramaki, K. Okonogi, Y. Ogawa, K. Meguro, and M. Fujino.
1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. USA96:5698–5703. 2.Bozzette, S., J. McCutchan, S. Spector, B. Wright, and D. Richman.1993. A
cross-sectional comparison of persons with syncytium- and non-syncytium-inducing human immunodeficiency virus. J. Infect. Dis.168:1374–1379. 3.Brumme, Z. L., J. Goodrich, H. B. Mayer, C. J. Brumme, B. M. Henrick, B.
Wynhoven, J. J. Asselin, P. K. Cheung, R. S. Hogg, J. S. Montaner, and P. R. Harrigan. 2005. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J. Infect. Dis.
192:466–474.
4.Bunnik, E. M., L. Pisas, A. C. van Nuenen, and H. Schuitemaker.2008. Autologous neutralizing humoral immunity and evolution of the viral enve-lope in the course of subtype B human immunodeficiency virus type 1 infection. J. Virol.82:7932–7941.
5.Cann, A. J., M. J. Churcher, M. Boyd, W. O’Brien, J. Q. Zhao, J. Zack, and I. S. Chen.1992. The region of the envelope gene of human immunodefi-ciency virus type 1 responsible for determination of cell tropism. J. Virol.
66:305–309.
6.Chesebro, B., K. Wehrly, J. Nishio, and S. Perryman.1996. Mapping of independent V3 envelope determinants of human immunodeficiency virus type 1 macrophage tropism and syncytium formation in lymphocytes. J. Vi-rol.70:9055–9059.
7.Cho, M. W., M. K. Lee, M. C. Carney, J. F. Berson, R. W. Doms, and M. A. Martin.1998. Identification of determinants on a dualtropic human immu-nodeficiency virus type 1 envelope glycoprotein that confer usage of CXCR4. J. Virol.72:2509–2515.
8.Cilliers, T., J. Nhlapo, M. Coetzer, D. Orlovic, T. Ketas, W. C. Olson, J. P. Moore, A. Trkola, and L. Morris.2003. The CCR5 and CXCR4 coreceptors are both used by human immunodeficiency virus type 1 primary isolates from subtype C. J. Virol.77:4449–4456.
9.Coetzer, M., T. Cilliers, M. Papathanasopoulos, G. Ramjee, S. A. Karim, C. Williamson, and L. Morris.2007. Longitudinal analysis of HIV type 1 sub-type C envelope sequences from South Africa. AIDS Res. Hum. Retrovir.
23:316–321.
10.Coetzer, M., T. Cilliers, L. H. Ping, R. Swanstrom, and L. Morris.2006. Genetic characteristics of the V3 region associated with CXCR4 usage in HIV-1 subtype C isolates. Virology356:95–105.
11.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau.1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mono-nuclear phagocytes. Virology206:935–944.
12.Connor, R. I., H. Mohri, Y. Cao, and D. D. Ho.1993. Increased viral burden and cytopathicity correlate temporally with CD4⫹T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected indi-viduals. J. Virol.67:1772–1777.
13.Deng, H. K., D. Unutmaz, V. N. KewalRamani, and D. R. Littman.1997. Expression cloning of new receptors used by simian and human immunode-ficiency viruses. Nature388:296–300.
14.Don, R. H., P. T. Cox, B. J. Wainwright, K. Baker, and J. S. Mattick.1991. “Touchdown” PCR to circumvent spurious priming during gene amplifica-tion. Nucleic Acids Res.19:4008.
15.Donzella, G. A., D. Schols, S. W. Lin, J. A. Este, K. A. Nagashima, P. J. Maddon, G. P. Allaway, T. P. Sakmar, G. Henson, E. De Clercq, and J. P. Moore.1998. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med.4:72–77.
16.Farzan, M., S. Chung, W. Li, N. Vasilieva, P. L. Wright, C. E. Schnitzler, R. J. Marchione, C. Gerard, N. P. Gerard, J. Sodroski, and H. Choe.2002. Tyrosine-sulfated peptides functionally reconstitute a CCR5 variant lacking a critical amino-terminal region. J. Biol. Chem.277:40397–40402. 17.Ferna`ndez, G., A. Llano, M. Esgleas, B. Clotet, J. A. Este, and M. A.
