JOURNALOFVIROLOGY,May 1970,p.639-650 Vol.5,No.5 Copyright©1970 AmericanSociety for Microbiology Printed inU.S.A.
Replication
of
Sendai
Virus
II.
Steps
in
Virus
Assembly
CAROL D. BLAIR' AND WILLIAM S. ROBINSON
VirusLaboratory, University of California, Berkeley, California 94720, and Departmentof Medicine,
Stanford
University,Stanford,
California
94305Received forpublication29January 1970
Chick embryo fibroblast culturesinfectedwith Sendai virus wereincubatedwith
3H-uridine in the presence ofactinomycinDbeginningat 18 hrafterinfection.The
35 and 185 virus-specific ribonucleic acid (RNA) components were found in a
ribonuclease-sensitiveforminthecell and appearedtobeassociated with
polyribo-somes. Newlysynthesized 575 viral RNAwasrapidly coated with protein to form
intracellularviral nucleocapsid, and no 57S RNA was found "free"
(ribonuclease-sensitive) in the 2,000 X gsupematant fraction of
disrupted
cells.Thenucleocap-sid from detergent-disrupted Sendai virus and that fromdisrupted cellswere
indis-tinguishable in ultrastructure and buoyant density, and neither was found to be
infectious or have hemagglutinating activity. Kinetic studies ofnucleocapsid and virus formation indicated a relative block in conversion ofviral nucleocapsid to complete enveloped virus inthese cells, resultinginaccumulation oflargeamounts ofnucleocapsid in the cellcytoplasm.
In apreviouspaper (2),weshowedthat several
distinct components of virus-specific ribonucleic
acid (RNA) are synthesized in chick embryo
fibroblast cultures infected with Sendai virus, a
subgroup 2 myxovirus. One component,
repre-senting about one-fourth ofthenewly synthesized
RNA,appears tobeidenticaltothe575 RNA
re-covered from whole virus. The remaining
virus-specific RNA consists of three distinct
com-ponents
sedimenting
at35, 22,and 18S, and 90%or more of this RNA appears to be
comple-mentary in base sequence to viral RNA, as
indi-cated by its annealing to viral RNA. Previous
studies with a similar subgroup 2 myxovirus,
Newcastle disease virus (NDV), indicated that
virus-specific RNA was synthesized in the cell
cytoplasm
and that much ofthe complementaryRNA was associated with polyribosomes of the
cell (3). The state ofthe 57SviralRNAafter its
synthesis
in cells infected with Sendai virus orNDV is not known, although some or all of it
may enterthe viralnucleocapsid, as was demon-strated in the cytoplasm of cells infected with
severalsubgroup2myxoviruses (5-7, 10, 13),and
some or all may eventually enter whole virus
whichisformedatthe cell surface (7, 10).
In this study, weattempted to determine the
IPartof this work was taken from a thesis submittedby C. D. B. in partial satisfaction of the requirements for the Doctor of Philosophy degree fromtheUniversityofCalifornia, Berkeley.
locationandstateof the newly synthesizedRNA
components within Sendai virus-infected chick
embryo cells and to study the
incorporation
ofviralRNAinto whole virus. No 57S viral RNA
was found free in the cell cytoplasm in these
experiments. Instead, the 575 RNAappeared to
beimmediately coatedwith
protein
after itssyn-thesis,
making
itinsusceptible
todigestion
byribonuclease.Itwasquantitativelyrecoveredfrom
thecellcytoplasm aspartof the helical viral
nu-cleocapsid. The smaller RNA components were
found in the cell
cytoplasm
in aribonuclease-susceptible form, and they sedimented in the
re-gion of polyribosomes and single ribosomes.
Kinetic studies showed that 3H-uridine first
ap-pears in the 57S RNA of the nucleocapsid
re-covered from the cell
cytoplasm;
second, innucleocapsid released from cells with trypsin;
third,
in whole virus released from cells withtrypsin; and
finally
inwhole virus in the culturemedium.
Quantitation
of the 575 RNA inthesepools
indicates thatmorethan95% ofallofthenewly synthesized 57S RNAremains in the form
of
nucleocapsid
in the cellcytoplasm,
and that lessthan5%enterswholevirus.MATERIALS
AND METHODSMaterials. Uridine-5-3H(25c/mmole) and carrier-free H332PO4 were purchased from New England
Nuclear Corp., Boston, Mass. Bovine pancreatic ribonuclease was purchased from Worthington Bio-chemical Corp. Trypsin was purchased from Difco
639
on November 11, 2019 by guest
http://jvi.asm.org/
and wasprepared as a0.25% solution (w/v) in
Tris-(hydroxymethyl)aminomethane(Tris)-buffered saline. Vibrio choleraeneuraminidase (500 units/ml)was pur-chased fromCalbiochem, Los Angeles, Calif. Tween
80 (polyoxyethylene sorbitan-mono-oleate) was pur-chased from J. T. BakerChemical Co. Actinomycin D was the gift of Merck & Co., Inc., Rahway, N.J. Nonidet 40(NP40)wasthegiftof Shell Chemical Co. Tobacco mosaic virus (TMV) was the gift ofC. A. Knight.
Standard buffer. Thebuffer contained 0.1 M NaCl,
0.01 M Tris-hydrochloride (pH 7.4), and 0.001 M
ethylenediaminetetraacetic acid (EDTA).
Viruses. TheHarris strainof Sendai virus and the
L-Kansas 48 strainofNDV weregrown in
embryo-nated eggs, and the chorioallantoic fluid containing
108 to 109 egg infectious units/mlwas usedto infect
cellsin tissue culture(2).
Cellcultures. Secondary cultures of chick embryo
fibroblasts were cultured and infected with virus as previously described (2).
Assays forinfectivityandhemagglutination. Sendai virusinfectivitywasmeasuredbyanendpointdilution method inembryonatedeggsas previouslydescribed
(2).
Quantitative assay for hemagglutination was done bypreparingserial twofold dilutions of the fluidtobe tested in phosphate-buffered saline (PBS), mixing
with chicken erythrocytes, and incubating as previ-ouslydescribed(2).
