Comparative Analysis of Hepatitis B Virus Polymerase Sequences
Required for Viral RNA Binding, RNA Packaging, and Protein
Priming
Scott A. Jones,aDaniel N. Clark,aFeng Cao,b* John E. Tavis,cJianming Hua
Department of Microbiology and Immunology, The Pennsylvania State University College of Medicine, Hershey, Pennsylvania, USAa
; VirRx, Inc., St. Louis, Missouri, USAb ; Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, Saint Louis, Missouri, USAc
Hepatitis B virus replicates a DNA genome through reverse transcription of a pregenomic RNA (pgRNA) by using a multifunc-tional polymerase (HP). A critical function of HP is its specific association with a viral RNA signal, termed(H), located on pgRNA, which is required for specific packaging of pgRNA into viral nucleocapsids and initiation of viral reverse transcription. HP initiates reverse transcription by using itself as a protein primer (protein priming) and Has the obligatory template. HP is made up of four domains, including the terminal protein (TP), the spacer, the reverse transcriptase (RT), and the RNase H do-mains. A recently developed, H-dependent,in vitroprotein priming assay was used in this study to demonstrate that almost the entire TP and RT domains and most of the RNase H domain were required for protein priming. Specific residues within TP, RT, and the spacer were identified as being critical for HP-Hbinding and/or protein priming. Comparison of HP sequence require-ments for Hbinding, pgRNA packaging, and protein priming allowed the classification of the HP mutants into five groups, each with distinct effects on these complex and related processes. Detailed characterization of HP requirements for these related and essential functions of HP will further elucidate the mechanisms of its multiple functions and aid in the targeting of these functions for antiviral therapy.
H
epatitis B virus (HBV), a member of theHepadnaviridae fam-ily, chronically infects over 350 million people worldwide and causes a million mortalities per year due to hepatic failure, cirrho-sis, and hepatocellular carcinoma (1). HBV encodes a multifunc-tional polymerase (HP), a specialized reverse transcriptase (RT) which replicates the viral 3.2-kb, partially double-stranded DNA genome via an RNA intermediate termed pregenomic RNA (pgRNA) (2–4). HP is composed of four domains, including, from the N terminus to the C terminus, the terminal protein (TP), the spacer that is nonessential for HP functions, the RT, and the RNase H domains (Fig. 1) (3,5–7). The RT domain of HP, with its YMDD active site, and the RNase H domain, with its D-E-D-D motif, are homologous to the retroviral counterparts, while TP is unique to hepadnaviruses (6–9).Both the initiation of viral reverse transcription and the assem-bly of replication-competent nucleocapsids in HBV depend criti-cally on the specific interaction between HP and pgRNA via an RNA element, termed epsilon (Hε), near the 5=end of pgRNA in a host chaperone-dependent process (10–16). Thus, HP-Hε bind-ing is required for packagbind-ing of both HP and pgRNA into nucleo-capsids, where viral DNA synthesis occurs (17–21). Furthermore, the HP-Hεinteraction also triggers initiation of viral reverse tran-scription in a process termed protein priming, which occurs inde-pendently of nucleocapsids (22–30). In protein priming, HP func-tions both as a protein primer, with the hydroxyl group of a specific tyrosine residue (Y63) on its TP domain used to initiate viral DNA synthesis, as well as the catalyst, with the RT active site being required to form a phosphotyrosyl linkage between Y63 and the first nucleotide residue of the viral minus-strand DNA. Hεalso serves two essential roles during protein priming, acting as the obligatory template for viral DNA synthesis and also as an activa-tor of HP catalytic activity.
Analyses of the structural requirements of both HP and Hεfor
HP-Hεinteractions have revealed the sequences of both HP and Hεto be required for this process (13,14,26). Minimum require-ments of HP for binding Hεinclude the central region of TP and the N-terminal region of RT, while the RNase H domain is com-pletely dispensable. Hε, which features a conserved lower stem, an internal bulge, an upper stem, and an apical loop, requires only its central portion adjacent to and including the internal bulge for HP binding, while its apical loop is entirely dispensable (13,26,
31–35). While the HP-Hεinteraction is required for packaging
of HP and pgRNA into nucleocapsids, pgRNA packaging has additional requirements beyond HP-Hε binding. In particular, pgRNA packaging appears to require the entire length of HP and all the structural elements of Hε plus a closely spaced 5= cap (26,36).
Biochemical and genetic studies to identify HP sequences required for protein priming have been limited, until very re-cently, by the lack ofin vitroassays that faithfully recapitulate HP protein priming under cell-free conditions. By employing HP expressed in insect cells, which displays Hε-independent protein priming activityin vitro, it was shown that the YMDD active-site motif in RT and Y63 in TP are both required for protein priming in this system, as are TP, RT, and most of the RNase H domain (27,37–39). However, this Hε-independent
Received27 September 2013Accepted10 November 2013 Published ahead of print13 November 2013
Address correspondence to Jianming Hu, [email protected].
* Present address: Feng Cao, John Cochran Division, Department of Veteran’s Affairs Medical Center, St. Louis, Missouri, USA.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02852-13
on November 7, 2019 by guest
http://jvi.asm.org/
priming activity may represent the template-independent, pro-tein-primed terminal transferase activity of HP that we discov-ered recently (28) and may not faithfully recapitulate the HP requirements for Hε-dependent protein priming. Genetic studies in cell culture have identified residues within TP and RT that are required for viral replication, although their spe-cific roles in Hε binding and protein priming remain to be defined. R105 and Y173 in TP are both important for RNA packaging (40), and W74 and Y147 in TP are important for viral DNA synthesis but not pgRNA packaging (41). Three cys-teine residues near the C terminus of the spacer and one at the N terminus of RT are also critical for RNA packaging (42). In addition, the polymerase from the related duck HBV (DHBV) has been used extensively as a model system for HP. The DHBV polymerase (DP) requires the central portion of TP and the N-terminal part of RT for DP-DHBVε(Dε) binding and pro-tein priming, with the remaining sequences, including the en-tire RNase H domain, being dispensable (12,43,44). Further analysis of DP led to the identification of the T3 and RT1 motifs in TP and RT, respectively, which are highly conserved between DP and HP and are critical for DP-Dε binding and protein priming (Fig. 1) (45,46).
