0022-538X/07/$08.00⫹0 doi:10.1128/JVI.02075-06
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Capsid Protein of Eastern Equine Encephalitis Virus
Inhibits Host Cell Gene Expression
䌤
Patricia V. Aguilar,
1Scott C. Weaver,
2,3and Christopher F. Basler
1*
Department of Microbiology, Mount Sinai School of Medicine, New York, New York 10029,1and
Center for Biodefense and Emerging Infectious Diseases2and Department of Pathology,3
University of Texas Medical Branch, Galveston, Texas 77555-0609
Received 21 September 2006/Accepted 18 January 2007
Eastern equine encephalitis virus (EEEV) causes sporadic but often severe cases of human and equine neurological disease in North America. To determine how EEEV may evade innate immune responses, we screened individual EEEV proteins for the ability to rescue the growth of a Newcastle disease virus expressing green fluorescent protein (NDV-GFP) from the antiviral effects of interferon (IFN). Only expression of the EEEV capsid facilitated NDV-GFP replication. Inhibition of the antiviral effects of IFN by the capsid appears to occur through a general inhibition of cellular gene expression. For example, the capsid inhibited the expression of several reporter genes under the control of RNA polymerase II promoters. In contrast, capsid did not inhibit expression from a T7 RNA polymerase promoter construct, suggesting that the inhibition of gene expression is specific and is not a simple manifestation of toxicity. The inhibition correlated both with capsid-induced phosphorylation of eukaryotic initiation factor 2 alpha and with capsid-mediated inhibition of cellular mRNA accumulation. Mapping analysis identified the N terminus as the region important for the inhibition of host gene expression, suggesting that this inhibition is independent of capsid protease activity. Finally, when cell lines containing EEEV replicons encoding capsid were selected, replicons consistently acquired mutations that deleted all or part of the capsid, for example, amino acids 18 to 135. Given that the amino terminus of the capsid is required to inhibit host cell gene expression, these data suggest that capsid expression from the replicons is ultimately toxic to host cells, presumably because of its ability to inhibit gene expression.
Eastern equine encephalitis virus(EEEV), a mosquito-borne
member of theAlphavirusgenus of theTogaviridaefamily, is among the deadliest of the mosquito-borne viruses. In humans, the fatality rate following symptomatic infection approaches 80%, and many survivors develop crippling sequelae, such as mental retardation, convulsions, and paralysis (18, 37).
EEEV possesses a single-stranded, positive-sense RNA ge-nome of approximately 11.7 kb. The four viral nonstructural proteins (nsP1-4) are produced from the full-length genomic RNA as a polyprotein and carry out functions important for viral RNA synthesis and polyprotein processing. A second polyprotein, translated from the 26S subgenomic mRNA, gives rise to the three major structural proteins, namely, the capsid and the two envelope proteins, E1 and E2. The envelope pro-teins are involved in receptor recognition, virus attachment, penetration, and membrane fusion during viral entry. These proteins also cooperate with the capsid during viral assembly and release. Based on observations made with the capsids of other alphaviruses, the EEEV capsid is expected to encapsu-late the viral genomic RNA, to interact with the viral glyco-proteins during assembly, and to function as a protease to release itself from the nascent structural polypeptide chain (54, 63, 65).
The alpha/beta interferon (IFN-␣/) system plays a major role in early innate immune responses and constitutes the first line of defense against viral infection. Production of IFN-␣/is mediated by the activation of transcription factors, such as NF-B, ATF-2/c-Jun, and interferon regulatory factors (IRFs). IFN-␣/ released from cells binds to the IFN-␣/ receptor, activating a Jak-STAT signaling cascade that leads to the phos-phorylation and activation of STAT-1 and STAT-2 transcrip-tion factors. Activatranscrip-tion (phosphorylatranscrip-tion) of STAT-1 and STAT-2 leads to their heterodimerization, translocation to the nucleus, and association with p48/IRF-9 to form a complex that binds DNA sequences containing interferon-stimulated response elements and promotes the induction of specific gene expression (5, 29). IFN-␣/regulates the transcription of nu-merous genes, a number of which encode antiviral proteins, and leads to the induction of an antiviral state in cells.
Due to the importance of the IFN response as an antiviral defense, many viruses have developed mechanisms to inhibit this response by means of blocking or inhibiting IFN produc-tion, IFN-mediated signaling, and/or the activity of IFN-in-duced gene products (3, 12, 26). For example, the influenza A virus NS1 protein has been shown to suppress the initiation of the IFN response (23, 49, 53, 66, 67, 74). In another example, the highly virulent Ebola virus carries two IFN antagonist pro-teins: VP35 blocks IFN production, and VP24 blocks IFN signaling. The ability of these proteins to inhibit different steps in the IFN response likely plays a role in the extreme virulence of Ebola virus (6, 11, 32, 33, 59).
Among the positive-strand RNA viruses, some members of * Corresponding author. Mailing address: Department of
Microbi-ology, Box 1124, Mount Sinai School of Medicine, 1 Gustave L. Levy Place, New York, NY 10029. Phone: (212) 241-4847. Fax: (212) 534-1684. E-mail: chris.basler@mssm.edu.
䌤Published ahead of print on 31 January 2007.
3866
on November 8, 2019 by guest
http://jvi.asm.org/
theFlaviviridaefamily have been shown to carry proteins that block the IFN response. For example, the hepatitis C virus NS3-4A protease inhibits signals that lead to IFN-␣/ expres-sion (9, 20, 42, 43). In addition, other flaviviruses inhibit IFN-induced Jak-STAT signaling. For example, the dengue virus nonstructural protein NS4B strongly inhibits IFN signaling, whereas NS4A and NS2A partially block IFN signaling (14, 38, 50, 51). In contrast, the flaviviruses Japanese encephalitis virus and Langat virus use the NS5 protein to block IFN signaling, suggesting that viruses from the same family that cause differ-ent spectra of human disease may use differdiffer-ent mechanisms to overcome the IFN response (7, 44, 45).
Studies with alphaviruses have demonstrated the importance of the IFN system in controlling infection and disease (1, 22, 30, 60, 71, 73). Mice with a defect in the IFN-␣/ response succumb faster to infections with several alphaviruses than do wild-type mice (30, 60, 73). However, it remains unknown whether alphaviruses, specifically EEEV, carry proteins that inhibit the interferon response.
Previous studies with Sindbis virus (SINV), an Old World alphavirus, suggest that nsP2 is important for the suppression of the antiviral response, including the production of IFN-␣/ in infected cells (22). Whether the same applies to the New World encephalitic alphaviruses (i.e., EEEV and Venezuelan equine encephalitis virus), which cause a different and more severe spectrum of clinical disease in humans than that of the Old World viruses (65), has not been determined. Because of the evolutionary, genetic, biological, and human disease differ-ences between the New World and Old World alphaviruses, we hypothesized that the mechanisms by which Old and New World alphaviruses evade the innate immune response might also differ. We therefore sought to identify specific EEEV proteins with IFN antagonist properties, anticipating that such factors will play an important role in EEEV pathogenesis. Our results indicate that the EEEV capsid functions as a potent IFN antagonist protein by globally inhibiting host cell gene expression. Mechanistically, we demonstrate that the capsid protein inhibits the expression of RNA polymerase II-tran-scribed genes and also promotes phosphorylation of the eu-karyotic initiation factor 2␣(eIF2␣) subunit, suggesting cap-sid-mediated effects upon translation as well.