Virus purification. Virus was purified from tissue
culturemediumorchorioallantoicfluidbydifferential
centrifugationandsedimentation insucrosegradients
aspreviouslydescribed (2,3).
Isolation of nucleic acids. Nucleic acidfrom cellsor
viruswasisolatedbytheuseof sodiumdodecylsulfate (SDS) and phenol (3). Whenthe amount of nucleic acid in thesampletobeextractedwasverysmall,2mg of TMV carrierwasadded beforephenolextraction.
Preparation ofcytoplasmic extracts. Two methods
wereusedtodisruptchickembryocells. Onemethod has beenpreviously described (3).Cellsgrowingina monolayerwereremovedbyaddition of0.25% tryp-sin and incubatedfor5minatroomtemperaturewith
occasional agitation. Suspended cells were washed withPBS,suspendedinice-coldhypotonicbuffer con-taining 0.01 MNaCl, 0.01 M Tris-hydrochloride (pH
7.4), and 0.002 M MgCl2, and allowedtoswell for 5
min.Cellswerebrokenby 15to 16strokesofa
tight-fitting Dounce homogenizer (Kontes Glass), leaving
most nuclei intact. The broken-cell suspension was centrifugedat4,000rev/minfor 5minin theSorvall
SS34rotorto removeunbrokencells, nuclei,andlarge
membrane fragments. The supernatant fraction was
usedas thecytoplasmicextract. A second methodof
cell disruption used the detergent NP-40 (16). The medium of cell cultures was replaced with 1 to 2
ml of0.5%NP40 in 0.1 M NaCl, 0.01 M Tris-hydro-chloride,and0.002M
MgCl2.
After5min ofincubation withgentle agitation at roomtemperature, cellfrag-mentsremainingonthedishwere scraped off with a
rubber policeman. Nuclei, which remained intact,
andlargecellfragmentswere removed by
centrifuga-tion at 4,000rev/minfor5min, and the supernatant fluidwasusedasthecytoplasmicextract.
Electronmicroscopy. Infected cells were fixed as a monolayer in 1.6% glutaraldehyde in PBS (pH 7.2)
with0.005MMgCl2 and 0.0001 M CaCl2 (PBS MgCa).
Thecells were then scraped off the dish, centrifuged to apellet in McNaught protein tubes, and fixed as a pellet in1% osmic acid in PBS MgCa. After dehydra-tion in agraded ethylalcohol-0.15 M NaCl series, in-filtration with propylene dioxide, and embedding in Epon 812, thin sections were cut on an LKB ultra-microtome. Contrast was increased by staining with 1% aqueous uranyl acetate and 1% alkaline lead citrate (14).
Virus or nucleocapsid was pelleted in the Spinco SW50 rotorby spinning at 47,000rev/min for 90min. Thepellet was resuspended in a small volume of dis-tilled water, and a small drop of this suspension was delivered to a collodion-covered 200-mesh copper grid. Negative stain was2%ophosphotungstic acid
ad-justedto pH7with KOH.
Micrographs were taken on a SiemensElmiskop I electron microscope at either 60 or 80 kv with a
50-Mzm
objectiveaperture.Scintillation counting. Radioactivity precipitated in 5% trichloroacetic acidwascollected and washed on Millipore filters and counted in toluene scintillation fluid inaPackardTri-Carb scintillation spectrometer
(3).
RESULTS AND DISCUSSION
Fractionation of cytoplasmic extracts from Sendai virus-infected cells. Chick embryo fibro-blast cultures were incubated with 3H-uridine in
the presence ofactinomycinD from 18 to 20 hr after infection with Sendai virus, when virus-specific RNA synthesis is maximum (2). Under theseconditions,alllabeledRNAhas beenshown
to bevirus-specific RNA (2). A cytoplasmic ex-tract wasthenprepared byusing NP40 detergent and divided into three partsforsedimentation on
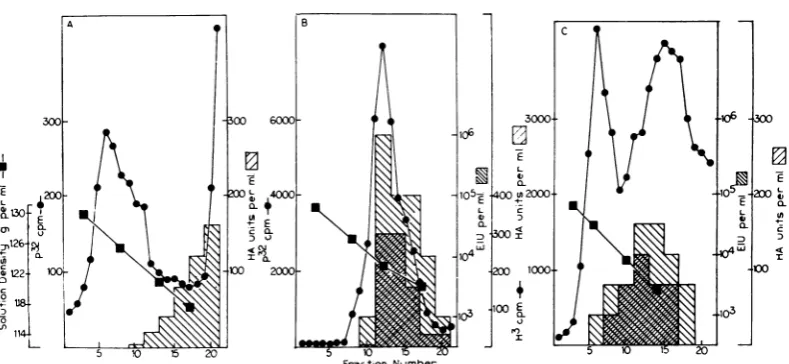
sucrosedensity gradients.Figure IA shows the dis-tribution in a sucrosegradient of trichloroacetic acid-precipitable radioactivity and cytoplasmic
materialabsorbing at 260nmaftersedimentation of a sample of the cytoplasmic extract layered
directly ona sucrose gradient. TheA260 peak in
fraction 23 representssingleribosomes(74S), and that in fraction 27, the 50S ribosomal subunit.
The ultraviolet-absorbing material sedimenting
morerapidly than single ribosomes (fractions1to 20) represents polyribosomes. A significant frac-tion of theradioactiveRNAsediments in a broad distribution with polyribosomes and the single
ribosomes.Adistinct radioactive component with
apeakinfraction 8 sediments morerapidlythan
the single ribosomes and most of the
polyribo-somes detected by ultraviolet absorbancy. Some
radioactive material sediments more slowly than
singleribosomes.