We recently developed an Hε-dependent, HPin vitropriming system using HP purified from human cells as a priming-active
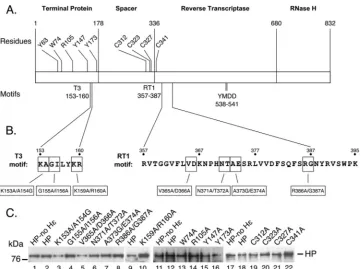
HP-Hεcomplex (26). Using this system, we demonstrated that the Hεrequirements to support HP protein priming are remarkably similar to those for pgRNA packaging (26). This same system al-lows the simultaneous measurement of HP-Hεbinding in living cells (26). The exact same HP proteins used in the HP-Hεbinding and protein priming reactions can also be used intrans -comple-mentation assays to determine their ability to support pgRNA packaging in the same host cells as those used for the other assays. These systems therefore afford the opportunity to carry out com-parative analysis of HP and Hε requirements for protein-RNA interactions, pgRNA packaging, and protein priming, all in the same host cells and using the same HP and Hεconstructs. Taking advantage of these systems, we have characterized two major groups of HP mutants for their effects on HP-Hεbinding and protein priming. The first set of mutants included single- and multiple-residue substitutions in TP and RT, both within and out-side the T3 and RT1 motifs, which are known to affect HP activi-ties and HBV replication. The second set of mutants included extensive domain truncations in HP in an attempt to map the domain boundaries of HP for protein priming. Furthermore, Hε binding and pgRNA packaging activities of selected HP mutants were also determined by using the same HP expression system to allow direct comparison of the HP requirements for all three re-FIG 1HP sequences and motifs important for HP functions and HP mutant expression. (A) HP is depicted, showing the TP, spacer, RT, and RNase H domains. Positions of previously identified residues and motifs important for HP function are highlighted. (B) The amino acid residues of the T3 and RT1 motifs are denoted, along with the mutations within these motifs used in this study. T3 and RT1 HP mutants were analyzed for RNA packaging in a previous study but were named differently because of the use of HP in the adw2 strain (48), which differs slightly in amino acid sequence from the HP in the ayw strain used in this study. For ease of comparison, the mutant names used in this study are shown with the alternate names, in parentheses, used in that study: K153A/A154G (HT3 155/156), G155A/I156A (HT3 157/158), K159A/R160A (HT3 161/162), V365A/D366A (HRT1 378 –379), N371A/T372A (HRT1 384 –385), A373G/E374A (HRT1 386 –387), and R386A/G387A (HRT1 399 – 400). The cysteine mutants were originally named C135A, C146A, C150A (within the spacer), and RTC06A (within RT) (42) but are designated in this study according to their positions in the context of full-length HP. (C) All mutant HP proteins were expressed and purified with Hε(lanes 3 to 8, 10, 13 to 16, and 19 to 22), while WT HP was expressed with (lanes 2, 9, 12, and 18) or without (lanes 1, 11, and 17) Hε. WT and mutant HPs were resolved by SDS-PAGE and detected by Western blot analysis using the anti-Flag antibody. Full-length HP and the 76-kDa protein molecular mass marker are indicated.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.113.474.63.332.2]lated HP functions, namely, Hεbinding, pgRNA packaging, and protein priming.
MATERIALS AND METHODS
Plasmids.Wild-type (WT) HP was expressed by using pcDNA-3FHP, which encodes an N-terminally 3⫻Flag-tagged HP gene (strain ayw) un-der the cytomegalovirus (CMV) promoter, while Hεwas coexpressed by using pCMV-HE, as previously described (26,28,47). For the evaluation of HBV T3 and RT1 motifs for their contribution toin vivoRNA binding, paired alanine-scanning mutations within the HP T3 and RT1 motifs (48) were transferred to pcDNA-3FHP. Plasmids for expressing point muta-tions affecting selected hydrophobic and charged residues in TP outside the T3 motif and the four cysteines in the spacer and RT along with the corresponding WT HP (40–42) were kindly provided by Wang-Shick Ryu. These constructs are in a different vector background from those used to express the T3 and RT1 mutants and contain additional HBV sequences downstream of the HP coding sequence such that the HP mRNAs produced contain a 3=copy of Hεafter the HP coding sequence. PCR-mediated mutagenesis was used to produce N-terminal, C-terminal, and internal HP deletions in the pcDNA-3FHP background.
Protein expression and purification,in vivoHbinding, and RNA packaging.WT and mutant HP proteins were expressed in HEK293T cells, with or without coexpression of the HεRNA, and purified by using protein A/G beads coupled to anti-Flag antibody, as previously described (26). Approximately 2g HP and 3 ng Hεwere expressed per 10-cm dish of transfected cells (26). RNA was extracted from purified HP proteins or cytoplasmic lysates and detected by Northern blot analysis following res-olution by urea-polyacrylamide gel electrophoresis (PAGE), as previously described (26). The RNA packaging assay was also conducted by using transiently transfected HEK293T cells, as previously outlined (26), using pCMVHBV-Pol⫺to express pgRNA and all viral proteins except HP and the HP expression plasmids to provide HPin trans.
In vitroprotein priming assay.WT HP and HP mutants were ana-lyzed for their protein priming activitiesin vitroas previously described (26,28,47), with minor modifications. Flag lysis buffer from HP purifi-cation was removed from aliquots of HP-bound anti-Flag affinity beads. TMgNK buffer (20 mM Tris-HCl [pH 7.0], 15 mM NaCl, 10 mM KCl, 4 mM MgCl2) with 1⫻EDTA-free protease inhibitor cocktail (Roche), 4
mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1 unit RNasin Plus RNase inhibitor (Promega) perl buffer was added to the beads. One microliter of [␣-32P]dGTP (10 mCi/ml and 3,000
Ci/mmol; PerkinElmer) was then added, and the reaction mixtures were incubated at 25°C for 4 h with shaking. Alternatively, protein priming was performed in TMnNK buffer (same as TMgNK buffer except containing 2 mM MnCl2instead of MgCl2). The beads were then washed in TNK buffer
(20 mM Tris-HCl [pH 7.0], 15 mM NaCl, 10 mM KCl) plus protease inhibitors (28M E-64, 1 mM PMSF, 5g/ml leupeptin) and 10 mM -mercaptoethanol. The washed beads were then boiled in 2⫻sodium dodecyl sulfate (SDS) sample buffer for 10 min to elute the bound pro-teins. Radiolabeled HP as a result of protein priming was resolved by running the eluate on an SDS–12.5% polyacrylamide gel and detected by autoradiography.