(Portions of the results of this study were presented at the 2006 National RCE Meeting, New York, NY, March 2006, and the American Society of Virology Meeting, Madison, WI, July 2006.)
MATERIALS AND METHODS
Cells and viruses.293T and A549 cells were maintained in Dulbecco’s mod-ified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). BSRT7 cells expressing the T7 RNA polymerase were maintained in DMEM-10% FBS with 400g/ml of G418. A Newcastle disease virus expressing green fluorescent protein (NDV-GFP) was previously described (55).
Construction of plasmids.The EEEV strain FL93-939, isolated from a mos-quito pool in Florida in 1993 and passaged once in Vero cells and once in a suckling mouse brain, was used for reverse transcription-PCR (RT-PCR) and cDNA cloning. Structural and nonstructural proteins were cloned into the pCAGGS mammalian expression plasmid and tagged with hemagglutinin (HA) in frame at the N terminus. The pCAGGS plasmid has been described previously (52), and it contains a chicken-actin promoter for strong transient expression of the inserted gene and a-globin poly(A) signal. For cloning of the individual nonstructural protein genes, the proteolytic cleavage sites in the nonstructural polyprotein genes were chosen as boundaries (65), and a stop codon was intro-duced at the end of each gene. For cloning of the structural proteins, a cassette
carrying the complete structural polyprotein gene was constructed, as well as a plasmid encoding the capsid protein and carrying the E3 through E1 genes. To map the specific region in the capsid protein able to inhibit gene expression, the coding regions for the N terminus (amino acids [aa] 1 to 126) and C terminus (aa 127 to 261) were also cloned into the pCAGGS mammalian expression plasmid. Expression of the proteins was confirmed by Western blotting, using antibodies against the HA tag (Sigma, St. Louis, MO). The pM1 plasmid (4) was used in this study to clone the firefly luciferase gene under the control of the T7 promoter. This low-copy-number, ampicillin-resistant plasmid (a modified version of the pBR322 vector) was selected because it lacks a T7 promoter, allowing us to introduce this promoter immediately upstream of the firefly luciferase gene. Also, a T7 terminator sequence was introduced at the end of the luciferase gene. Construction of the PR8 NS1 plasmid has been reported previously (66).
Construction of EEEV replicons.Schematic representations of the EEEV rep-licons generated in this study are shown in Fig. 7A. Maps and sequences are available upon request. To produce cDNA templates for RNA synthesis, plasmids were linearized using the NotI site located downstream of the poly(A) tail. In vitro transcription was performed by using an mMessage mMachine T7 kit (Ambion, Austin, TX) following the manufacturer’s protocol. In vitro-transcribed RNA was electroporated into BHK cells as previously described (4), and 1 day after electro-poration, the medium was replaced with DMEM-10% FBS containing 12.5g of puromycin. Single puromycin-resistant cells were isolated and expanded, and RNAs were extracted from the cells for PCR and sequencing purposes. Cells were lysed in reporter lysis buffer, and lysates were assayed for luciferase activity following the manufacturer’s protocol (Promega, Madison, WI).
NDV-GFP complementation assay.An NDV-GFP complementation assay was performed as previously described (55). Briefly, A549 cells were seeded into 24-well plates, and the next day the medium was replaced with 0.25 ml of DMEM with 10% FBS. Cells were transfected with 2g of plasmid DNA previously diluted to 50l in OptiMEM (Gibco, Carlsbad, CA). The plasmid was combined with 2l of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) diluted in 50l of OptiMEM, and the mixture was incubated at room temperature for 25 min and then added to the cells. Cells were incubated overnight at 37°C in 5% CO2, and
after incubation the plate was washed twice with phosphate-buffered saline, infected with NDV-GFP at a multiplicity of infection (MOI) of 1 to 2, and incubated at 37°C for 1 h. The virus inoculum was removed from the cells, and 0.5 ml of DMEM-10% FBS was added to the cells. The infected cells were incubated for 20 h at 37°C in 5% CO2prior to detection of green fluorescence
by microscopy.
Reporter gene assays.Reporter assays were performed as previously described (11). To determine whether the EEEV proteins block IFN induction, 293T cells (⬃1⫻106
cells) were transfected with 1g of expression plasmid DNA together with an IFN- promoter–chloramphenicol acetyltransferase (CAT) reporter (pIFN-CAT) and the indicated firefly luciferase plasmids, using the calcium phosphate mammalian transfection method (Stratagene, La Jolla, CA). One day after transfection, the cells were infected with Sendai virus at an MOI of⬃8 and incubated for 1 h at 37°C in 5% CO2. After incubation, the virus inoculum was
removed, 1 ml of DMEM-10% FBS was added to the cells, and the plates were incubated at 37°C in 5% CO2for 24 h. The cells were then lysed with reporter
lysis buffer (Promega). CAT activities were determined by standard methods (61), and luciferase activities were determined according to the manufacturer’s protocol (Promega). Firefly luciferase activity was used as a transfection control and to normalize the CAT activity where indicated. The results are represented asx-fold induction relative to the activity of an uninduced, empty vector-trans-fected control. To determine whether the EEEV proteins affect IFN signaling, Vero cells were transfected with 1g of expression plasmid together with an IFN-␣/-responsive ISG54 promoter-CAT reporter (kindly provided by David Levy, NYU) and a firefly luciferase plasmid, using Lipofectamine 2000 (Invitro-gen). One day after transfection, the cells were washed once with phosphate-buffered saline and then maintained in 10% DMEM containing 1,000 IU/ml of human IFN-(PBL, Piscataway, NJ). Mock controls were maintained in 10% DMEM without IFN. One day after treatment, cells were lysed by using a reporter lysis buffer, and the CAT and luciferase activities were determined. Firefly luciferase activity was used as a transfection control and to normalize the CAT activity. The results are shown asx-fold induction relative to the activity of an uninduced, empty vector-transfected control.
Transfection of the EEEV capsid with reporter plasmids.293T cells were transfected with 1g of a GFP or firefly luciferase pCAGGS mammalian ex-pression plasmid (whose exex-pression is driven by the RNA polymerase II pro-moter) in the presence or absence of the plasmid encoding the capsid protein. Twenty-four hours later, cells transfected with the firefly luciferase plasmid were lysed with reporter lysis buffer following the manufacturer’s protocol, and the firefly luciferase activity was measured in the cell lysates. Cells transfected with
on November 8, 2019 by guest
http://jvi.asm.org/
a GFP plasmid were observed under a fluorescence microscope for a GFP signal. In another set of experiments, BSRT7 cells were transfected with 1g of a firefly luciferase pCAGGS plasmid (under the control of an RNA polymerase II pro-moter) or 1g of a firefly luciferase pM1 plasmid (under the control of the T7 promoter) in the presence or absence of the plasmid carrying the capsid gene. Twenty-four hours later, cells were lysed and luciferase activity was measured as described above.