Figure1Bshowstheresults of sedimentation of
asecondsampleofcytoplasmicextractafter
incu-J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
SENDAI VIRUS REPLICATION
Fract,on Number
FIG. 1. Sedimentation of cell extractsfrom Sendai virus-infected cells. Four 150-mm tissue culture dishes
with2 X 107 cells eachwereinfected withSendai virus. Eighteenhours later, the culture mediumof each was
re-placed with 10mlof medium containing 2 ,ugofactinomycin D perml; 30 min later,300 ,uc of3H-uridinewas
addedtoeach culture. After2hr, theculturemedium was removed, and a cytoplasmic extract was preparedfrom the cellsofallfourculturesby using0.5%NP40. The 4,000 rev/minsupernatantfractionwasdividedintothree
parts. Thefirstportion (A) waslayered directlyonto agradientof5 to
30'7j
sucrose in0.1MNaCI, 0.01 M Tris(pH 7.4), and0.002 MMgCl2. Thesecond portion (B) was incubated with 5 ,ug of pancreatic ribonuclease for 5
min at roomtemperature beforelayeringon asecond sucrosegradient. ED TA wasadded to a third portion (C)
tomakeafinalconcentrationof0.004 Mbefore layeringthe extracton athird sucrosegradient.All three sucrose
gradients werecentrifuged for2.5 hr at 25,000rev/minand 4 C in aSpincoSW 25.3 rotorand analyzedfor
tri-chloroacetic acid-precipitableradioactivity andA260aspreviously described (2).
bation withpancreatic ribonuclease. The increase
in
ultraviolet-absorbing
material in thesingle-ribosome peak (around fraction 23) and the
de-crease in polyribosomes, indicated by the
de-creaseinabsorbancy around fraction15 (compare
Fig. 1A), resulted from ribonuclease action on
polyribosomes. It can be seen that ribonuclease
did not alter the sedimentation behavior of the
rapidly sedimenting radioactive component
around fraction8ortheamountoftrichloroacetic
acid-precipitable radioactivity
in this component.Figure 1C shows the results of sedimentation
ofathird
portion
ofcytoplasmic
extracttowhichEDTA had been added. The reduction in the
amount of
ultraviolet-absorbing
material infrac-tions 1 to 23 is consistent with dissociation by
EDTA ofpolyribosomes into more slowly
sedi-menting components. Italso appears thatEDTA
did not alter the sedimentation behavior or the
amountof trichloroaceticacid-precipitable
radio-activity in the rapidly sedimenting component
around fractions 9 and 10. The amount of this
RNA-containing component appears to be too
smalltobedetectedbyabsorbancyat260nm.
These results suggest that some newly
synthe-sized RNA in Sendai virus-infected cells
sedi-mentswithpolyribosomes, single ribosomes, and
a significant fraction ofnew RNA in a rapidly
sedimenting
structure. The sedimentationbe-havior of this structure isnotaltered by
ribonu-cleaseorEDTA, andthelabeledRNAassociated
withitisnotmadetrichloroacetic acid-soluble by
ribonuclease. In experiments in which infected
cells were disrupted by a Dounce homogenizer
without NP40detergent, aspreviously described,
the sucrose gradient profiles and the amount of
radioactiveRNAineach component inthe
gradi-ents werethesame asin theexperiment shown in
Fig. 1, indicating that the rapidly sedimenting
structure doesnotresult from theaction ofNP40
on infected cells or whole virus associated with
the cells.
Identification of RNA in cytoplasmic extracts.
To identify the RNA in different regions of the
sucrosegradient shown in Fig. 1A, the fractions
in each of the regions designated 1 to 4 were
pooled, and the RNA was recovered from each
byphenolextractionbyusing50,ugofTMVRNA
as carrier. The purified RNA from each region
was then fractionated by sucrose gradient
sedi-mentation (Fig. 2).
Figure 2A shows the results with the RNA fromregion1 inFig.1A.Sedimentationwasfrom
rightto left. The component of RNA detectedby
641
VOL. 5, 1970
on November 11, 2019 by guest
http://jvi.asm.org/
[image:3.486.48.435.67.262.2]Prac*on Niuber
FIG. 2. Sedimentation of RNA extractedfrom fourregionsof thesucrosegradient showninFig. IA.All
frac-tions ineachof the numbered regions of thesucrosegradientinFig. JAwerepooled,2mgofTMVwasaddedas
carrier, andRNAwas extractedfrom each. Each RNA sample wasthensedimentedin asucrose densitygradienlt
in aSpinco SW 50rotor at47,000 rev/min and4Cfor105 min aspreviously described (2).
A260 with a peak in fraction 18 (Fig. 2A) is the
small amountofTMV RNAused ascarrier.No
distinct component of radioactive RNA is
ap-parent.
The RNAfromregion2 inFig. IA, which
in-cludes the rapidly sedimenting radioactive
struc-ture, is shown inFig. 2B. The RNA component
detected byA260with a peak infraction 15 (Fig.
2B) representsTMV RNAandasmall amountof
28SribosomalRNAwhich sediments withTMV
RNA under these conditions. Almost all of the
radioactiveRNAsediments like 57SviralRNA,
and no significantamount ofradioactive 18, 22,
or35Svirus-specificRNAis present.
Theresults ofsedimentation of the RNAfrom
region 3 in Fig. 1A, which includesmost ofthe
polyribosomes and single ribosomes detected by
ultraviolet absorbancy,areshowninFig.2C.The
RNA componentsdetectedby A260,withpeaks in
fractions 14and 19 (Fig. 2C), consist mostly of
28and18Sribosomal RNA,respectively. Twoof
the components of radioactive RNA sediment like the355(fraction 9)and the 18S (fraction 19)
virus-specific RNA forms, which have been
showntobealmostcompletely complementaryin base sequence to viral RNA (2). Some
radioac-tive RNA (fraction 25) sediments more slowly than 18S and may represent virus-specific RNA
degraded duringthe preparationof the
cytoplas-mic extract. No labeled57S RNA was recovered
from this region of the gradient (e.g., region 3,
Fig. 1A) in severalexperiments.
The RNAfromregion 4inFig. 1A, which
in-cludes material sedimenting more slowly than
single ribosomes, is shown in Fig. 2D. Again,
ribosomal RNA is detected by A260, and almost
all of the radioactive RNA (fraction 25)
sedi-mentsmoreslowlythan 18S.