RESULTS
Expression and purification of HP substitution mutants.We se-lected four groups of single- and double-substitution HP mutants for analysis based on their phenotypes in previous studies: (i) T3 motif mutants, (ii) RT1 motif mutants, (iii) mutations of certain charged and hydrophobic residues in TP outside T3, and (iv) four cysteines in the spacer and RT (Fig. 1Aand B). A number of mutations in the T3 and RT1 motifs of HP were recently assayed for their HεRNA binding activity using anin vitroRNA binding assay in which purified HP was incubated with anin vitro -tran-scribed HεRNA (26,28,47), and many were found to be defective
in HP-Hε binding (48). Four RT1 mutants (V365A/D366A, N371A/T372A, A373G/E374A, and R386A/G387A), all compe-tent in Hεbindingin vitrobut differing in their pgRNA packaging activities (seeFig. 8Afor a summary), were selected here for fur-ther analysis to determine their Hεbindingin vivoand protein priming activities. Three T3 mutants, K153A/A154G, G155A/ I156A, and K159A/R160A, which retained ca. 20%, 10%, and
⬍5% of Hεbinding activity, respectively,in vitrocompared to WT HP and varying pgRNA packaging activity (seeFig. 8A), were also analyzed. Additional single-residue substitution mutants in TP outside T3 (W74A, R105A, Y147A, and Y173A), in the spacer (C312A, C323A, and C327A), and in RT (C341A) were selected because they are defective in viral RNA packaging or DNA synthe-sis, but their Hεbinding or protein priming activities were not defined (seeFig. 8A) (40–42).
The mutant HP constructs were cotransfected into HEK293T cells with a construct expressing Hεand purified as an HP-Hεcomplex
(Fig. 1C, lanes 3 to 8, 10, 13 to 16, and 19 to 22) (26). In parallel, WT
HP was also purified with or without Hε(Fig. 1C, lanes 1, 2, 9, 11, 12, 17, and 18). Western blot analysis of WT and mutant HP proteins showed that the HP mutants were expressed at least as well as WT HP, and they all migrated at ca. 90 kDa (Fig. 1C).
In vivoHbinding activity of HP substitution mutants.To
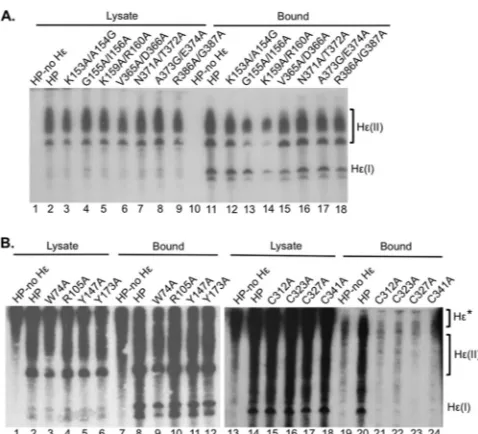
assess the ability of the HP mutants to bind to Hεin vivo, RNAs bound to purified HP proteins were extracted and detected by Northern blotting (26). Of the T3 mutants tested, the G155A/ I156A and K159A/R160A mutants showed reduced (by ca. 70%) but readily detectable HP-Hεbindingin vivo(Fig. 2A, lanes 13 and 14, and seeFig. 8A). All other T3 and RT1 motif mutants tested showed WT-level Hεbinding activity (Fig. 2A, lanes 12 and 15 to 18). Of the single-residue substitution mutants outside the T3 motif, three (R105A, Y147A, and Y173A) bound Hεat near-WT levels, and the remaining one (W74A) showed a modest (by ca. 40%) decrease in Hεbinding (Fig. 2B, lanes 9 to 12, and seeFig. 8A). All four cysteine mutants, C312A, C323A, C327A, and C341A, were severely defective in Hεbinding (Fig. 2B, lanes 21 to 24, and seeFig. 8A).
Protein priming activity of HP substitution mutants. Puri-fied HP proteins, with or without bound Hε, were tested for their ability to carry out protein priming by using a recently developed
in vitroprotein priming assay (26). Two mutants in the T3 motif, G155A/I156A and K159A/R160A, and two in the RT1 motif, V365A/D366A and R386A/G387A, were severely defective (de-ceased by ca. 90% to undetectable) in protein priming (Fig. 3A, lanes 4, 5, 6, and 9, and seeFig. 8A). The remaining T3 mutant, K153A/A154G, showed WT-level priming activity (Fig. 3A, lane 3, and seeFig. 8A). The remaining two RT1 motif mutants, N371A/ T372A and A373G/E374A, showed modestly reduced (by ca. 70% and 50%, respectively) protein priming activity (Fig. 3A, lanes 7 and 8, and seeFig. 8A). All the other HP mutants, with substitu-tions of the charged or hydrophobic residues in TP (outside T3) or the conserved cysteines in the spacer and RT for alanines, were completely defective in protein priming (Fig. 3A, lanes 12 to 15 and 17 to 20, and seeFig. 8A).