Detection of phosphorylated eIF2␣.Cell lysates were separated by 10% so-dium dodecyl sulfate-polyacrylamide gel electrophoresis under reducing condi-tions. After electrophoresis, proteins were transferred to a polyvinylidene difluo-ride membrane, blocked for 1 h in 5% nonfat dry milk dissolved in Tris-buffered saline, and then probed with antibodies against phospho-eIF2␣(serine 52) (Bio-source, Camarillo, CA), eIF2␣ (Biosource), or ␣-tubulin (Sigma, St. Louis, Missouri). Secondary antibodies conjugated to horseradish peroxidase and a chemiluminescence detection system from Perkin-Elmer (Wellesley, MA) were used to visualize the proteins. To determine the levels of eIF2␣phosphorylation in EEEV-infected cells, 293T cells were infected with EEEV strain FL93-939 (MOI⫽3), and supernatants and cell lysates were obtained at 5 and 24 h postinfection (p.i.). Cell lysates were then assayed for eIF2␣as described above, and the supernatants were assayed for virus titer by a plaque assay in Vero cells as previously described (1).
Quantitative RT-PCR.Quantitative RT-PCR was performed by using a pre-viously published SYBR green protocol with an ABI7900 HT thermal cycler (75). Briefly, 293T cells were transfected with 1g of PR8 NS1 plasmid together with 0, 0.25, 0.5, or 1g of the EEEV capsid plasmid. One day after transfection, proteins and RNAs were extracted simultaneously from the cells by using a PARIS kit (Ambion) following the manufacturer’s protocol. RNAs were treated with DNA-free(Ambion) following the manufacturer’s protocol, and 2g of RNA was used for cDNA synthesis, using 50M of oligo(dT)20primers. To
determine the effect of capsid on the endogenous, IFN-inducible ISG54 and STAT1 genes, 293T cells were transfected with 1g of empty plasmid or plasmid encoding full-length capsid, the capsid N terminus (aa 1 to 126), or the capsid C terminus (aa 127 to 261), and 24 h after transfection, the cells were treated with 1,000 IU of IFN-(PBL Biomedical Laboratories). Twelve hours after treat-ment, RNAs were extracted from the cells by using an RNeasy mini kit (QIAGEN, Valencia, CA) following the manufacturer’s protocol. RNAs were treated with DNA-free(Ambion) following the manufacturer’s protocol, and 1 g of RNA was used for cDNA synthesis using 50M of oligo(dT)20primers. To
quantify the data, a robust global normalization algorithm was used. Each tran-script in each sample was assayed three times, and the median cycle threshold (CT) values were used to calculate the transcript copy number by using the following formula: transcript copy number⫽2,500⫻1.93(HKi⫺TGi)
, where 2,500 is an empirical estimate of the number of actin mRNAs/cell (39), 1.93 reflects a typical reaction amplification efficiency of 93%, HKi is the housekeep-ing gene sample’s medianCTvalue, and TGi is the target CTvalue. Three housekeeping genes were used for global normalization in each experiment (actin, Rps11, and tubulin genes). For Western blot analysis, rabbit NS1 poly-clonal and mouse HA monopoly-clonal antibodies were used.
Selection of stable cell lines containing EEEV replicons.BHK-21 cells were electroporated with 5g of in vitro-synthesized RNA. Twenty-four hours after electroporation, the medium was replaced with DMEM-10% FBS containing 12.5g/ml of puromycin, and surviving cells were individually selected and expanded under puromycin selection. RNAs were extracted from the stable cell lines, and PCR and sequence analyses were performed to identify potential changes within the replicons.
RESULTS
Expression of EEEV capsid protein counteracts the antivi-ral effects of the IFN response.To identify EEEV genes that inhibit the IFN-␣/ response, we employed a previously de-scribed assay that measures the ability of individual viral pro-teins to complement the replication of NDV-GFP (51, 55). Briefly, A549 cells were transfected with an empty expression plasmid or a plasmid encoding Ebola virus VP35, Nipah virus W, or individual EEEV proteins. At 1 day posttransfection, cells were infected with NDV expressing GFP, and 1 day later, the cells were analyzed for GFP expression. As previously reported, transfection of empty plasmid induced the produc-tion of IFN, and therefore the replicaproduc-tion of NDV-GFP was
inhibited. In contrast, cells transfected with plasmids encoding the known IFN antagonist proteins Ebola virus VP35 and Nipah virus W supported NDV-GFP replication (55). Inter-estingly, when plasmids encoding the EEEV proteins were transfected, only the capsid protein was able to rescue NDV-GFP replication (Fig. 1). To further address the possibility that one of the nonstructural proteins might also influence the IFN-␣/response, the IFN response was also examined in cells stably transfected with EEEV replicons. However, in these cell lines, we did not detect either inhibition of IFN-promoter activation or inhibition of IFN-induced gene expression (data not shown), further supporting the view that the capsid is the primary inhibitor of IFN responses in EEEV-infected cells.
EEEV capsid is a general inhibitor of gene expression.Next, reporter gene-based assays were conducted with the goal of determining whether the capsid inhibits IFN-␣/ production and/or the cellular response to IFN-␣/. 293T cells or Vero cells were transfected with an IFN-promoter-CAT reporter plasmid or an interferon-responsive ISG54 promoter-CAT re-porter plasmid together with a constitutively expressed simian virus 40 promoter-luciferase reporter plasmid which served as a control for transfection efficiency. The 293T cells were then infected with Sendai virus to activate the IFN-promoter, and the Vero cells were treated with 1,000 units/ml IFN-to acti-vate the ISG54 promoter. The results showed that transfection of the capsid plasmid inhibited the expression of both Sendai virus- and IFN--induced reporter genes (Fig. 2A and C). However, when the results of the reporter assay were normal-ized to the luciferase controls, we observed that the capsid protein also down-regulated the activity of the transfection control protein (firefly luciferase), suggesting that capsid may generally inhibit gene expression (Fig. 2B and D). To deter-mine whether the inhibition of the reporters was due to a global inhibition of gene expression, we transfected the capsid together with either luciferase plasmid alone or with a GFP expression plasmid. The results demonstrated that transfection of the capsid inhibited the expression of both luciferase and GFP (Fig. 2E and F, respectively). These results suggest that expression of the capsid protein broadly inhibits the expression of multiple genes expressed from a variety of promoters.
To further characterize the inhibition of gene expression, the capsid was coexpressed in BSRT7 cells (which stably ex-press T7 RNA polymerase) with a plasmid carrying the firefly luciferase gene under the control of either an RNA polymerase II promoter or a T7 promoter. A dose-dependent inhibition of luciferase activity was observed only in cells transfected with the RNA polymerase II-driven promoter plasmid, but this in-hibition was not observed when the luciferase gene was under the control of the T7 promoter (Fig. 3). These data suggest that capsid inhibition is selective and can target RNA polymerase II gene expression. The insensitivity of the T7 promoter plasmid also suggests that the inhibition of gene expression in reporter gene assays was not due to toxic effects of the capsid.