Theseresults suggestthatall newly synthesized
57S virus-specific RNA from Sendai
virus-in-fected cells can be recovered from the rapidly
sedimenting ribonuclease-resistant structure (Fig.
1), and the35and 18Svirus-specificRNAforms
sediment in a ribonuclease-sensitive form with
polyribosomes and single ribosomes. The latter
finding is in agreement with experiments with
NDV-infectedcells, in whichthe 18 and 35S
virus-specific RNA components sedimented with
polyribosomes (3).
Identification of the rapidly sedimenting
ribo-nuclease-resistant structure in cytoplasmic
ex-tracts as viral nucleocapsid. A number of the
properties of the
rapidly
sedimentingribo-nuclease-resistant structure from infected cells
(Fig. 1) indicate that it is a viral nucleocapsid.
Consistent with this arethe observationsthatits
sedimentation was not altered by ribonuclease
(Fig. 1B)orEDTA(Fig.IC),thattheradioactive
RNAassociatedwiththe structure was not made
trichloroacetic acid-soluble byribonuclease (Fig.
1B),andthatRNAwith sedimentation character-istics of viral RNA (57S) was exclusively
re-covered from this component in many
experi-ments. Kingsbury and Darlington (12) have shown that the nucleocapsid recovered from
de-tergent-disrupted NDV contains high molecular
weight RNA,andthat ribonucleasedoesnotalter
thesedimentationrateof the nucleocapsidor de-grade the RNA to a trichloroacetic acid-soluble
form.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:4.486.65.450.63.228.2]SENDAI VIRUS REPLICATION
The buoyant density of the rapidly sedimenting
structure from Sendai virus-infected cells,
de-termined by equilibrium centrifugation in a
pre-formedsucrose density gradient, was 1.26to 1.27 g/ml. (Fig. 3). This value is significantly greater than the buoyant density value of whole virus, which is 1.22 g/ml under the same conditions.
Compans and Choppin (5) found a buoyant
density of1.30g/mlinaCsCldensity gradientfor
the nucleocapsid isolated from SV-5-infected BHKcells.
Figure 4 shows the results of rate zonal sedi-mentation of the rapidly sedimenting component
(closed circles) in a 5 to 20% sucrose density gradient. TMV (opentriangles),which hasa
sedi-mentation coefficient of about 160 S20,w under these conditions, was used as a sedimentation
marker in the experiment. The radioactive
cyto-plasmic component clearly sediments more
rapidly than TMV and can be estimated to
haveasedimentation coefficient ofapproximately 200S under these conditions. The sedimentation
coefficient of intact NDV, a virus with almost identical size andshapeas Sendaivirus,hasbeen estimated to be 1,100S (15). Thus, whole virus
has a much greater rate of sedimentation than that of the cytoplasmic component in Fig. 4.
3,000
, 2,000
0
E
E
H9-1,000
5 10 15
f raction number
20
FIG. 3. Equilibrium centrifugation of viral
nucleo-capsid. 3H-labelednucleocapsid, preparedasdescribed
inthelegendtoFig.IC,waslayeredoverapreformed
15 to65% sucrosegradient containingstandardbuffer
andcentrifugedat47,000 rev/minl for4 hrat4 C ina SpinicoSW50rotor.
0
09 C\J
0-U
l<
Fraction Number
FIG.4. Velocity sedimentation of nucleocapsid.
3H-labeled nucleocapsid,preparedas describedinFig.
IC, wasmixedwith unilabeledTMV, anzdthe mixture
waslayeredovera5 to 20% sucrosedensity gradient
andcentrifugedat45,000rev/minzand 4 C for 15 min inanS W50rotor.
Kingsbury and Darlington (12) have estimated
the sedimentation coefficient of the nucleocapsid from detergent-disruptedNDVtobe200S.
Todetermine themorphological appearance of
the200S cytoplasmic structure, it was examined
by electron microscopy after negative staining.
Whole virus grown in the chorioallantoic cavity
was also examined for comparison. Figure 5A
shows that typical viral nucleocapsid is present
in thesucrosegradient in the position of the 200S
structure. The nucleocapsid is indistinguishable
from that described in detail from SV-5-(5) and NDV-(6) infectedcellsandfrom disrupted Sendai
virus (4, 9) and NDV (12).
Figure SB shows whole Sendai virus. The
nu-cleocapsid within virions and that lying outside
have the same morphology as the cytoplasmic cleocapsidinFig. 5A.
Comparison of cytoplasmic nucleocapsid with
wholevirus anddetergent-disruptedvirus. Further
evidence that therapidly sedimenting cytoplasmic
structure is viral nucleocapsid was obtained by
testing its infectivity and hemagglutinating
ac-tivity (HA) andby comparingit with whole virus and detergent-disrupted virus. Figure 6A shows theresults ofequilibrium sedimentation Of32PO4_ labeled Sendai virus after disruption with 0.3%
VOL. 5, 1970
643
on November 11, 2019 by guest
http://jvi.asm.org/
[image:5.486.242.432.65.304.2] [image:5.486.39.229.347.582.2]sodium deoxycholate (DOC) and layering over a preformed sucrosedensity gradient. The heteroge-neous32P-labeled component with apeakin
frac-tion6has the buoyantdensity of the nucleocapsid
isolated from cells (1.27 g/ml). It was demon-strated that almost all of the 32p in this
com-ponent afterphenol extractionwas in theform of
57S RNA,which is consistent with its being viral
FIG. 5. Morphology ofcellularnucleocapsidand virus. (A) Nucleocapsidwaspreparedfrom infectedcells as
describedinFig. 1,andsucrosegradientfractionscontaining 3H-nucleocapsidwerecentrifuged to make a pellet
forelectron microscopyasdescribedinthetext. X240,000. (B) Sendai virus growninembryontatedeggs
andpuri-fiedin sucrosegradientswaspreparedforelectronmicroscopyasdescribedinthe text. X 100,000.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:6.486.93.425.113.602.2]SENDAI VIRUS REPLICATION
Fraction Number
FIG. 6. Comparison of nucleocapsidfrom cells, nucleocapsid from virus, and intact virus. Sendai virus-labeled with32PO4 waspreparedinembryonatedeggs andpurified as previously described (2). To one sample Of 32pO4 virus (A), DOCwasaddedto afinalconcentrationof0.3%,and the mixture was shaken for5min at room tem-peratureandthenlayeredover a15to65% sucrosegradient.Asecondsampleof 32PO4 virus (B) was layered di-rectlyon an identicalsucrosegradient. A cytoplasmic extract was preparedfrom seveninifected150-mm cultures after incubationz with 3H-uridineandactinomycinD asdescribedinFig.1, and the extract (C) was layered over a third 15to65%' sucrose densitygradient.