We recently discovered a novel, template-independent but protein-primed, terminal transferase activity of HP, which is ac-tivated when Mn2⫹instead of Mg2⫹is used as the metal ion co-factor (28). The synthesis of long stretches of DNA by this trans-ferase activity depends on the RT active site and Y63 in TP as the primer, similar to Hε-templated viral DNA synthesis. The novel Jones et al.
on November 7, 2019 by guest
http://jvi.asm.org/
transferase activity of HP thus provides a system to dissect re-quirements of protein-primed DNA synthesis, such as primer po-sitioning and catalysisper se, that are independent of the require-ments for an Hε interaction. Therefore, we determined the transferase activities of the various HP mutants in the presence of Mn2⫹instead of Mg2⫹(Fig. 3B). As we reported recently (28), the protein priming signal with Mn2⫹(i.e., transferase activity) in the presence or absence of Hε(Fig. 3B, lanes 1, 2, 10, 11 and 16) was stronger than that with Mg2⫹. This was probably due to the incor-poration of multiple radiolabeled dGMP residues into the DNA strand that is attached to each HP molecule and also because more HP molecules were active in the transferase activity than in the Hε-dependent priming with Mg2⫹(28). The relative priming ac-tivities with Mn2⫹of most HP mutants, compared to WT HP, were similar to those with Mg2⫹(Fig. 3B, lanes 3 to 9, 12, 13 15, and 17 to 20, and seeFig. 8A). However, the Y147A mutant clearly had a protein-primed transferase activity with Mn2⫹that was sim-ilar to that of WT HP (Fig. 3B, lane 14, and seeFig. 8A). This stood in sharp contrast to its severe defect in Hε-dependent protein priming with Mg2⫹(Fig. 3A, lane 14 and seeFig. 8A). Mapping of HP domain boundaries for Hbinding, protein priming, and pgRNA packaging.To define the boundaries of the TP, RT, and RNase H domains required for HP functions in Hε
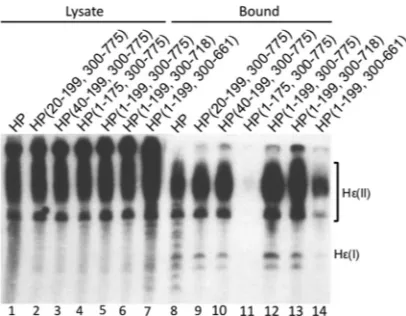
binding, protein priming, and pgRNA packaging, we created N-terminal, internal (within the spacer), and C-terminal HP trunca-tions (Fig. 4A). All truncation mutants contained an internal de-letion in the spacer region from positions 200 to 299 except for HP(1–175, 300 –775), which had a deletion from positions 176 to 299. The mutants were all expressed at least as well as WT HP (Fig. 4B). When the truncation mutants were tested for their ability to bind Hεin vivo, all the mutants had WT-like Hεbinding activity except for HP(1–199, 300 – 661), which showed ca. 30% of WT activity, and HP(1–175, 300 –775), which strikingly lost all Hε binding activity (Fig. 5). When these same mutants were tested for their ability to carry out protein priming in the presence of Mg2⫹, only one mutant, HP(1–199, 300 –775), showed priming activity that was similar to that of WT HP (Fig. 6A, lane 5). All the other truncation mutants were completely defective in Hε-dependent protein priming (Fig. 6A). When the mutants were tested for their ability to carry out protein priming in the presence of Mn2⫹(i.e., Hε-independent transferase activity), HP(1–199, 300 –775) again had WT-like priming activity (Fig. 6B, lane 5). HP(1–199, 300 – 718) showed a low level (ca. 10% of the WT level) of priming activity (Fig. 6B, lanes 6 and 13, and seeFig. 8A), and very weak priming signals (⬍5% of the WT level) were detectable with HP(20 –199, 300 –775), HP(40 –199, 300 –775), and HP(1–175, 300 –775) (Fig. 6B, lanes 9 to 11, and seeFig. 8A). No priming signal could be detected with HP(1–199, 300 – 661) (Fig. 6B, lane 14).
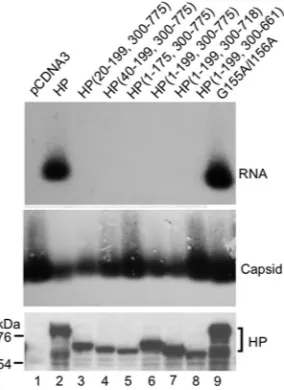
We also measured the ability of the truncated HP constructs to support pgRNA packaging using the trans-complementation RNA packaging assay. We found that all the truncation mutants, including the minimal C-terminal truncation mutant HP(1–175, 300 –775), were completely defective in pgRNA packaging (Fig. 7, lanes 3 to 8). Capsid assembly or HP expression was not impaired by the HP truncations (Fig. 7). The T3 G155A/I156A mutant was FIG 2Northern blot detection ofin vivoHP-Hεinteractions by HP
substitu-tion mutants. (A) T3 and RT1 mutants. (B) Non-T3 TP mutants and cysteine mutants. HP expressed alone (A, lanes 1 and 10; B, lanes 1, 7, 13, and 19) or HP or HP mutants coexpressed with Hε(A, lanes 2 to 9 and 11 to 18; B, lanes 2 to 6, 8 to 12, 14 to 18, and 20 to 24) were purified as outlined in Materials and Methods. RNAs were extracted from cytoplasmic lysates (A, lanes 1 to 10; B, lanes 1 to 6 and 13 to 18) or from the immunoaffinity-purified HP pellets (A, lanes 11 to 20; B, lanes 7 to 12 and 19 to 24) and were resolved on a urea– 6% PAGE gel. A riboprobe specific for Hεwas then used to detect lysate or bound Hε. Hε(II) represents the Hε-containing RNA produced from the transfected plasmid that is polyadenylated by using the strong bovine growth hormone poly(A) site ca. 220 nucleotides downstream from the weak HBV poly(A) site, which is used to produce the shorter Hε-containing RNA, designated Hε(I) (26). Hε* denotes the Hε-containing RNA expressed as part of the much longer mRNA that encodes the WT or mutant HP proteins used in panel B. The HP expression plasmids used in panel B are different from those used in panel A and contain a 3=copy of Hεdownstream of the HP coding sequence. This RNA also showed binding to HP (B, lanes 7 and 19).