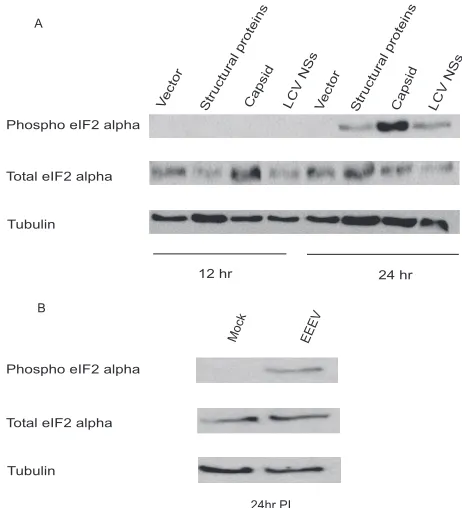
Capsid protein induces phosphorylation of the␣subunit of eIF2.The previous results suggest that the EEEV capsid pro-tein targets cellular mRNA synthesis. However, some alphavi-ruses are known to activate protein kinase R (PKR) and con-sequently induce the phosphorylation of the␣subunit of the translation initiation factor eIF2 (16). Therefore, to determine whether the capsid might induce the phosphorylation of eIF2␣,
on November 8, 2019 by guest
http://jvi.asm.org/
293T cells were transfected with a luciferase reporter plas-mid in the absence or presence of a plasplas-mid encoding the capsid protein, the entire EEEV structural polyprotein, or the LaCrosse virus (LCV) NSs protein, previously described as a general inhibitor of host cell transcription (58). Twelve or 24 h after transfection, cells were lysed and assayed for the presence of phosphorylated eIF2␣. Phosphorylation of eIF2␣ was evi-dent at 1 day posttransfection in cells expressing the EEEV capsid, EEEV structural proteins (including capsid), and LCV NSs but was strongest in cells where the capsid alone was expressed (Fig. 4A). The ability of the capsid to strongly induce eIF2␣ phosphorylation may contribute to the inhibition of gene expression noted above. How the capsid stimulates eIF2␣ phosphorylation remains to be determined. However, it is no-table that LCV NSs, which like the EEEV capsid inhibits mRNA expression (see below), also promotes eIF2␣ phosphor-ylation. To determine whether capsid-induced eIF2␣ phosphor-ylation might be relevant to EEEV infection, 293T cells were infected with EEEV (MOI ⫽ 3), and supernatants and cell lysates were collected at 5 and 24 h p.i. Phosphorylation of eIF2␣was more evident at 24 h p.i. than at 5 h p.i. (Fig. 4B and data not shown) and also correlated with higher virus titers than those at the 5-h time point (8.6⫾0.04 log10versus 5.3⫾
0.07 log10, respectively).
[image:4.585.77.508.68.400.2]Capsid protein inhibits host mRNA accumulation. Al-though inhibition of protein synthesis through the PKR path-way is a hallmark of alphavirus infection, a second described mechanism for the shutdown of host protein synthesis by Old World alphaviruses is the inhibition of host cell transcription (27, 28). Therefore, we evaluated whether the capsid also af-fects RNA levels by transfecting an influenza A/PR/8/34 (H1N1) virus NS1 protein expression plasmid under the con-trol of the RNA polymerase II promoter. We selected the influenza virus NS1 plasmid because of the availability of an established quantitative RT-PCR assay for the NS1 gene and of antibodies for the detection of the NS1 protein by Western blotting. For this experiment, cells were transfected with the NS1 protein expression plasmid in the absence or presence of increasing amounts of capsid expression plasmid, and relative NS1 mRNA copy numbers were determined by quantitative RT-PCR. In the presence of the capsid gene, the PR8 NS1 mRNA copy number was decreased⬎7-fold compared to the control copy number (Fig. 5A), and this reduction in RNA levels correlated with a substantial reduction in NS1 protein levels (Fig. 5B). In conclusion, these results suggest that the capsid inhibits protein synthesis by inhibiting host cell tran-scription.
FIG. 1. Expression of the EEEV capsid protein counteracts the antiviral effects of the IFN response. A549 cells were transfected with empty plasmid or a plasmid encoding GFP (as a control for transfection efficiency), Ebola virus VP35, Nipah virus W, or the indicated EEEV proteins, and 1 day later, cells were infected with NDV-GFP. Only plasmids expressing Ebola virus VP35, Nipah virus W, and EEEV capsid rescued NDV-GFP replication. The results shown are representative of at least three different experiments. The NDV-GFP panel shows replication of this virus in untransfected cells.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 2. EEEV capsid is a general inhibitor of gene expression. (A and B) Vero cells were cotransfected with the ISG54 promoter-CAT reporter plasmid, a constitutively expressed luciferase reporter plasmid, and empty vector or a plasmid expressing EEEV capsid or Nipah virus V protein. At 24 hours posttransfection, the cells were mock treated or treated with 1,000 U human IFN-/ml for 24 h. Cells were harvested and then assayed for CAT and luciferase activities. (A) Nonnormalized CAT activity, with the untreated empty vector control value set to 1.
on November 8, 2019 by guest
http://jvi.asm.org/
The N terminus of the EEEV capsid mediates the inhibition of gene expression.To map the specific area of the capsid gene required for the inhibition of protein synthesis, the coding regions for the N-terminal (aa 1 to 126) and C-terminal (aa 127 to 261) regions of the capsid protein were cloned into a mam-malian expression plasmid and transfected into 293T cells along with a GFP or luciferase reporter plasmid. A decrease in GFP and luciferase expression was observed only in cells trans-fected with the N-terminal half of the capsid protein, whereas the C-terminal half, which contains the protease active site, did not produce a significant decrease in the expression of the reporter genes (Fig. 6A and B). Consistent with this observa-tion, only the N-terminal half of the capsid was able to induce the phosphorylation of eIF2␣(Fig. 6C). To determine whether the effects of the capsid extend to endogenous gene expression, empty vector or plasmids encoding full-length capsid, the N terminus, and the C terminus were transfected into 293T cells, and the relative mRNA copy numbers of the ISG54 and STAT-1 genes were measured by quantitative RT-PCR after IFN treatment. In the presence of the full-length capsid or the capsid N terminus, the relative copy numbers of ISG54 and STAT-1 decreased two- and fourfold, respectively, compared
to those of the control, whereas the effect of overexpressing the C-terminal half of the capsid had less of an effect on mRNA accumulation (Fig. 6D). It should be noted that these experi-ments, which rely upon transient transfection and leave some percentage of cells untransfected, likely underestimate the ef-fects of capsid on endogenous gene expression. When the expression of each of the capsid constructs was analyzed by Western blotting (for the HA tag), the carboxy-terminal half was expressed at much higher levels than either the full-length capsid or the amino-terminal half, possibly reflecting the ability of the latter two constructs to inhibit gene expression (data not
[image:6.585.304.535.68.322.2](B) Relative CAT activity upon normalization to the luciferase control. (C and D) 293T cells were cotransfected with the IFN-promoter–CAT reporter plasmid, a constitutively expressed luciferase reporter plasmid, and empty vector or a plasmid expressing EEEV capsid, Ebola virus VP35, or Nipah virus W. At 24 hours posttransfection, the cells were mock infected or infected with Sendai virus (SeV) for 24 h. Cells were harvested and then assayed for CAT and luciferase activities. (C) Nonnormalized CAT activity, with the untreated empty vector control value set to 1. (D) CAT activity upon normalization to the luciferase control. The results are representative examples from a set of at least three separate experiments. (E) 293T cells were cotransfected with a luciferase reporter plasmid and either empty vector or a plasmid expressing EEEV capsid. At 24 h posttransfection, the cells were harvested and assayed for luciferase activity. The data represent the means⫾standard errors among samples from three separately transfected wells. The experiment was repeated at least twice, with consistent results. (F) An experiment was performed as described for panel E, except that a plasmid expressing GFP was used and cells were examined by microscopy for GFP expression. The experiment was repeated at least twice, with consistent results.