All threesampleswerecentrifugedfor 14 hrat25,000rev/min and 4 CinaSpinco SW 25.3 rotor. Fractions werecollected dropwisefrom the bottom of the tube, and trichloroacetic acid-precipitable radioactivity was
deter-minedfor 0.1-ml samples. A 0.05-mlamount wasremovedfrom each fraction, diluted in serum-free 199A, and
frozenfor laterinfectivity and HA assays as describedin the text. Densities were determined by weighing
100-pliterportionsinamicropipette.
nucleocapsid released from DOC-treated virus.
No viral infectivity was associated with
DOC-treated virus, and HA was only associated with
material sedimentingnearthetopofthe gradient
(fractions 10 to21). Figure6B showstheresults
of sedimentation of 32P-labeled whole virus before
treatment with DOC and demonstrates that
radioactivity, infectivity, and HA coincide at a
density
of 1.23g/ml (around
fraction12).Figure
6C shows the results of equilibriumsedimenta-tion ofa cytoplasmic extract from infected cells
prepared asdescribedfor the experiment in Fig.
1. Thetritium-labeled componentwith apeak in
fraction6has thebuoyantdensityofnucleocapsid
(1.27 g/ml). The material with HA has a peak
witha
density
around1.22to1.23g/mlandrepre-sentswholevirus. Infectivity andHA do not
ap-peartobespecificallyassociated with the
nucleo-capsid in this experiment, and purified
nucleo-capsid in otherexperiments failed to show
infec-tivityorHA.
These
experiments
suggest that nucleocapsidsfrom detergent-disrupted Sendai virus and from
the cytoplasm of infected cells are similar in
buoyant density, contain 57S RNA, and do not
have infectivity or HA. Although Bukrinskaya
etal. (4) havereportedinfectivity associated with
nucleocapsid from Sendai virus-infectedcells, we
have beenunable to demonstrateinfectivity with
ournucleocapsid preparations.
Formation of nucleocapsid. To determine the
time required for nucleocapsid assembly
after viral RNA synthesis, an experiment was
done in which the rate of 3H-uridine
incorpora-tion into total 575 RNA and into intracellular
nucleocapsid was measured, starting at 18 hr
afterinfection.Different cultures ofinfected cells
wereincubated with high specific activity
3H-uri-dinefor 5, 10,and 15 min inthepresenceof
acti-nomycinD, and theamounts ofradioactivity in
total57S RNA ofthecells and theradioactivity
inintracellular nucleocapsidweredetermined for
each incubation time. It was shown earlier (Fig.
2B) that all of the radioactive RNA extracted
fromthenucleocapsid is57SRNA.
The results in Table 1 show that, after only 5
min ofincubation with
3H-uridine,
all of the la-beled 57S RNAin the cell cytoplasm canbeac-counted for by radioactivity in nucleocapsid.
Similarly, after 10 and 15 minofincubation with
3H-uridine, essentially all of the radioactivity in
57S RNAcanbe accounted for in
nucleocapsid.
The differences between theamounts of
radioac-tivity in the two fractions at all three times are
10% or less, a value which is within the
experi-mental error of the methods used. Two conclu-645
VOL. 5, 1970
on November 11, 2019 by guest
http://jvi.asm.org/
[image:7.486.44.439.67.249.2]BLAIR AND ROBINSON
TABLE 1. Incorporation of 3H-uridine into total 57S RNA in infected cells and into viral
nucleocapsida
Counts/min of BHlabel in Time of 3H-uridine
incubation
57S RNA Nucleocapsid
min
5 822 856
10 1,828 1,724
15 8,548 7,718
a
Six
150-mm culture dishes containing2 X 107cells each wereinfectedwith Sendai virus.
Eight-een hours later, the medium of each culture was replaced with 10 ml of medium containing 2ug of actinomycin D per ml. Thirty minutes later,
400 scof3H-uridinewasadded to eachand,atthe subsequent times designated, two culture dishes
wereplaced inan ice-waterbath and themedium
replaced with ice-cold phosphate-buffered solu-tion.Cells wereremoved from thedishes with con-centratedtrypsinin thecold, and the cell
suspen-sionswere divided intotwoparts.From onepart,
RNA wasextractedwith sodium dodecyl sulfate andphenol, and theradioactivityin575RNA was
determined after sucrose gradient sedimentation
of the RNA (2). The second part of each cell
suspension was disrupted with NP40, and the
radioactivity in nucleocapsid was determined
after sucrose gradient sedimentationasdescribed forFig.1.
sions can bemade from this experiment. First,
57S RNAbecomes partoftheviralnucleocapsid
very rapidly after its synthesis. The time is
un-doubtedly significantly
less than 5 min, sincees-sentially all57S RNAis found innucleocapsidat
5 min. This rapid assembly of Sendai virus
nu-cleocapsid is similar to the maturation ofpolio
virus, which has been estimatedto occurwithin3
min orless of thecompletion of synthesisofviral
RNA (1). Second, not only is57S RNA rapidly converted tonucleocapsid after its synthesis, but
all or avery
large
fraction of it entersviralnu-cleocapsid. Free57S RNAisnotdetected byour
methods in cell cytoplasm at this stage of the
virus growth cycle. The results described inFig.
1A and 2 arein accordwiththeseconclusions.
The resultshere suggest that thesmallerRNA
componentswhich appear to be associated with
polyribosomes and not the 57SRNAmay serve
asviralmessenger. It is notexcluded thatthe 57S
RNAdoes enterpolyribosomesat an
earlier
stagein the virus
replication
cycle. Ithasbeen shownthatmost of
newly
synthesized poliovirus RNA becomes associated with polyribosomes early inthepolio growth cycle, andmost enterscomplete
virions latein thecycle (1).