FIG 3In vitroprotein priming activities of HP substitution mutants. HP purified without Hε(lanes 1 and 10), or HP or HP mutants copurified with Hε (lanes 2 to 9 and 11 to 20), were incubated in the presence of Mg2⫹(A) or
Mn2⫹(B) and [␣-32P]dGTP as the nucleotide substrate to allow protein prim-ing. Following priming, the labeled HP proteins were resolved on an SDS– 12.5% PAGE gel. The gel was dried, and primed HP proteins were detected by autoradiography. The primed HP products are indicated along with the posi-tions of the protein molecular mass markers.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.43.282.63.280.2] [image:4.585.301.540.65.258.2]also included in the pgRNA packaging assay and was found to be as active as WT HP (Fig. 7, lane 9), as we reported recently (48).
DISCUSSION
Using our recently developedin vivoHεRNA binding and Hε -dependent and -in-dependentin vitroprotein priming assays, we analyzed specific residues, motifs, and domain boundaries of HP that are important for HP-Hεbinding and protein priming and further compared their roles in these processes to their role in pgRNA packaging (Fig. 8). Our results demonstrate that specific residues in both the TP and RT domains, including those within the T3 and RT1 motifs and elsewhere, are critical for Hεbinding, protein priming, and pgRNA packaging. We have also found that almost the entire TP and RT domains and most of the RNase H
domain were required for protein priming. Furthermore, pgRNA packaging required even more sequences from the RNase H do-main than did protein priming. In contrast, only portions of TP and RT were needed forin vivoHεbinding.
Comparison of HP sequence requirements for Hε binding, protein priming, and pgRNA packaging allows five distinct groups of HP mutants tested here to be classified (Fig. 8A): competent for all the HP activities tested (K153A/A154G at the N-terminal boundary of the T3 motif and N371A/T372A and A373G/E374A within the RT1 motif) (group 1), competent for Hεbinding and pgRNA packaging but defective for protein priming (W74A, Y147A, and G155A/I156A in T3, and V365A/D366A in RT1) (group 2), competent for Hεbinding and protein priming but defective for pgRNA packaging [HP(1-199, 300-775)] (group 3), competent for Hεbinding but defective in pgRNA packaging and protein priming [R105A and Y173A in T3, R386A/G387A in RT1, HP(20-199, 775), HP(40-199, 775), HP(1-199, 300-718), and HP(1-199, 300-661)] (group 4), and defective in Hε binding, pgRNA packaging, and protein priming [K159A/R160A in T3, all four cysteine mutants in the spacer and RT, and HP(1-175, 300-775)] (group 5). Thus, our results clearly show that both protein priming and pgRNA packaging require additional HP se-quences beyond those for Hεbinding and furthermore that there are distinct HP requirements for pgRNA packaging and protein priming that can be genetically separated.
Our recent studies identified multiple T3 motif residues as im-portant for Hεbinding using thein vitroRNA binding assay (48). We found here that two T3 mutants (K159A/R160A and, to a lesser degree, G155A/I156A) also showed reduced Hεbindingin vivo. This confirms an important role of T3 in Hεbinding, al-though the extent of the reduction was not as great as that found in thein vitroRNA binding assay. The four cysteines targeted here are conserved in the mammalian (but not the avian) hepadnavi-ruses and are known to be critical for pgRNA packaging (42). The requirement of these residues for Hεbinding provides an expla-nation for their role in pgRNA packaging observed previously and in protein priming found here. The domain truncation mutant in group 5 [HP(1-175, 300-775)] further suggests that the 24 amino FIG 4Expression of HP domain truncation mutants. (A) Diagram of HP mutants truncated at the N terminus, within the spacer region, and at the C terminus. Full-length HP is depicted at the top, with primer Y63 and the RT active-site motif YMDD highlighted. (B) HP was expressed in and purified from HEK293T cells. Immunopurified HP and HP mutants coexpressed with Hεwere resolved on an SDS–12.5% PAGE gel and detected by Western blot analysis. The HP proteins and the antibody heavy chain (HC) are indicated, as are the positions of the protein molecular mass markers.
FIG 5In vivoHεbinding activity of HP domain truncation mutants. WT or mutant HP was expressed together with Hεand purified as outlined in Mate-rials and Methods. RNAs were extracted from cytoplasmic lysates (lanes 1 to 7) or from the immunoaffinity-purified HP pellets (lanes 8 to 14) and were re-solved on a urea– 6% PAGE gel. A riboprobe specific for Hεwas then used to detect lysate or bound Hε. Hε(II) represents the Hε-containing RNA pro-duced from the transfected plasmid that is polyadenylated by using the strong bovine growth hormone poly(A) site ca. 220 nucleotides downstream from the weak HBV poly(A) site, which is used to produce the shorter Hε-containing RNA, designated Hε(I) (26).
Jones et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.94.485.64.235.2] [image:5.585.61.264.471.629.2]acid residues at the junction between TP and the spacer (positions 176 to 299) may also be critical for Hεbinding. Thus, T3, the C-terminal region of TP, the conserved cysteines in the spacer and RT, and the RT1 motif in RT (48) are all required for Hεbinding. Whether any of these sequences are involved directly in RNA binding or play a more indirect role (e.g., affecting the folding of local regions involved in Hεbinding) remains to be determined.
We have identified a large number of HP mutants (group 4)
distributed in TP, RT, and the RNase H domain that were defec-tive in both protein priming and RNA packaging while remaining competent in Hε binding. This result supports the notion that both of these processes share similar requirements post-RNA binding, as also suggested by our previous observation that the Hε RNA requirements for pgRNA packaging and protein priming are similar (26). How the sequences involved facilitate both of these HP functions beyond RNA binding remains to be elucidated. One possibility is that they may help establish an HP conformation, induced upon Hεbinding, that is required for activation of the HP enzymatic activity as well as its RNA packaging function. Indeed, DP interaction with its cognate DεRNA has been shown to induce DP conformational changes required for protein priming (23,24). On the other hand, the identification of the group 2 mutants de-fective in protein priming but competent for pgRNA packaging and the group 3 mutant competent for protein priming but defec-tive for pgRNA packaging, with both groups being competent in Hεbinding, clearly indicates that there are also distinct require-ments for these two related processes, which are both triggered by HP-Hεbinding. Similarly, DP mutants defective in pgRNA pack-aging but competent in protein priming have also been reported (49).