Martinez. 2006. Purifying selection of CCR5-tropic human
immunodefi-ciency virus type 1 variants in AIDS subjects that have developed syncytium-inducing, CXCR4-tropic viruses. J. Gen. Virol.87:1285–1294.
18.Goodenow, M. M., and R. G. Collman.2006. HIV-1 coreceptor preference is distinct from target cell tropism: a dual-parameter nomenclature to define viral phenotypes. J. Leukoc. Biol.80:965–972.
19.Groenink, M., R. A. Fouchier, S. Broersen, C. H. Baker, M. Koot, A. B. van’t Wout, H. G. Huisman, F. Miedema, M. Tersmette, and H. Schuitemaker.
1993. Relation of phenotype evolution of HIV-1 to envelope V2 configura-tion. Science260:1513–1516.
20.Harouse, J. M., C. Buckner, A. Gettie, R. Fuller, R. Bohm, J. Blanchard, and C. Cheng-Mayer.2003. CD8⫹T cell-mediated CXC chemokine receptor 4-simian/human immunodeficiency virus suppression in dually infected rhesus macaques. Proc. Natl. Acad. Sci. USA100:10977–10982.
21.Harrington, P. R., J. A. Nelson, K. M. Kitrinos, and R. Swanstrom.2007. Independent evolution of human immunodeficiency virus type 1envV1/V2 and V4/V5 hypervariable regions during chronic infection. J. Virol.81:5413– 5417.
22.Harrowe, G., and C. Cheng-Mayer.1995. Amino acid substitutions in the V3 loop are responsible for adaptation to growth in transformed T-cell lines of a primary human immunodeficiency virus type 1. Virology210:490–494. 23.Hartley, O., H. Gaertner, J. Wilken, D. Thompson, R. Fish, A. Ramos, C.
Pastore, B. Dufour, F. Cerini, A. Melotti, N. Heveker, L. Picard, M. Alizon, D. Mosier, S. Kent, and R. Offord.2004. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. USA101:16460–16465.
24.Hartley, O., P. J. Klasse, Q. J. Sattentau, and J. P. Moore.2005. V3: HIV’s switch-hitter. AIDS Res. Hum. Retrovir.21:171–189.
25.He´bert, B., J. Bergeron, E. F. Potworowski, and P. Tijssen.1993. Increased PCR sensitivity by using paraffin wax as a reaction mix overlay. Mol. Cell. Probes7:249–252.
26.Herbeck, J. T., D. C. Nickle, G. H. Learn, G. S. Gottlieb, M. E. Curlin, L. Heath, and J. I. Mullins.2006. Human immunodeficiency virus type 1env evolves toward ancestral states upon transmission to a new host. J. Virol.
80:1637–1644.
27.Ho, S. H., S. Tasca, L. Shek, A. Li, A. Gettie, J. Blanchard, D. Boden, and C. Cheng-Mayer.2007. Coreceptor switch in R5-tropic simian/human immuno-deficiency virus-infected macaques. J. Virol.81:8621–8633.
28.Hu, Q. X., A. P. Barry, Z. X. Wang, S. M. Connolly, S. C. Peiper, and M. L. Greenberg.2000. Evolution of the human immunodeficiency virus type 1 envelope during infection reveals molecular corollaries of specificity for coreceptor utilization and AIDS pathogenesis. J. Virol.74:11858–11872. 29.Huang, W., S. H. Eshleman, J. Toma, S. Fransen, E. Stawiski, E. E. Paxinos,
J. M. Whitcomb, A. M. Young, D. Donnell, F. Mmiro, P. Musoke, L. A. Guay, J. B. Jackson, N. T. Parkin, and C. J. Petropoulos.2007. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J. Vi-rol.81:7885–7893.
30.Hwang, S. S., T. J. Boyle, H. K. Lyerly, and B. R. Cullen.1991. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science253:71–74.
31.Jensen, M. A., F. S. Li, A. B. van ’t Wout, D. C. Nickle, D. Shriner, H. X. He, S. McLaughlin, R. Shankarappa, J. B. Margolick, and J. I. Mullins.2003. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 envV3 loop sequences. J. Virol.77:13376–13388.
32.Karlsson, I., L. Antonsson, Y. Shi, M. Oberg, A. Karlsson, J. Albert, B. Olde, C. Owman, M. Jansson, and E. M. Fenyo.2004. Coevolution of RANTES sensitivity and mode of CCR5 receptor use by human immunodeficiency virus type 1 of the R5 phenotype. J. Virol.78:11807–11815.