Virus formation at the cell surface. The final
steps inmyxovirus assembly are thoughtto take place atthe cell surface by theprocessdescribed
asbudding (7, 11).We havefound thattreatment
of Sendai virus-infected cells with trypsin quanti-tativelyremovescellassociated virus without
dis-rupting cells. This was shown by an experiment
in which cells, after incubation with 8H-uridine
in the presenceof2,ug of actinomycin D per ml
between 18 and 20 hr after infection with Sendai virus, were gently removed from culture dishes
withtrypsin solution and centrifuged, and the
re-sulting supernatant fluid was analyzed by
equi-librium centrifugation in a preformed sucrose
density gradient. Figure 7 shows the distribution in the gradient of the radioactive particulate
ma-terial released from the infected cellsby trypsin.
Two incompletely separated radioactive
com-ponentsappearin the gradient. One has the
buoy-antdensity of whole virus (1.23 g/ml) and is
asso-ciated with infectivity and HA. The other
L 130 Q 0) 0N26 U) c122 °~1,14 200~ E 0I to 100 104_. L_ a) D _-*£ 14 Vo E
0.
120I 4024 6 8 10 12 14 16 Fraction N\umber
FIG. 7. Particulate material releasedfrom cellsby
trypsin. Seven 150-mm culturesofSendaivirus-infected
cellswereincubated with 100,c of3H-uridineper
cul-tureand actinomycin D asdescribed in thelegendto
Fig. 1. The culture mediumwasthenremoved,thecells
werewashedwithPBSwithout CaorMg,and1 mlof
0.25%trypsin solution wasaddedtoeachfor5 minof
incubationatroom temperaturewithoccasionalgentle
agitation. The trypsinsolutionandfloatingcellswere
removed,and each dishwaswashedwithImlof PBS.
Thepooledtrypsin and washsolutionswerecentrifuged at4,000rev/minfor5 mintoremovewhole cells; the supernatant fraction was layered over two sucrose layers (Imlof65% sucroseinD20and 5 mlof15% sucrose) in acentrifuge tube andcentrifuged for2hr at25,000rev/min and 4 C in theSpincoS W25.3rotor as previously described for viruspurification (3). All
oftheparticulate material which included whole virus
andnucleocapsidatthe interface between the sucrose layers wasrecovered andlayeredover a linear 15 to
65%sucrosedensity gradientandcentrifugedfor12hr
at25,000rev/min and 4 CinaSpincoS W 25.3rotor.
646
J. VIROL.on November 11, 2019 by guest
http://jvi.asm.org/
[image:8.486.52.241.66.185.2]SENDAI VIRUS REPLICATION
component has the buoyant density of viral
nucleocapsid, and electron microscopyofmaterial
from this region of the gradient reveals typical
structures of viralnucleocapsid.
When 3H-RNA was isolated from each com-ponent, it was found to sediment as 57S viral RNA. Thus, it appears that the action of trypsin
resulted in release ofboth infectious virus and
viral nucleocapsid from infected cells. After
incu-bation ofinfected cells with H3-uridine for 2 hr,
significantly more radioactivity appeared in the
RNAof thetrypsin-released nucleocapsid than in
trypsin-released wholevirus.
Figure 8 shows a comparison of the effects of
trypsin, neuraminidase, and EDTA on release of
virusandnucleocapsidfrominfectedcells.
Identi-calcultures ofinfectedcells afterincubationwith
3H-uridine in the presence ofactinomycin D, as
described forFig.7, were treatedwithtrypsin
so-lution (Fig. 8A), V. cholerae neuraminidase at a
concentration 10timesthatrequiredtoinhibit
ag-glutination of an equivalent number of chicken erythrocytes (Fig.8B), or a versenatesolutionat a
concentrationwhich removesepithelial cellsfrom
culture dishes (Fig. 8C). The particulate material
released by trypsin canagainbe seen to be
frac-tionated by sucrose gradient centrifugation into
twocomponentscorresponding towhole virus at
a density of 1.23 g/ml and nucleocapsid at a
density
of 1.27 g/ml (Fig. 8A). Very smallamountsof whole virusandalmost nodetectable
nucleocapsid were released from cells by
neura-minidase
(Fig. 8B)
or EDTA(Fig. 8C),
indicat-ing that the whole virus and nucleocapsidreleased
withtrypsinarenotbound to cellsby bonds
dis-ruptedby neuraminidase or EDTA.
Incubation of purified whole 3H-virus with trypsin at room temperature and subsequent frac-tionation ofthe 3H-labeled virus preparation by
sucrose gradient sedimentation, as described for Fig. 7, demonstrated that nonucleocapsid was re-leased from whole virus by trypsin alone. This in-dicates that nucleocapsid is released from cells rather than whole virus after incubation of in-fected cells with trypsin. Whether the nucleocap-sid in these experiments comes from a few cells which are mechanically disrupted during the
trypsin treatment and centrifugation, or whether
trypsin more selectively releases nucleocapsid
from sites near or at the surface of infected cells, is not completely clear. An experiment to test
these possibilities suggests the latter. Cells were
incubated with 3H-uridine between 11 and 25 hr afterinfection in the absence of actinomycin D to
permit cellular RNA, such as ribosomal RNA
and virus-specific RNA, to become radioactive.