Previous studies showed that HP mutants truncated at posi-tion 744 (20) or position 729 (50) were defective in pgRNA pack-aging. Our analysis here has extended the C-terminal boundary of HP required for pgRNA packaging to beyond residue 775. The limited spacer (positions 200 to 299) deletion did not affect pgRNA packaging (S. A. Jones and J. Hu, unpublished results), in agreement with a previous report (20). One interpretation for the phenotype of the HP(1-199, 300-775) mutant is that the RNase H sequences C terminal to residue 775 are specifically required for pgRNA packaging but not for protein priming, e.g., via an inter-action with the viral capsid protein during nucleocapsid assembly. The sequences involved in the group 2 mutants, especially the TP sequences (W74, Y147, and G155/I156 in T3), may play a role in positioning Y63 in the RT active site to allow priming. The defect of W74A and Y147A mutants in protein priming could also ex-FIG 6In vitroprotein priming activity of HP domain truncation mutants. Immunopurified full-length HP or HP truncation mutants coexpressed with Hεwere assayed for protein priming activity, as outlined in the legend ofFig. 3, in the presence of Mg2⫹(A) or Mn2⫹(B). Lanes 8 to 14 of panel B represent a longer
exposure of lanes 1 to 7. The primed HP products are indicated along with the positions of the protein molecular mass markers.
FIG 7RNA packaging activity of domain truncation mutants. HEK293T cells were cotransfected with pCMVHBV-Pol⫺, an HBV replication construct en-coding pgRNA and all viral proteins except HP, and the indicated HP con-struct or the pCDNA3 vector control. Viral RNA packaged into nucleocapsids was detected by resolving the capsids on an agarose gel, after which they were transferred onto a nitrocellulose membrane. A riboprobe specific for Hεwas used to detect packaged RNA (top), while the same membrane was probed subsequently with an anti-HBV core antibody (middle). Cell lysates were also resolved by SDS-PAGE, and HP expression was detected by Western blotting using the anti-Flag antibody (bottom). The positions of HP and the protein molecular mass markers are indicated.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.115.466.67.262.2] [image:6.585.93.235.427.622.2]FIG 8HP requirements for Hεbinding, pgRNA packaging, and protein priming. (A) The abilities of the various HP mutants to bind Hεin vivo(this study), to mediate pgRNA packaging (this and previous studies), and to carry out protein priming (this study) are summarized. ** indicates that the RNA packaging results designated “positive” or “negative” were obtained in previous analyses (40–42,48). All results reported here represent the averages of at least three independent experiments.⫺,⬍10% of WT activity;⫹, 10 to 30% of WT activity;⫹⫹, 30 to 70% of WT activity;⫹⫹⫹,⬎70% of WT activity; ***, differential priming activity of the Y147A mutant with Mn2⫹versus Mg2⫹(see the text for details). Group designations of HP mutants are as follows: 1, competent for Hεbinding, protein
priming, and RNA packaging; 2, competent for Hεbinding and RNA packaging but defective in protein priming; 3, competent for Hεbinding and pro-tein priming but defective in RNA packaging; 4, competent for Hεbinding but defective in protein priming and RNA packaging; 5, defective in Hεbinding, protein priming, and RNA packaging. (B) HP is depicted with its domain boundaries. Residues and motifs are highlighted with different symbols to indicate that mutations at these sites affected HP-Hεbinding (RNA binding), RNA packaging, and/or protein priming. (C) Minimal domain requirements of HP for RNA binding (13; this study) and Hε-dependent protein priming (this study) are indicated by thick bars.
Jones et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.77.510.64.624.2]plain the previously reported observation that these mutants could package pgRNA but could not make viral DNA (41). The RT1 V365/D366 sequences could also be involved in primer posi-tioning or perhaps more directly in catalysis.
Our mapping results for HP domain boundaries involved in Hεbindingin vivoare fully consistent with our previous results obtained using anin vitroRNA binding assay with bacterially ex-pressed and chaperone-reconstituted HP (13). Both studies showed that the N-terminal portion of TP, the C-terminal portion of RT, and the entire RNase H domain are dispensable for Hε binding (Fig. 8C). This is similar to the requirement of DP in Dε binding (12,44). On the other hand, the HP domain requirements for protein priming are dramatically different from those for DP. Almost the entire TP and RT domains and the majority of the RNase H domain were required for protein priming in the pres-ence of either Mg2⫹(Hεdependent) or Mn2⫹(Hεindependent). The sequences at the junction between TP and the spacer (posi-tions 176 to 199) were found to be critical for the Hε-independent transferase activity both in this study and in a previous report (37), in addition to its essential role in Hεbinding, as discussed above. In contrast, DP can carry out protein priming efficiently without the RNase H domain entirely and with significant truncations at the N terminus of TP and the C terminus of RT (43,51). Also, R105 in HP is critical for both protein priming (this study) and pgRNA packaging (40). In contrast, the equivalent DP residue, R128, is not required for DP protein priming or pgRNA packaging (52). These discordances between HP and DP highlight important differences in the sequence requirements for protein priming and RNA packaging between these two related proteins and suggest caution in extrapolating observations made with DP to HP.
The strong, Mn2⫹-dependent, and template-independent pro-tein priming (i.e., transferase) activity of the Y147A mutant, in contrast to its severe defect in Mg2⫹-dependent and Hε-templated protein priming, indicates that it is possible to separate genetically these activities (Fig. 3and8A). This may also be true, to a lesser extent, for the W74A and R386A/G387A mutations. These muta-tions also had little effect on HP-Hεbinding. Assuming that the Y63 primer is positioned into the RT active site similarly for catal-ysis of the covalent phosphotyrosyl linkage between the nucleo-tide and Y63 in both priming reactions, the deleterious effect of these mutations preferentially on Hε-dependent protein priming versus Hε-independent transferase activity may suggest that they are specifically involved in properly aligning the HεRNA template relative to the RT active site and/or the Y63 primer. On the other hand, the HP requirements for both priming reactions are quite similar, including the need for the YMDD RT active site and the Y63 primer, the domain boundary requirements, and the fact that most HP mutations tested here affected the two priming reactions similarly. The domain requirements of protein priming using HP expressed in insect cells are also very similar to what we have defined here (37). Although the protein priming activity of the insect cell-derived HP was conducted with Mg2⫹, it was Hε inde-pendent, and its nucleotide selectivity also mimicked the trans-ferase activity that we detected using human cell-derived HP and Mn2⫹. Thus, the transferase activity of HP may also manifest itself in the presence of Mg2⫹when it is expressed in heterologous host cells.