33.Kaslow, R. A., D. G. Ostrow, R. Detels, J. P. Phair, B. F. Polk, and C. R. Rinaldo, Jr.1987. The Multicenter AIDS Cohort Study: rationale, organi-zation, and selected characteristics of the participants. Am. J. Epidemiol.
126:310–318.
34.Kiselyeva, Y., R. Nedellec, A. Ramos, C. Pastore, L. B. Margolis, and D. E. Mosier.2007. Evolution of CXCR4-using human immunodeficiency virus type 1 SF162 is associated with two unique envelope mutations. J. Virol.
81:3657–3661.
35.Koot, M., A. B. van ’t Wout, N. A. Kootstra, R. E. de Goede, M. Tersmette, and H. Schuitemaker. 1996. Relation between changes in cellular load, evolution of viral phenotype, and the clonal composition of virus populations in the course of human immunodeficiency virus type 1 infection. J. Infect. Dis.173:349–354.
36.Korber, B. T. M., B. Brander, B. F. Haynes, R. Koup, J. P. Moore, B. D. Walker, and D. I. Watkins (ed.).2006–2007. HIV molecular immunology, vol. LA-UR 07–4752. Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, NM.
37.Kozak, S. L., J. M. Heard, and D. Kabat.2002. Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mech-anism for membrane destabilization by human immunodeficiency virus. J. Virol.76:1802–1815.
38.Kozak, S. L., E. J. Platt, N. Madani, F. E. Ferro, Jr., K. Peden, and D. Kabat.
1997. CD4, CXCR-4, and CCR-5 dependencies for infections by primary
on November 8, 2019 by guest
http://jvi.asm.org/
patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J. Virol.71:873–882.
39.Kuiken, C. L., J. J. de Jong, E. Baan, W. Keulen, M. Tersmette, and J. Goudsmit.1992. Evolution of the V3 envelope domain in proviral sequences and isolates of human immunodeficiency virus type 1 during transition of the viral biological phenotype. J. Virol.66:4622–4627.
40.Labrosse, B., C. Treboute, A. Brelot, and M. Alizon.2001. Cooperation of the V1/V2 and V3 domains of human immunodeficiency virus type 1 gp120 for interaction with the CXCR4 receptor. J. Virol.75:5457–5464. 41.Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan,
H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thomp-son, T. J. GibThomp-son, and D. G. Higgins.2007. Clustal W and Clustal X version 2.0. Bioinformatics23:2947–2948.
42.Learn, G. H., D. Muthui, S. J. Brodie, T. Zhu, K. Diem, J. I. Mullins, and L. Corey.2002. Virus population homogenization following acute human im-munodeficiency virus type 1 infection. J. Virol.76:11953–11959.
43.Liu, Y., J. McNevin, J. Cao, H. Zhao, I. Genowati, K. Wong, S. McLaughlin, M. D. McSweyn, K. Diem, C. E. Stevens, J. Maenza, H. He, D. C. Nickle, D. Shriner, S. E. Holte, A. C. Collier, L. Corey, M. J. McElrath, and J. I. Mullins.2006. Selection on the human immunodeficiency virus type 1 pro-teome following primary infection. J. Virol.80:9519–9529.
44.Long, E. M., H. L. Martin, Jr., J. K. Kreiss, S. M. Rainwater, L. Lavreys, D. J. Jackson, J. Rakwar, K. Mandaliya, and J. Overbaugh.2000. Gender differences in HIV-1 diversity at time of infection. Nat. Med.6:71–75. 45.Low, A. J., D. Marchant, C. J. Brumme, Z. L. Brumme, W. Dong, T. Sing,
R. S. Hogg, J. S. Montaner, V. Gill, P. K. Cheung, and P. R. Harrigan.2008. CD4-dependent characteristics of coreceptor use and HIV type 1 V3 se-quence in a large population of therapy-naive individuals. AIDS Res. Hum. Retrovir.24:219–228.
46.Mansky, L. M., and H. M. Temin.1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol.69:5087–5094.
47.Moyle, G. J., A. Wildfire, S. Mandalia, H. Mayer, J. Goodrich, J. Whitcomb, and B. G. Gazzard.2005. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J. Infect. Dis.191:866–872.