The cells were then gently removed from the dishes with trypsin and collected by centrifuga-tion. The totalradioactive RNA in the cells was determined, a sample of the trypsin solution was used to determine totalradioactivity released by trypsin, and a second sample was used to
deter-mine theradioactivityin the RNA in
nucleocap-sid and whole virus which were isolated from the
E
0I
Fraction Number
FIG. 8. Particulate material releasedfrom cells by trypsin, neuraminidase, andED TA. Six 100-mm cultures
with 6 X 106cellseach wereincubatedwith 100 xsc of3H-uridineperculture andactinomycin Dbetween 18 and
20hrafterinfection, asdescribedinthelegendtoFig.1. (A) The cells oftwocultureswereincubatedwithtrypsin
andwashedasdescribed in thelegendtoFig.7. (B)Fiftyunitsof Vibrio cholerae neuraminidaseinImlwasadded
to thesecondpairof cultures, whichwere then incubatedat room temperaturefor20 min. Neuraminidase and
floating cells were removed, and I mlofPBS was used to washeachplateasinpart A. (C)A
1-ml
amountof0.001M sodium versenate in PBS without Ca2+ andMg'+ wasaddedtothe thirdpairofcultures. Thecultures
were incubatedfor20 min at room temperature, and the disheswere washedasforparts Aand B. The
super-natant fraction ofeachsample, whichwascentrifugedat4,000rev/mintoremovewholecells,wasthen centrifuged
insucrosegradientsasdescribedinthelegendtoFig. 7.
VOL.5, 1970 647
on November 11, 2019 by guest
http://jvi.asm.org/
[image:9.486.103.396.400.548.2]AND
trypsin solution in a sucrose density
gradient,
asinFig. 7. Table 2 shows that about 1.5% ofthe total radioactive RNA of the cells was released into the trypsin solution, and, of the
trypsin-re-leased material, about 20%wasRNA in nucleo-capsid. We have also shown that the amount of radioactive nucleocapsid and virus released by trypsin is 15 to 20% of the amount of
intra-cellular radioactive
nucleocapsid
(Fig. 9). Thus,amuch greaterproportionof totalcell-associated
radioactive
nucleocapsid
than totalradioactive-cellRNA, mostof which is ribosomal and trans-fer RNA, is released by trypsin. This indicates that trypsin
preferentially
orselectively
freesnucleocapsidfrom infectedcellsandsuggeststhat
nucleocapsid may accumulate at or near the cell
surface. In the case of SV5-infectedmonkey
kid-neycells,viralnucleocapsidhas been shown to
ac-cumulate immediately under some areas of the cellsurface (7).
Kinetics ofvirus
assembly.
We have described four structures containing 57S RNA which canbe recovered from Sendai-infected cells or cell culture medium: intracellular nucleocapsid,
nucleocapsid, virus released from cells by
trypsin, and virus in the culture medium. An
experiment was then done to determine the time course of 3H-uridine
incorporation
into theRNA of each ofthese structures.Actinomycin
D-treated cells were incubated with 3H-uridine from 18 to22 hr after
infection,
and the four particulate componentsnamed above were isolated atvarioustimes throughout this period. The cells were
removed withtrypsin asdescribedfor
Fig. 7,
andTABLE 2. Release of tritium-labeled RNA from
infected cellsbytrypsina
Total acid-Fraction precipitable 8H-uridine (counts/min)
Total cell RNA
.4,204,000
Total RNA released by trypsin 64,210 Nucleocapsid RNA released by
trypsin.13,428
aFour 100-mm cultures were each incubated
with 100,c of3H-uridine in 5 ml of medium
with-outactinomycinDfrom11 to 25hr afterinfection
with Sendai virus. Cellswere then removed with
trypsin and collected by centrifugation as de-scribed in Fig. 7. The total RNA was extracted from cells and from one-half of the trypsin so-lution by the sodium dodecyl sulfate-phenol method; a sample was used for trichloroacetic
acidprecipitationandcounting.The other half of the trypsin solution was centrifuged in sucrose
density gradients for isolation of nucleocapsid
andvirus,asdescribed in thelegendtoFig. 7.
E 14,000
-10,000
-6,000
-2,000- T
20 40 60 80 100 120 140 60 180 200 220 240
Time,minutes
FIG.9. Incorporation of3H-uridine intofour viral
structurescontaining 57S RNA. Five milliliters of
me-dium with2 ,ug ofactinomycinDperml was addedto
each
offive
100-mmculturesat17.5hrafterinfectionwithSendai virus; 30 min later, 100 Muc of3H-uridine
was added to each culture, and incubation was
con-tinuedat37C. Theincubation of individual cultureswas
stopped5,30, 60,120,and 240minlater by placing the
dishinanicebath andreplacingthemedium with
ice-cold PBS.Cellswereremovedwith0.25%trypsin in the
cold, collected by centrifugation, and disrupted by
Dounce homogenization as described in the text.The culturemedium, trypsin solution, and cytoplasmic
ex-tract for each incubation time were cenztrifuged
in sucrose density gradients to isolate niucleocapsid
and intact virus. The sum of the trichloroacetic
acid-precipitable
radioactivityin thenucleocapsidfromthecytoplasmic extract or homogenate (H),
nucleo-capsidfrom
thetrypsinsolution(T,),intact virusfrom thetrypsinsolution (T2),andintactvirusfrom thecul-turemedium
(V)
areplottedforeachincubationtime.thencellswere
disrupted
by
a Douncehomoge-nizer without NP40. Virus in the medium and
whole virusand
nucleocapsid
released by trypsinat each time were separated (Fig. 7), and the
nucleocapsid
was recovered from thedisrupted
cells
(Fig. IC).
Figure
9showsthe resultsofthisexperiment,
and it can be seen that 3H-uridinefirstappearsincytoplasmic nucleocapsid; second,
in
nucleocapsid
releasedbytrypsin; third, invirusreleased
by
trypsin;
and finally in virus in theculturemedium. Thisorderiscompatible withthe
structures
being
formed in a stepwise sequenceduring
virus assembly.In addition, during theex-periment, intracellular
nucleocapsidprogressivelyaccumulates,
since the rate of its formation issignificantly
greaterthan itsrate ofconversiontovirus.
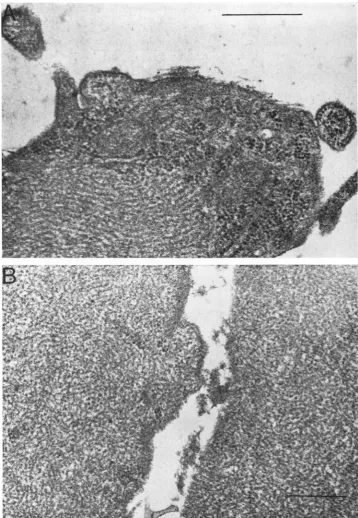
Serial electron micrographs ofSendai-infected
cells
(Fig.