All current direct-acting antiviral drugs used to treat HBV in-fections are nucleos(t)ide analogs that inhibit the DNA polymer-ase activity of HP (2,53,54). Furthermore, with few exceptions,
these drugs inhibit the elongation stage of viral DNA synthesis, with no effect on protein priming (2,47). Thus, the protein prim-ing function as well as the Hε binding and pgRNA packaging functions of HP represent un- or underexploited targets for anti-viral development against HBV. Our analysis of the HP require-ments for these essential and related viral processes has revealed additional HP targets for developing novel antivirals to inhibit HP functions beyond its DNA polymerase activity.
ACKNOWLEDGMENTS
We thank Wang-Shick Ryu for kindly providing HP expression plasmids. This work was supported by Public Health Service grants (R01 AI074982 to J.H. and R43 AI084232 to F.C. at VirRx, Inc.) from the Na-tional Institutes of Health. S.A.J. was supported by Viruses and Cancer training grant 2 T32 CA60395 from the National Cancer Institute.
REFERENCES
1.Seeger C, Mason WS, Zoulim F.2007. Hepadnaviruses, p 2977–3029.In
Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
2.Jones SA, Hu J. 2013. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg. Microbes Infect.2:e56.http://dx.doi.org/10.1038/emi.2013.56. 3.Hu J, Seeger C.1996. Expression and characterization of hepadnavirus
reverse transcriptases. Methods Enzymol.275:195–208.http://dx.doi.org /10.1016/S0076-6879(96)75013-9.
4.Summers J, Mason WS.1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell29:403– 415.http://dx.doi.org/10.1016/0092-8674(82)90157-X.
5.Chang LJ, Hirsch RC, Ganem D, Varmus HE.1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus poly-merase. J. Virol.64:5553–5558.
6.Radziwill G, Tucker W, Schaller H.1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol.64:613– 620.
7.Toh H, Hayashida H, Miyata T. 1983. Sequence homology between retroviral reverse transcriptase and putative polymerases of hepatitis B virus and cauliflower mosaic virus. Nature305:827– 829.http://dx.doi.org /10.1038/305827a0.
8.Bartenschlager R, Schaller H.1988. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked pro-tein) believed to prime reverse transcription. EMBO J.7:4185– 4192. 9.Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R,
Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG.2013. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunode-ficiency virus ribonuclease H and integrase enzymes. PLoS Pathog. 9:e1003125.http://dx.doi.org/10.1371/journal.ppat.1003125.
10. Stahl M, Beck J, Nassal M.2007. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J. Vi-rol.81:13354 –13364.http://dx.doi.org/10.1128/JVI.01196-07.
11. Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J.16:59 – 68.http://dx.doi.org/10 .1093/emboj/16.1.59.
12. Hu J, Anselmo D.2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J. Virol. 74:11447–11455.http://dx.doi.org/10.1128/JVI.74.24.11447-11455.2000. 13. Hu J, Boyer M.2006. Hepatitis B virus reverse transcriptase and epsilon
RNA sequences required for specific interaction in vitro. J. Virol.80:2141– 2150.http://dx.doi.org/10.1128/JVI.80.5.2141-2150.2006.
14. Hu J, Flores D, Toft D, Wang X, Nguyen D.2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase func-tion. J. Virol.78:13122–13131.http://dx.doi.org/10.1128/JVI.78.23.13122 -13131.2004.
15. Hu J, Toft D, Anselmo D, Wang X.2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol. 76:269 –279. http://dx.doi.org/10.1128/JVI.76.1.269 -279.2002.
on November 7, 2019 by guest
http://jvi.asm.org/
16. Hu J, Seeger C.1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A.93:1060 –1064. http://dx.doi.org/10.1073/pnas.93.3.1060.
17. Hirsch RC, Loeb DD, Pollack JR, Ganem D.1991. cis-Acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol.65:3309 –3316.
18. Pollack JR, Ganem D.1993. An RNA stem-loop structure directs hepa-titis B virus genomic RNA encapsidation. J. Virol.67:3254 –3263. 19. Pollack JR, Ganem D.1994. Site-specific RNA binding by a hepatitis B
virus reverse transcriptase initiates two distinct reactions: RNA packaging and DNA synthesis. J. Virol.68:5579 –5587.
20. Bartenschlager R, Junker-Niepmann M, Schaller H.1990. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol.64:5324 –5332.
21. Bartenschlager R, Schaller H.1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J.11:3413–3420.
22. Tavis JE, Ganem D.1995. RNA sequences controlling the initiation and transfer of duck hepatitis B virus minus-strand DNA. J. Virol.69:4283– 4291.
23. Tavis JE, Ganem D.1996. Evidence for activation of the hepatitis B virus polymerase by binding of its RNA template. J. Virol.70:5741–5750. 24. Tavis JE, Massey B, Gong Y.1998. The duck hepatitis B virus polymerase
is activated by its RNA packaging signal, epsilon. J. Virol.72:5789 –5796. 25. Wang GH, Seeger C.1992. The reverse transcriptase of hepatitis B virus
acts as a protein primer for viral DNA synthesis. Cell71:663– 670.http: //dx.doi.org/10.1016/0092-8674(92)90599-8.
26. Jones SA, Boregowda R, Spratt TE, Hu J.2012. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymer-ase. J. Virol.86:5134 –5150.http://dx.doi.org/10.1128/JVI.07137-11. 27. Lanford RE, Notvall L, Lee H, Beames B.1997. Transcomplementation
of nucleotide priming and reverse transcription between independently expressed TP and RT domains of the hepatitis B virus reverse transcrip-tase. J. Virol.71:2996 –3004.