FIG. 3. Expression of EEEV capsid inhibits RNA polymerase II-directed gene expression. BSRT7 cells were cotransfected with a firefly luciferase reporter plasmid expressed from an RNA polymerase II promoter (in the plasmid pCAGGS) (52) or from a T7 promoter and with increasing concentrations of a plasmid expressing EEEV capsid. At 24 h posttransfection, the cells were harvested and assayed for luciferase activity. Average relative luciferase activities are reported, and the no-capsid control value was set to 100%. The error bars represent standard errors among samples from three separately trans-fected wells. The experiment was repeated at least twice, with consis-tent results.
FIG. 4. Expression of EEEV capsid protein induces phosphoryla-tion of the␣subunit of eIF2. (A) 293T cells were cotransfected with a luciferase reporter plasmid and empty vector (vector) or a plasmid expressing EEEV capsid, EEEV structural proteins, or the LCV NSs protein. Twelve or 24 h after transfection, the cells were harvested, and Western blotting was performed to detect the phosphorylation of eIF2␣, total eIF2␣, and␣-tubulin as a loading control. Reductions in luciferase activity were observed for cells transfected with the capsid, the EEEV structural proteins, and LCV NSs (data not shown). (B) 293T cells were mock infected or infected with EEEV strain FL93-939 (MOI⫽3), and at 5 and 24 h p.i., cells were lysed and Western blotting was performed to detect the phospho-eIF2␣, total eIF2-␣, and␣-tubulin as a loading control.
on November 8, 2019 by guest
http://jvi.asm.org/
shown). These results suggest that the inhibition of protein synthesis mediated by the EEEV capsid is encoded within the amino-terminal half of the protein and is independent of its protease function.
An EEEV replicon harboring the capsid gene inhibits pro-tein synthesis.We finally turned to EEEV replicons to deter-mine whether an untagged EEEV capsid expressed in the context of EEEV nonstructural proteins would inhibit the ex-pression of endogenous cellular mRNAs and proteins. Two EEEV replicons harboring the EEEV nonstructural protein gene region, a luciferase gene, and the puromycin resistance gene were constructed. One of the replicons also expressed the capsid gene as a direct fusion to the luciferase gene such that capsid protease activity would cleave and separate the capsid from luciferase (Fig. 7A). BHK cells were electroporated with equal amounts of in vitro-transcribed replicon RNA, and 24 h after electroporation, selection with puromycin was applied. Interestingly, cells electroporated with replicon RNA without the capsid gene formed colonies more efficiently than did rep-licon-infected cells with the capsid gene (data not shown). Also, it was evident that the replicon with the capsid gene was more cytopathic than the replicon without the capsid gene, making it more difficult to establish persistent replication and suggesting that expression of the capsid gene has a profound effect in the host cell (data not shown). Sequence analysis of stable cell lines electroporated with the replicon harboring the capsid gene revealed a partial or complete deletion of the capsid gene from the replicon RNA a few weeks after selec-tion, further supporting our findings that expression of the
capsid gene was detrimental for the cells (Fig. 7B). One dele-tion mutant that was recovered from a stable cell line con-tained a deletion of aa 18 to 135. Given that the amino termi-nus of the capsid is required to inhibit host cell gene expression, these data suggest that, ultimately, capsid expres-sion from replicons is toxic to host cells, presumably because of its ability to inhibit gene expression.
DISCUSSION
Several viral mechanisms for the inhibition of the IFN re-sponse have been reported. However, to date, the mechanisms by which New World encephalitic alphaviruses inhibit the IFN response have not been described. Most of our knowledge about alphaviruses comes from studies with SINV and Semliki Forest virus (SFV), which cause a different spectrum of clinical disease in humans than the New World encephalitic viruses. Specifically, studies with SINV suggested that the nsP2 protein is involved in inhibiting the IFN response by interfering with host protein synthesis at the levels of transcription and trans-lation (22, 24, 27). During our attempts to study viral proteins involved in the inhibition of the IFN response by the New World encephalitic viruses, specifically EEEV, we identified the capsid protein as a potent IFN antagonist. Interestingly, overexpression of the EEEV nsP2 protein did not show signif-icant inhibition of the antiviral IFN response, suggesting that the strategies used by the Old World and New World alpha-viruses to overcome the IFN response are different. Similar to previous reports for nsP2 of SINV (24, 27), the inhibition of the IFN response by EEEV capsid was mediated by a global shutdown of host protein synthesis.
[image:7.585.80.246.66.287.2]Previous studies have defined roles for alphavirus capsid proteins in proteolytic cleavage of the structural polyprotein, RNA encapsidation, nucleocapsid formation, and binding to ribosomes to facilitate the uncoating process during viral rep-lication (25, 35, 40, 46, 56, 62, 64, 68, 69, 72). In addition, it was reported that the capsid of SFV induces phosphorylation of PKR, leading to inhibition of protein synthesis (16). Therefore, we first investigated whether the EEEV capsid might induce phosphorylation of the␣subunit of eIF2, which would suggest an activation of PKR and a means of globally inhibiting protein synthesis. Overexpression of the EEEV capsid gene in 293T cells induced the phosphorylation of eIF2␣. However, attempts to establish persistent replication of an EEEV replicon har-boring the capsid gene in PKR⫺/⫺mouse embryo fibroblasts were unsuccessful (data not shown), suggesting that the ab-sence of PKR does not relieve the toxic effects of the capsid-expressing replicon. Therefore, it seems possible that other mechanisms are involved in the inhibition of host protein syn-thesis. Thus, we sought to determine whether the capsid gene also inhibits the accumulation of host cell mRNAs, which would be a function analogous to that of the Old World al-phavirus nsP2 proteins (24, 27). Cotransfection of the capsid inhibited the expression of reporter genes driven by RNA polymerase II but not by the T7 reporter construct, suggesting a mechanism whereby the capsid targets some aspect of RNA polymerase II synthesis or induces host cell RNA degradation. This result also suggests that capsid-induced inhibition of re-porter gene expression is not simply due to toxic effects of capsid overexpression.