10) document
this accumulation. By 18 hrpostinfection (Fig. 10A),largeaggregatesofviral
nucleocapsid
are evident in infected cellcytoplasm. By
4days
after infection (Fig. lOB), the size ofnucleocapsid
aggregates hasenlarged
J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:10.486.256.447.69.212.2] [image:10.486.48.239.443.525.2]SENDAI VIRUS REPLICATION
FIG. 10. Electronmicroscopy of Sendaivirus-infectedchick embryofibroblasts. Cellsat 18 hr(A) and4days (B)werepreparedfor electron microscopy as describedin the text.Magnlification:A, X 54,000; B,44,000.
VOL. 5, 1970 649
on November 11, 2019 by guest
http://jvi.asm.org/
[image:11.486.58.417.66.585.2]BLAIR AND ROBINSON
until much of the normal cytoplasmisdisplaced.
In other subgroup 2 myxovirus-infected cells, such as SV5-infected BHK cells (7) and
para-influenzatype2-infectedFLand HeLa cells (10), similar accumulation of viral nucleocapsid in
infected cell cytoplasm has beenshown.
These results indicate that, relative to the rate
ofviral RNA synthesis and its rapid conversion
to nucleocapsid in Sendai virus-infected chick
embryo
fibroblasts,
conversion of nucleocapsidto matureenvelopedvirus isaveryslowor
ineffi-cient step in virus formation. The radioactivity
data suggest that only approximately 5% of
newly synthesized viral RNA leaves the cell in
whole virus and the remainder progressively accumulatesinthe cellintheform ofviral
nucleo-capsid. The low yield of infectious virus by
cul-turesinwhichalmost allcellsareinfected (2) and
the serial electron micrographs showing large
amounts of nucleocapsid within infected cell
cytoplasm are in accord with this. With SV-5, another subgroup 2 myxovirus, there isevidence
that thisbehavioriscell-specific (7, 8).Thereason
for the relative block in virus formation is not
clear, but it could be due to a limiting rate of
synthesis ofanessential component of the virus,
such as a viral envelope protein. Experiments
areinprogresstocharacterize thevirionproteins
and attempts will be made to learn whether any
virus component is synthesized in limiting
amounts.
ACKNOWLEDGMENTS
Thisinvestigationwassupportedby Public HealthService re-searchgrants CA 08557andCA 10467from theNationalCancer Institute andtraininggrantTIGM 1389from theNational Insti-tuteof GeneralMedicine.
We thank Jeana Levinthal, Robley C. Williams, Charlotte Lizarraga,andJoseph Toby for assistance with electron
micros-copy.
LITERATURE CITED
1. Baltimore,D.,M.Girard,andJ. E.Darnell. 1966.Aspectsof thesynthesisofpoliovirusRNA and theformationofvirus particles. Virology 29:179-189.
2.Blair, C. D.,andW. S.Robinson.1968.ReplicationofSendai
virus.I.Comparisonof viral RNA andvirus-specific RNA
synthesistoNewcastle disease virus. Virology 35:537. 3. Bratt, M. A., andW. S. Robinson. 1967. Ribonucleic acid
synthesisin cellsinfected withNewcastlediseasevirus. J. Mol. Biol. 23:1-21.
4.Bukrinskaya, A. G.,S.M.Klimenlco,J.A.Smirnov,and B. V. Guschin. 1968. Infective substructures of Sendai virusfrom infectedEhrlich ascitestumorcells. J. Virol. 2:752-758. 5.Compans,R.W.,andP. W. Choppin. 1967a. Isolationand
propertiesof thehelicalnucleocapsidof theparainfluenza virusSV5.Proc. Nat. Acad. Sci. U.S.A.57:949-956. 6.Compans,R.W.,and P.W. Choppin. 1967b. The lengthof
the helicalnucleocapsidofNDV.Virology 33:344-346. 7.Compans,R.W.,K.V.Holmes,S.Dasel,andP.W.Choppin.
1966. An electron microscopic study of moderate and virulent virus-cell interactions of the parainfluenza virus SV5. Virology 30:411-426.
8.Holmes, K. V., and P.W.Choppin. 1966. On the role ofthe
response of the cellmembrane indetermining virus viru-lence;contrasting effect of the parainfluenza virus SV5 in
twocelltypes.J.Exp. Med.124:501-519.
9. Hosaka,Y.,H.Kitano,andS.Ikeguchi. 1966.Studiesonthe
pleomorphismof HVJ virions.Virology 29:205-221. 10. Howe, C., C. Morgan,C.deVaux St. Cyr,K.C. Hsu,and
H.M. Rose. 1967.Morphogenesisoftype2parainfluenza virusexamined by light and electron microscopy. J. Virol. 1:215-237.
11. Hoyle, L. 1952. Structure of the influenza virus. Therelation between biologicalactivity and chemicalstructureofvirus fractions. J. Hyg. 50:229-245.
12. Kingsbury,D. W., and R. W.Darlington. 1968. Isolationand properties of Newcastle disease virus nucleocapsid. J. Virol. 2:248-255.
13. Kuhn, N. O., and C. G. Harford. 1963. Electron microscopic examination of cytoplasmic inclusion bodies in cells in-fected withparainfluenza virus, type2. Virology 21:527-530.
14. Reynolds,E.S.1963. Theuseof lead citrateathighpHasan
electron-opaquestain inelectronmicroscopy. J. Cell Biol. 17:208-212.
15. Rott, R., 1.M. Reda, and W. Schafer. 1962. Isolationand
characterization ofhemagglutinating non-infectious par-ticles produced during multiplication of NDV. Virology 16:207-209.
16.Weinberg, R.A., V.Loening, M.Willems, and S. Penman. 1967. Acrylamide gel electrophoresis of HeLa cellnucleolar RNA. Proc. Nat. Acad. Sci.U.S.A.58:1088-1095.