48.Overbaugh, J., and C. R. Bangham.2001. Selection forces and constraints on retroviral sequence variation. Science292:1106–1109.
49.Pastore, C., R. Nedellec, A. Ramos, O. Hartley, J. L. Miamidian, J. D. Reeves, and D. E. Mosier.2007. Conserved changes in envelope function during human immunodeficiency virus type 1 coreceptor switching. J. Virol.
81:8165–8179.
50.Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier.
2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J. Vi-rol.80:750–758.
51.Pastore, C., G. R. Picchio, F. Galimi, R. Fish, O. Hartley, R. E. Offord, and D. E. Mosier.2003. Two mechanisms for human immunodeficiency virus type 1 inhibition by N-terminal modifications of RANTES. Antimicrob. Agents Chemother.47:509–517.
52.Pastore, C., A. Ramos, and D. E. Mosier.2004. Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J. Virol.78:7565–7574. 53.Patel, M. B., N. G. Hoffman, and R. Swanstrom. 2008. Subtype-specific conformational differences within the V3 region of subtype B and subtype C human immunodeficiency virus type 1 Env proteins. J. Virol.82:903–916. 54.Peeters, M., R. Vincent, J. L. Perret, M. Lasky, D. Patrel, F. Liegeois, V.
Courgnaud, R. Seng, T. Matton, S. Molinier, and E. Delaporte.1999. Evi-dence for differences in MT2 cell tropism according to genetic subtypes of HIV-1: syncytium-inducing variants seem rare among subtype C HIV-1 vi-ruses. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol.20:115–121. 55.Peters, P. J., J. Bhattacharya, S. Hibbitts, M. T. Dittmar, G. Simmons, J.
Bell, P. Simmonds, and P. R. Clapham.2004. Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. J. Virol.78:6915–6926. 56.Ping, L. H., J. A. Nelson, I. F. Hoffman, J. Schock, S. L. Lamers, M. Goodman, P. Vernazza, P. Kazembe, M. Maida, D. Zimba, M. M. Goodenow, J. J. Eron, Jr., S. A. Fiscus, M. S. Cohen, and R. Swanstrom.1999. Char-acterization of V3 sequence heterogeneity in subtype C human immunode-ficiency virus type 1 isolates from Malawi: underrepresentation of X4 vari-ants. J. Virol.73:6271–6281.
57.Rinaldo, C. R., Jr., P. Gupta, X. L. Huang, Z. Fan, J. I. Mullins, S. Gange, H. Farzadegan, R. Shankarappa, A. Munoz, and J. B. Margolick.1998. Anti-HIV type 1 memory cytotoxic T lymphocyte responses associated with changes in CD4⫹T cell numbers in progression of HIV type 1 infection. AIDS Res. Hum. Retrovir.14:1423–1433.
58.Rolland, M., C. Brander, D. C. Nickle, J. T. Herbeck, G. S. Gottlieb, M. S. Campbell, B. S. Maust, and J. I. Mullins.2007. HIV-1 over time: fitness loss or robustness gain? Nat. Rev. Microbiol.5:C1.
59.Sagar, M., E. Kirkegaard, E. M. Long, C. Celum, S. Buchbinder, E. S. Daar, and J. Overbaugh.2004. Human immunodeficiency virus type 1 (HIV-1) diversity at time of infection is not restricted to certain risk groups or specific HIV-1 subtypes. J. Virol.78:7279–7283.
60.Schuitemaker, H., M. Koot, N. A. Kootstra, M. W. Dercksen, R. E. de Goede, R. P. van Steenwijk, J. M. Lange, J. K. Schattenkerk, F. Miedema, and M. Tersmette.1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is asso-ciated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol.66:1354–1360.
61.Shankarappa, R., J. B. Margolick, S. J. Gange, A. G. Rodrigo, D. Upchurch, H. Farzadegan, P. Gupta, C. R. Rinaldo, G. H. Learn, X. He, X. L. Huang, and J. I. Mullins.1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol.
73:10489–10502.
62.Shriner, D., A. G. Rodrigo, D. C. Nickle, and J. I. Mullins.2004. Pervasive genomic recombination of HIV-1 in vivo. Genetics167:1573–1583. 63.Smyth, R. J., Y. Yi, A. Singh, and R. G. Collman.1998. Determinants of entry
cofactor utilization and tropism in a dualtropic human immunodeficiency virus type 1 primary isolate. J. Virol.72:4478–4484.