FIG. 5. Expression of EEEV capsid inhibits host mRNA accumula-tion. 293T cells were cotransfected with a plasmid expressing the influenza A/PR8/34 (H1N1) virus NS1 protein and either empty vector or increas-ing amounts of a plasmid expressincreas-ing EEEV capsid. At 24 h posttransfec-tion, RNAs were extracted from the cells for quantitative real-time RT-PCR (A), and cell lysates were used for Western blot analyses of NS1, HA capsid, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control (B). The data represent means⫾standard errors among samples from two independently transfected wells. The experiment was repeated twice and gave consistent results.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 6. The N terminus of the EEEV capsid mediates the inhibition of gene expression. (A) 293T cells were cotransfected with a plasmid expressing the GFP reporter and either empty vector or a plasmid expressing the N terminus (aa 1 to 126) or the C terminus (aa 127 to 261) of the EEEV capsid. At 24 h posttransfection, cells were examined by microscopy for GFP expression. (B) The experiment was performed as described for panel A, except that a luciferase reporter plasmid was used and the cells were harvested and assayed for luciferase activity. (C) 293T cells were cotransfected with a luciferase reporter plasmid and empty vector (vector) or a plasmid expressing EEEV full-length capsid or the N terminus (aa 1 to 126) or C terminus (aa 127 to 261) of the capsid. Twenty-four hours after transfection, the cells were harvested and Western blotting was performed to detect the phosphorylation of eIF2␣, total eIF2␣, and␣-tubulin as a loading control. (D) 293T cells were transfected with empty plasmid or with a plasmid encoding the full-length capsid or the N-terminal or C-terminal half of the capsid. At 24 h posttransfection, the cells were treated with human IFN-, and 12 h after treatment, RNAs were extracted from the cells for quantitative real-time RT-PCR of STAT-1 and ISG54 mRNAs. The data represent the means⫾standard errors among samples from three wells in two experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
To further determine the effects of untagged capsid in the context of viral replication, we evaluated host protein synthesis of EEEV replicons, with and without the capsid gene. As previously reported for other EEEV replicons (57), no adap-tive viral mutations were identified in our EEEV replicon-bearing cells that did not carry capsid. However, only the EEEV replicon without the capsid gene was readily able to establish persistent replication in IFN-deficient cells, whereas capsid-containing replicons that did yield foci contained a com-plete or partial deletion of the N-terminal half of the capsid. This observation is reminiscent of previous findings with SINV replicons, in which adaptive mutations in nsP2 that reduced the cytopathic effect caused by this protein were required to establish persistent replication (21, 22, 24). These data support the conclusion that EEEV capsid is a general inhibitor of host cell gene expression. They also suggest that the capsid may contribute to cell death during the course of EEEV replication. Our results demonstrate that the expression of EEEV capsid in cells is sufficient to counteract the antiviral effects of IFN. They therefore suggest a novel mechanism by which EEEV inhibits the IFN response, through capsid-mediated suppres-sion of host cell gene expressuppres-sion, and suggest an additional and important function for the capsid protein in viral pathogenesis. These results also identify the N-terminal region of the capsid
[image:9.585.131.454.72.365.2]as responsible for this unique function, providing evidence that this function is protease independent. In this regard, EEEV appears to evade IFN responses in a manner different from hepatitis C virus, where the NS3-4A protease cleaves IPS-1 (13, 34, 42, 48) and TRIF (43) to inhibit activation of IRF3. Instead, this mechanism is similar to that proposed for the Old World alphavirus SINV, where nsP2 acts as a general inhibitor of gene expression (24, 27). However, further studies are nec-essary to map specific residues involved in the inhibition of host gene expression. The N terminus of the EEEV capsid is positively charged (rich in lysine and arginine residues) and also rich in proline and is believed to be important for protein-protein interactions and encapsidation of viral RNA (65). Whether these positively charged residues play a role in the capsid-mediated inhibition of the cellular response, perhaps by specifically binding host proteins or host RNAs or by localizing the capsid protein in specific cell compartments, needs to be determined. Interestingly, the capsid protein of SFV (an Old World alphavirus) has been suggested to have two nuclear localization signals (17), and computer-based analysis has identified potential nuclear localization signals within the N-terminal half of the EEEV capsid. Further experiments should explore whether the EEEV capsid localizes to the nucleus and/or sites of mRNA translation and whether localization is FIG. 7. The capsid gene is mutated in stably selected EEEV replicons. (A) Schematic representation of the EEEV replicons constructed for this study. nsP1 to nsP4, genes for nonstructural proteins 1 to 4; Luc, luciferase gene; PAC, puromycin acetyltransferase gene (encoding puromycin resistance); C, capsid gene; arrows with 26S, 26S subgenomic promoters. (B) BHK cells were electroporated with equal amounts of in vitro-transcribed replicon RNA, and 12 h after electroporation, puromycin selection was applied to the cells. Surviving cells were expanded, and RNAs were extracted from individual clones for RT-PCR amplification and sequence analysis. The figure shows the deletions found in the capsid and luciferase genes in the stable cell lines containing the EEEV replicons with the capsid gene. EEE rep capsid, EEEV replicon encoding capsid; EEE rep, EEEV replicon lacking capsid. The gray areas indicate the deleted regions in the stable capsid replicon cell lines.
on November 8, 2019 by guest
http://jvi.asm.org/
important for capsid inhibition of gene expression. Further studies should also be carried out to evaluate the importance of eIF2␣ phosphorylation in capsid inhibition of protein ex-pression, perhaps by using cells expressing a nonphosphory-lated form of eIF2␣. Such experiments will clarify whether or not the inhibition of host cell mRNA accumulation and eIF2␣ phosphorylation separately contribute to capsid-mediated in-hibition of gene expression or whether phosphorylation of eIF2␣occurs as a consequence of the inhibition of host cell gene expression. It will also be of interest to determine whether capsid that enters the host cell with incoming viral particles may contribute to suppression of host cell gene ex-pression.
The inhibition of protein synthesis as a way to inhibit the IFN response by alphaviruses is a mechanism shared with several other arboviruses, including members of theBunyaviridaefamily (70). LCV and Bunyamwera and Rift Valley fever viruses have previously been shown to inhibit the IFN response by mediating inhibition of host protein synthesis (8, 10, 36, 70). Mechanistically, they interfere with RNA polymerase II transcription (8, 41, 70). Interestingly, vesicular stomatitis virus, which is also transmitted by insects, also inhibits IFN responses by globally targeting host cell gene expression (2, 15, 19).
These observations contrast with those made recently for arthropod-borne flaviviruses, such as West Nile and dengue viruses, which target specific components of the IFN induction or signaling pathway (7, 31, 47, 50, 51). Further experiments are needed to define the exact mechanism by which New World alphaviruses inhibit cellular transcription and the full consequences of this inhibition for viral replication.
Our results raise interesting questions regarding Old World and New World alphavirus pathogenesis and the different means of inhibiting gene expression. It will be of interest to determine whether the capsid protein is used to inhibit host protein synthesis by all New World alphaviruses or whether this property correlates with a given virus’s ability to cause encephalitis. Future studies using capsid and nsP2 constructs de-rived from other alphaviruses, including Venezuelan equine en-cephalitis virus and Western equine enen-cephalitis virus, are neces-sary to address this question. This study also provides important information about the specific mechanisms used by EEEV to evade the host response, and more importantly, we present evi-dence of a novel mechanism for the capsid protein in viral patho-genesis. This information may provide insights that facilitate the development of effective measures to control EEEV infection.
ACKNOWLEDGMENTS
This study was supported by NIH grants to C.F.B., including grant U54 AI057158 (Northeast Biodefense Center-Lipkin), and by a grant to S.C.W. through the Western Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research. P.V.A. was supported by a fellowship awarded through grant AI057158 (Northeast Biodefense Center-Lipkin).
We thank Robert Tesh (University of Texas Medical Branch) for providing reagents and Larry Leung (Mount Sinai School of Medicine) for critically reading the manuscript.
REFERENCES
1.Aguilar, P. V., S. Paessler, A. S. Carrara, S. Baron, J. Poast, E. Wang, A. C. Moncayo, M. Anishchenko, D. Watts, R. B. Tesh, and S. C. Weaver.2005. Variation in interferon sensitivity and induction among strains of Eastern equine encephalitis virus. J. Virol.79:11300–11310.
2.Ahmed, M., M. O. McKenzie, S. Puckett, M. Hojnacki, L. Poliquin, and D. S. Lyles.2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol.77:4646–4657. 3.Alcami, A., and U. H. Koszinowski.2000. Viral mechanisms of immune
evasion. Immunol. Today21:447–455.