28. Jones SA, Hu J.2013. Protein-primed terminal transferase activity of hepatitis B virus polymerase. J. Virol.87:2563–2576.http://dx.doi.org/10 .1128/JVI.02786-12.
29. Weber M, Bronsema V, Bartos H, Bosserhoff A, Bartenschlager R, Schaller H.1994. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol.68:2994 –2999. 30. Zoulim F, Seeger C.1994. Reverse transcription in hepatitis B viruses is
primed by a tyrosine residue of the polymerase. J. Virol.68:6 –13. 31. Girard FC, Ottink OM, Ampt KA, Tessari M, Wijmenga SS. 2007.
Thermodynamics and NMR studies on duck, heron and human HBV encapsidation signals. Nucleic Acids Res.35:2800 –2811.http://dx.doi.org /10.1093/nar/gkm131.
32. Knaus T, Nassal M.1993. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res.21:3967–3975.http://dx.doi.org/10.1093/nar /21.17.3967.
33. Nassal M, Rieger A.1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol.70:2764 –2773.
34. Tavis JE, Perri S, Ganem D.1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol.68:3536 –3543.
35. Hu J, Lin L.2009. RNA-protein interactions in hepadnavirus reverse transcription. Front. Biosci. 14:1606 –1618. http://dx.doi.org/10.2741 /3328.
36. Jeong JK, Yoon GS, Ryu WS.2000. Evidence that the 5=-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J. Vi-rol.74:5502–5508.http://dx.doi.org/10.1128/JVI.74.12.5502-5508.2000. 37. Lanford RE, Kim YH, Lee H, Notvall L, Beames B.1999. Mapping of the
hepatitis B virus reverse transcriptase TP and RT domains by transcomplementation for nucleotide priming and by proteprotein in-teraction. J. Virol.73:1885–1893.
38. Lanford RE, Notvall L, Beames B.1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol.69:4431– 4439.
39. Urban M, McMillan DJ, Canning G, Newell A, Brown E, Mills JS, Jupp R.1998. In vitro activity of hepatitis B virus polymerase: requirement for distinct metal ions and the viral epsilon stem-loop. J. Gen. Virol.79(Part 5):1121–1131.
40. Shin YC, Park S, Ryu WS.2011. A conserved arginine residue in the terminal protein domain of hepatitis B virus polymerase is critical for RNA pre-genome encapsidation. J. Gen. Virol.92:1809 –1816.http://dx .doi.org/10.1099/vir.0.031914-0.
41. Shin YC, Ko C, Ryu WS.2011. Hydrophobic residues of terminal protein domain of hepatitis B virus polymerase contribute to distinct steps in viral genome replication. FEBS Lett.585:3964 –3968.http://dx.doi.org/10.1016 /j.febslet.2011.11.003.
42. Kim S, Lee J, Ryu WS.2009. Four conserved cysteine residues of the hepatitis B virus polymerase are critical for RNA pregenome encapsida-tion. J. Virol.83:8032– 8040.http://dx.doi.org/10.1128/JVI.00332-09. 43. Wang X, Qian X, Guo HC, Hu J.2003. Heat shock protein 90-independent
activation of truncated hepadnavirus reverse transcriptase. J. Virol.77:4471– 4480.http://dx.doi.org/10.1128/JVI.77.8.4471-4480.2003.
44. Wang GH, Zoulim F, Leber EH, Kitson J, Seeger C.1994. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol.68:8437– 8442.
45. Cao F, Badtke MP, Metzger LM, Yao E, Adeyemo B, Gong Y, Tavis JE. 2005. Identification of an essential molecular contact point on the duck hepatitis B virus reverse transcriptase. J. Virol.79:10164 –10170.http://dx .doi.org/10.1128/JVI.79.16.10164-10170.2005.
46. Badtke MP, Khan I, Cao F, Hu J, Tavis JE.2009. An interdomain RNA binding site on the hepadnaviral polymerase that is essential for reverse transcription. Virology 390:130 –138. http://dx.doi.org/10.1016/j.virol .2009.04.023.
47. Jones SA, Murakami E, Delaney W, Furman P, Hu J.2013. Noncompetitive inhibition of hepatitis B virus reverse transcriptase protein priming and DNA synthesis by the nucleoside analog clevudine. Antimicrob. Agents Che-mother.57:4181– 4189.http://dx.doi.org/10.1128/AAC.00599-13. 48. Cao F, Jones SA, Li W, Cheng X, Hu Y, Hu J, Tavis JE.Sequences in the
terminal protein and reverse transcriptase domains of the hepatitis B virus polymerase contribute to RNA binding and encapsidation. J. Viral Hepat., in press.
49. Chen Y, Robinson WS, Marion PL.1994. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. J. Virol.68:5232–5238. 50. Roychoudhury S, Faruqi AF, Shih C.1991. Pregenomic RNA
encapsi-dation analysis of eleven missense and nonsense polymerase mutants of human hepatitis B virus. J. Virol.65:3617–3624.
51. Boregowda R, Adams C, Hu J.2012. TP-RT domain interactions of duck hepatitis B virus reverse transcriptase in cis and in trans during protein-primed initiation of DNA synthesis in vitro. J. Virol.86:6522– 6536.http: //dx.doi.org/10.1128/JVI.00086-12.
52. Seeger C, Leber EH, Wiens LK, Hu J.1996. Mutagenesis of a hepatitis B virus reverse transcriptase yields temperature-sensitive virus. Virology 222:430 – 439.http://dx.doi.org/10.1006/viro.1996.0440.
53. De Clercq E, Ferir G, Kaptein S, Neyts J.2010. Antiviral treatment of chronic hepatitis B virus (HBV) infections. Viruses2:1279 –1305.http: //dx.doi.org/10.3390/v2061279.
54. Zoulim F, Locarnini S.2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology137:1593–1608.e2.http://dx.doi.org/10.1053/j .gastro.2009.08.063.
Jones et al.