64.Sougrat, R., A. Bartesaghi, J. D. Lifson, A. E. Bennett, J. W. Bess, D. J. Zabransky, and S. Subramaniam.2007. Electron tomography of the contact between T cells and SIV/HIV-1: implications for viral entry. PLoS Pathog.
3:e63.
65.van Rij, R. P., H. Blaak, J. A. Visser, M. Brouwer, R. Rientsma, S. Broersen, A. M. de Roda Husman, and H. Schuitemaker.2000. Differential coreceptor expression allows for independent evolution of non-syncytium-inducing and syncytium-inducing HIV-1. J. Clin. Investig.106:1039–1052.
66.van Rij, R. P., M. D. Hazenberg, B. H. van Benthem, S. A. Otto, M. Prins, F. Miedema, and H. Schuitemaker.2003. Early viral load and CD4⫹T cell count, but not percentage of CCR5⫹or CXCR4⫹CD4⫹T cells, are associated with R5-to-X4 HIV type 1 virus evolution. AIDS Res. Hum. Retrovir.19:389–398.
67.Wang, R. F., and J. I. Mullins.1995. Mammalian cell/vaccinia virus expres-sion vectors with increased stability of retroviral sequences in Escherichia coli: production of feline immunodeficiency virus envelope protein. Gene
153:197–202.
68.Westby, M., M. Lewis, J. Whitcomb, M. Youle, A. L. Pozniak, I. T. James, T. M. Jenkins, M. Perros, and E. van der Ryst.2006. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol.
80:4909–4920.
69.Whitcomb, J. M., W. Huang, S. Fransen, K. Limoli, J. Toma, T. Wrin, C. Chappey, L. D. Kiss, E. E. Paxinos, and C. J. Petropoulos.2007. Develop-ment and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Anti-microb. Agents Chemother.51:566–575.
70.Wilke, C. O., J. L. Wang, C. Ofria, R. E. Lenski, and C. Adami.2001. Evolution of digital organisms at high mutation rates leads to survival of the flattest. Nature412:331–333.
71.Wilkin, T. J., Z. Su, D. R. Kuritzkes, M. Hughes, C. Flexner, R. Gross, E. Coakley, W. Greaves, C. Godfrey, P. R. Skolnik, J. Timpone, B. Rodriguez, and R. M. Gulick. 2007. HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin. Infect. Dis.44:591–595. 72.Wolinsky, S. M., C. M. Wike, B. T. Korber, C. Hutto, W. P. Parks, L. L.
Rosenblum, K. J. Kunstman, M. R. Furtado, and J. L. Munoz.1992. Selec-tive transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science255:1134–1137.
73.Yi, Y., F. Shaheen, and R. G. Collman.2005. Preferential use of CXCR4 by R5X4 human immunodeficiency virus type 1 isolates for infection of primary lymphocytes. J. Virol.79:1480–1486.
74.Yi, Y., A. Singh, F. Shaheen, A. Louden, C. Lee, and R. G. Collman.2003. Contrasting use of CCR5 structural determinants by R5 and R5X4 variants within a human immunodeficiency virus type 1 primary isolate quasispecies. J. Virol.77:12057–12066.
75.Zhang, L. Q., P. MacKenzie, A. Cleland, E. C. Holmes, A. J. Brown, and P. Simmonds.1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J. Virol.67:3345–3356.
76.Zhang, M., B. Gaschen, W. Blay, B. Foley, N. Haigwood, C. Kuiken, and B. Korber.2004. Tracking global patterns of N-linked glycosylation site varia-tion in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology14:1229–1246.
77.Zhu, T., H. Mo, N. Wang, D. S. Nam, Y. Cao, R. A. Koup, and D. D. Ho.1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science261:1179–1181.
![FIG. 6. Coreceptor (CoR) preference results for Env clones from year 5.67 p.i. (A) Entry of Env-pseudotyped viruses into MT-2.CD4.CCR5hirelative light units [RLU]) versus U87.CD4.CXCR4 target cells (log CXCR4 RLU)](https://thumb-us.123doks.com/thumbv2/123dok_us/159564.36708/7.585.138.467.80.621/coreceptor-preference-results-entry-pseudotyped-viruses-hirelative-versus.webp)