4.Anishchenko, M., S. Paessler, I. P. Greene, P. V. Aguilar, A. S. Carrara, and S. C. Weaver. 2004. Generation and characterization of closely related epizootic and enzootic infectious cDNA clones for studying interferon sen-sitivity and emergence mechanisms of Venezuelan equine encephalitis virus. J. Virol.78:1–8.
5.Basler, C. F., and A. Garcia-Sastre.2002. Viruses and the type I interferon antiviral system: induction and evasion. Int. Rev. Immunol.21:305–337. 6.Basler, C. F., A. Mikulasova, L. Martinez-Sobrido, J. Paragas, E.
Muhl-berger, M. Bray, H. D. Klenk, P. Palese, and A. Garcia-Sastre.2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol.77:7945–7956.
7.Best, S. M., K. L. Morris, J. G. Shannon, S. J. Robertson, D. N. Mitzel, G. S. Park, E. Boer, J. B. Wolfinbarger, and M. E. Bloom.2005. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol.79:12828–12839. 8.Billecocq, A., M. Spiegel, P. Vialat, A. Kohl, F. Weber, M. Bouloy, and O. Haller.2004. NSs protein of Rift Valley fever virus blocks interferon pro-duction by inhibiting host gene transcription. J. Virol.78:9798–9806. 9.Breiman, A., N. Grandvaux, R. Lin, C. Ottone, S. Akira, M. Yoneyama, T.
Fujita, J. Hiscott, and E. F. Meurs.2005. Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon. J. Virol.79:3969–3978.
10.Bridgen, A., F. Weber, J. K. Fazakerley, and R. M. Elliott.2001. Bunyam-wera bunyavirus nonstructural protein NSs is a nonessential gene product that contributes to viral pathogenesis. Proc. Natl. Acad. Sci. USA98:664– 669.
11.Cardenas, W. B., Y. M. Loo, M. Gale, Jr., A. L. Hartman, C. R. Kimberlin, L. Martinez-Sobrido, E. O. Saphire, and C. F. Basler.2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol.80:5168–5178.
12.Cebulla, C. M., D. M. Miller, and D. D. Sedmak.1999. Viral inhibition of interferon signal transduction. Intervirology42:325–330.
13.Cheng, G., J. Zhong, and F. V. Chisari.2006. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA103:8499–8504. 14.Diamond, M. S., and E. Harris.2001. Interferon inhibits dengue virus
in-fection by preventing translation of viral RNA through a PKR-independent mechanism. Virology289:297–311.
15.Enninga, J., D. E. Levy, G. Blobel, and B. M. Fontoura.2002. Role of nucleoporin induction in releasing an mRNA nuclear export block. Science
295:1523–1525.
16.Favre, D., E. Studer, and M. R. Michel.1996. Semliki Forest virus capsid protein inhibits the initiation of translation by upregulating the double-stranded RNA-activated protein kinase (PKR). Biosci. Rep.16:485–511. 17.Favre, D., E. Studer, and M. R. Michel.1994. Two nucleolar targeting signals
present in the N-terminal part of Semliki Forest virus capsid protein. Arch. Virol.137:149–155.
18.Feemster, R. F., and W. Haymaker.1958. Eastern equine encephalitis. Neu-rology8:882–883.
19.Ferran, M. C., and J. M. Lucas-Lenard.1997. The vesicular stomatitis virus matrix protein inhibits transcription from the human beta interferon pro-moter. J. Virol.71:371–377.
20.Foy, E., K. Li, C. Wang, R. Sumpter, Jr., M. Ikeda, S. M. Lemon, and M. Gale, Jr.2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science300:1145–1148.
21.Frolov, I., E. Agapov, T. A. Hoffman, Jr., B. M. Pragai, M. Lippa, S. Schlesinger, and C. M. Rice.1999. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J. Virol.73:3854– 3865.
22.Frolova, E. I., R. Z. Fayzulin, S. H. Cook, D. E. Griffin, C. M. Rice, and I. Frolov.2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in determining the outcome of Sindbis virus infection. J. Virol.76:11254– 11264.
23.Garcia-Sastre, A.2001. Inhibition of interferon-mediated antiviral responses by influenza A viruses and other negative-strand RNA viruses. Virology
279:375–384.
24.Garmashova, N., R. Gorchakov, E. Frolova, and I. Frolov.2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol.80:5686–5696.
25.Glanville, N., and I. Ulmanen.1976. Biological activity of in vitro synthesised protein: binding of Semliki Forest virus capsid protein to the large ribosomal subunit. Biochem. Biophys. Res. Commun.71:393–399.
26.Goodbourn, S., L. Didcock, and R. E. Randall.2000. Interferons: cell sig-nalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol.81:2341–2364.
on November 8, 2019 by guest
http://jvi.asm.org/
27.Gorchakov, R., E. Frolova, and I. Frolov.2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol.79:9397–9409. 28.Gorchakov, R., E. Frolova, B. R. Williams, C. M. Rice, and I. Frolov.2004.
PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol.78:8455–8467.
29.Grandvaux, N., B. R. tenOever, M. J. Servant, and J. Hiscott.2002. The interferon antiviral response: from viral invasion to evasion. Curr. Opin. Infect. Dis.15:259–267.
30.Grieder, F. B., and S. N. Vogel.1999. Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology257:106–118.
31.Guo, J. T., J. Hayashi, and C. Seeger.2005. West Nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol.79:1343–1350. 32.Hartman, A. L., J. E. Dover, J. S. Towner, and S. T. Nichol.2006. Reverse
genetic generation of recombinant Zaire Ebola viruses containing disrupted IRF-3 inhibitory domains results in attenuated virus growth in vitro and higher levels of IRF-3 activation without inhibiting viral transcription or replication. J. Virol.80:6430–6440.
33.Hartman, A. L., J. S. Towner, and S. T. Nichol.2004. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology
328:177–184.
34.Hiscott, J., J. Lacoste, and R. Lin.2006. Recruitment of an interferon molecular signaling complex to the mitochondrial membrane: disruption by hepatitis C virus NS3-4A protease. Biochem. Pharmacol.72:1477–1484. 35.Hong, E. M., R. Perera, and R. J. Kuhn.2006. Alphavirus capsid protein
helix I controls a checkpoint in nucleocapsid core assembly. J. Virol.80:
8848–8855.
36.Ikegami, T., S. Won, C. J. Peters, and S. Makino.2006. Rescue of infectious Rift Valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J. Virol.80:2933–2940.
37.Johnston, R. E., and C. J. Peters.1996. Alphaviruses, p. 843–898.InD. M. Knipe, B. N. Fields, and P. M. Howley (ed.), Fields virology, 3rd ed. Lip-pincott-Raven Publishers, Philadelphia, PA.
38.Jones, M., A. Davidson, L. Hibbert, P. Gruenwald, J. Schlaak, S. Ball, G. R. Foster, and M. Jacobs.2005. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol.79:5414–5420.
39.Kislauskis, E. H., X. Zhu, and R. H. Singer.1997.-Actin messenger RNA localization and protein synthesis augment cell motility. J. Cell Biol.136:
1263–1270.
40.Lee, S., K. E. Owen, H. K. Choi, H. Lee, G. Lu, G. Wengler, D. T. Brown, M. G. Rossmann, and R. J. Kuhn.1996. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure4:531–541.
41.Le May, N., S. Dubaele, L. Proietti De Santis, A. Billecocq, M. Bouloy, and J. M. Egly.2004. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell116:541–550.
42.Li, K., E. Foy, J. C. Ferreon, M. Nakamura, A. C. Ferreon, M. Ikeda, S. C. Ray, M. Gale, Jr., and S. M. Lemon.2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA102:2992–2997.
43.Li, X. D., L. Sun, R. B. Seth, G. Pineda, and Z. J. Chen.2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA
102:17717–17722.
44.Lin, R. J., B. L. Chang, H. P. Yu, C. L. Liao, and Y. L. Lin.2006. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol.80:
5908–5918.
45.Lin, R. J., C. L. Liao, E. Lin, and Y. L. Lin.2004. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J. Virol.78:9285–9294.
46.Linger, B. R., L. Kunovska, R. J. Kuhn, and B. L. Golden.2004. Sindbis virus nucleocapsid assembly: RNA folding promotes capsid protein dimerization. RNA10:128–138.
47.Liu, W. J., X. J. Wang, V. V. Mokhonov, P. Y. Shi, R. Randall, and A. A. Khromykh.2005. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol.79:1934–1942. 48.Loo, Y. M., D. M. Owen, K. Li, A. K. Erickson, C. L. Johnson, P. M. Fish,
D. S. Carney, T. Wang, H. Ishida, M. Yoneyama, T. Fujita, T. Saito, W. M. Lee, C. H. Hagedorn, D. T. Lau, S. A. Weinman, S. M. Lemon, and M. Gale, Jr.2006. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc. Natl. Acad. Sci. USA103:6001–6006. 49.Lu, Y., M. Wambach, M. G. Katze, and R. M. Krug.1995. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the eIF-2 translation initiation factor. Virology214:222–228.
50.Munoz-Jordan, J. L., M. Laurent-Rolle, J. Ashour, L. Martinez-Sobrido, M. Ashok, W. I. Lipkin, and A. Garcia-Sastre.2005. Inhibition of alpha/beta
interferon signaling by the NS4B protein of flaviviruses. J. Virol.79:8004– 8013.
51.Munoz-Jordan, J. L., G. G. Sanchez-Burgos, M. Laurent-Rolle, and A. Gar-cia-Sastre.2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA100:14333–14338.
52.Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene108:193– 199.
53.Noah, D. L., K. Y. Twu, and R. M. Krug.2003. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3⬘end processing of cellular pre-mRNAs. Virology307:386–395.
54.Owen, K. E., and R. J. Kuhn.1997. Alphavirus budding is dependent on the interaction between the nucleocapsid and hydrophobic amino acids on the cytoplasmic domain of the E2 envelope glycoprotein. Virology230:187–196. 55.Park, M. S., M. L. Shaw, J. Munoz-Jordan, J. F. Cros, T. Nakaya, N. Bouvier, P. Palese, A. Garcia-Sastre, and C. F. Basler.2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol.
77:1501–1511.
56.Perera, R., K. E. Owen, T. L. Tellinghuisen, A. E. Gorbalenya, and R. J. Kuhn.2001. Alphavirus nucleocapsid protein contains a putative coiled coil alpha-helix important for core assembly. J. Virol.75:1–10.
57.Petrakova, O., E. Volkova, R. Gorchakov, S. Paessler, R. M. Kinney, and I. Frolov.2005. Noncytopathic replication of Venezuelan equine encephalitis virus and Eastern equine encephalitis virus replicons in mammalian cells. J. Virol.79:7597–7608.
58.Raju, R., and D. Kolakofsky.1988. La Crosse virus infection of mammalian cells induces mRNA instability. J. Virol.62:27–32.
59.Reid, S. P., L. W. Leung, A. L. Hartman, O. Martinez, M. L. Shaw, C. Carbonnelle, V. E. Volchkov, S. T. Nichol, and C. F. Basler.2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol.80:5156–5167.
60.Ryman, K. D., K. C. Meier, E. M. Nangle, S. L. Ragsdale, N. L. Korneeva, R. E. Rhoads, M. R. MacDonald, and W. B. Klimstra.2005. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J. Virol.79:1487–1499. 61.Sambrook, J., E. F. Fritsch, and T. Maniatis.1989. Molecular cloning: a
laboratory manual, 2nd ed., vol. 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
62.Singh, I., and A. Helenius.1992. Role of ribosomes in Semliki Forest virus nucleocapsid uncoating. J. Virol.66:7049–7058.
63.Soderlund, H.1973. Kinetics of formation of the Semliki Forest virus nu-cleocapsid. Intervirology1:354–361.
64.Soderlund, H., and I. Ulmanen.1977. Transient association of Semliki For-est virus capsid protein with ribosomes. J. Virol.24:907–909.
65.Strauss, J. H., and E. G. Strauss.1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev.58:491–562.
66.Talon, J., C. M. Horvath, R. Polley, C. F. Basler, T. Muster, P. Palese, and A. Garcia-Sastre.2000. Activation of interferon regulatory factor 3 is inhib-ited by the influenza A virus NS1 protein. J. Virol.74:7989–7996. 67.Talon, J., M. Salvatore, R. E. O’Neill, Y. Nakaya, H. Zheng, T. Muster, A.
Garcia-Sastre, and P. Palese.2000. Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc. Natl. Acad. Sci. USA97:
4309–4314.
68.Tellinghuisen, T. L., A. E. Hamburger, B. R. Fisher, R. Ostendorp, and R. J. Kuhn.1999. In vitro assembly of alphavirus cores by using nucleocapsid protein expressed inEscherichia coli. J. Virol.73:5309–5319.
69.Tellinghuisen, T. L., R. Perera, and R. J. Kuhn.2001. In vitro assembly of Sindbis virus core-like particles from cross-linked dimers of truncated and mutant capsid proteins. J. Virol.75:2810–2817.
70.Thomas, D., G. Blakqori, V. Wagner, M. Banholzer, N. Kessler, R. M. Elliott, O. Haller, and F. Weber.2004. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J. Biol. Chem.279:31471– 31477.
71.Trgovcich, J., J. F. Aronson, J. C. Eldridge, and R. E. Johnston.1999. TNFalpha, interferon, and stress response induction as a function of age-related susceptibility to fatal Sindbis virus infection of mice. Virology263:
339–348.
72.Ulmanen, I., H. Soderlund, and L. Kaariainen.1976. Semliki Forest virus capsid protein associates with the 60S ribosomal subunit in infected cells. J. Virol.20:203–210.
73.White, L. J., J. G. Wang, N. L. Davis, and R. E. Johnston.2001. Role of alpha/beta interferon in Venezuelan equine encephalitis virus pathogenesis: effect of an attenuating mutation in the 5⬘untranslated region. J. Virol.
75:3706–3718.
74.Yoshida, T., M. W. Shaw, J. F. Young, and R. W. Compans.1981. Charac-terization of the RNA associated with influenza A cytoplasmic inclusions and the interaction of NS1 protein with RNA. Virology110:87–97.
75.Yuen, T., E. Wurmbach, R. L. Pfeffer, B. J. Ebersole, and S. C. Sealfon.2002. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res.30:e48.