Tissue-specific Inhibition of Apolipoprotein B mRNA Editing in the
Liver by Adenovirus-mediated Transfer of a Dominant Negative
Mutant APOBEC-1 Leads to Increased Low Density Lipoprotein in
Mice*
(Received for publication, April 23, 1996, and in revised form, October 17, 1996)
Kazuhiro Oka‡, Kunihisa Kobayashi‡, Merry Sullivan‡, Julie Martinez‡, Ba-Bie Teng‡, Kazumi Ishimura-Oka§, and Lawrence Chan‡
From the ‡Departments of Cell Biology, ‡Medicine, and §Pediatrics, Baylor College of Medicine, Houston, Texas 77030-3498 and the §United States Department of Agriculture/Agricultural Research Service Children’s Nutrition Research Center, Houston, Texas 77030
APOBEC-1 is a catalytic subunit of an apolipoprotein B (apoB) mRNA editing enzyme complex. In humans it is expressed only in the intestine, whereas in mice it is expressed in both the liver and intestine. APOBEC-1 exists as a spontaneous homodimer (Lau, P. P., Zhu, H.-J., Baldini, A., Charnsangavej, C., and Chan, L. (1994) Proc. Natl. Acad. Sci. U. S. A. 91, 8522– 8526). We tested the editing activity and dimerization potential of three different mouse APOBEC-1 mutants usingin vitro edit-ing activity assay and immunoprecipitation in the pres-ence of epitope-tagged APOBEC-1. One catalytically in-active mutant, mu1 (H61K/C93S/C96S), that retains its capacity to dimerize with wild-type APOBEC-1 was found to inhibit the editing activity of the latter and was thus a dominant negative mutant. Two other inactive mutants that dimerized poorly with APOBEC-1 failed to inhibit its activity. Intravenous injection of a mu1 ade-novirus, Admu1, in C57BL/6J mice in vivo resulted in liver-specific expression of mu1 mRNA. On days 4 and 9 after virus injection, endogenous hepatic apoB mRNA editing was 23.3 6 5.0 and 36.8 6 5.7%, respectively, compared with 65.3 611 and 71.365.2%, respectively, for luciferase adenovirus-treated animals. Plasma apoB-100 accounted for 95 and 93% of total plasma apoB in Admu1 animals on days 4 and 9, respectively, compared with 78 and 72% in luciferase adenovirus animals. Plasma cholesterol on day 9 was 98 617 mg/dl in the mu1-treated animals, substantially higher than phos-phate-buffered saline-treated (57 69 mg/dl) or lucifer-ase-treated (71612 mg/dl) controls. Fast protein liquid chromatography analysis of mouse plasma showed that the intermediate density/low density lipoprotein frac-tions in the animals treated with the dominant negative mutant adenovirus were much higher than those in con-trols. We conclude that active APOBEC-1 functions as a dimer and its activity is inhibited by a dominant nega-tive mutant. Furthermore, apoB mRNA editing deter-mines the availability of apoB-100, which in turn limits the amount of intermediate density/low density lipopro-tein that can be formed in mice. Liver-specific inhibition of apoB mRNA editing is an important component of any strategy to enhance the value of mice as a model for human lipoprotein metabolism.
The mouse is a useful animal model for lipoprotein metabo-lism and atherosclerosis (1, 2). Its value as a model for human disease, however, is limited by the fact that there is a substan-tial difference in lipoprotein metabolism between the two spe-cies. One major difference is the presence of high levels of apolipoprotein B (apoB)1mRNA editing in the liver in mice but
not in humans.
ApoB mRNA editing is a process by which apoB-100 mRNA is converted to apoB-48 mRNA (3, 4) (reviewed in Ref. 5). It involves the conversion of the first base of the codon CAA, encoding glutamine 2153 in apoB-100, to UAA, a stop codon, in apoB-48 mRNA. In humans, editing occurs exclusively in the small intestine but not in the liver. Therefore, the human liver produces apoB-100 and the small intestine produces apoB-48. In mice, apoB mRNA editing occurs in both the small intestine and the liver. Therefore, the amount of apoB-100 produced by the liver is very small, and mice have very low levels of circu-lating apoB-100; consequently, they have low levels of interme-diate density lipoprotein (IDL) and low density lipoprotein (LDL), atherogenic lipoproteins that require apoB-100 as an essential component.
ApoB mRNA editing is mediated by a multiprotein enzyme complex. APOBEC-1 is a cytidine deaminase-like protein that has been identified as a catalytic component of the complex (6 –10) that efficiently edits synthetic apoB mRNA in vitro in the presence of complementation factors. Lau et al. (10) showed that APOBEC-1 exists as a spontaneous homodimer. It shows sequence similarity to Escherichia coli cytidine deaminase, which also exists as a homodimer (11). In the case of APO-BEC-1, Lau et al. (10) postulated that dimerization may be mediated by hydrophobic interactions in a leucine-rich domain in the C-terminal third of the molecule. It is not known, how-ever, if the active form of APOBEC-1 is in the form of a homodimer.
The crystallographic structure of E. coli cytidine deaminase suggests that the active enzyme functions as a dimer (11). We reasoned that active APOBEC-1 might also function as a dimer and enzymatically inactive APOBEC-1 mutants (e.g. those that have substitutions in the zinc-coordinating residue in the ac-tive site (11)) might under appropriate conditions inactivate the wild-type enzyme by forming an inactive heterodimer with
* This work was supported by National Institutes of Health Grants HL51586 and HL56668. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1The abbreviations used are: apoB, apolipoprotein B; APOBEC-1,
apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1; IDL, intermediate density lipoprotein; LDL, low density lipoprotein; PCR, polymerase chain reaction; FPLC, fast protein liquid chromatography; PBS, phosphate-buffered saline; HA-1, Haemophilus influenzae hemag-glutinin epitope.
Printed in U.S.A.
This paper is available on line at http://www-jbc.stanford.edu/jbc/
1456
by guest on November 7, 2019
http://www.jbc.org/
observation is consistent with hepatic apoB mRNA editing being a limiting factor in the production of IDL/LDL in mice.
MATERIALS AND METHODS
Site-directed Mutagenesis by Polymerase Chain Reaction—Mouse
APOBEC-1 mutants (mu1, H61K/C93S/C96S; mu2, APOBEC-1 1–172 that misses the C-terminal 57 residues containing the leucine-rich domain; mu3, L182R/L187R) were produced by the polymerase chain reaction (PCR). In brief, pBluescript II KS (Stratagene) containing the wild-type mouse APOBEC-1 cDNA (7) was amplified using the reverse primer (59-GGAAACAGCTATGACCATG-39), forward primer (59 -GTA-AAACGACGGCCAGT-39), and specific mutagenesis primers. The spe-cific mutagenesis oligonucleotides used for PCR are listed below. The underlined nucleotides represent restriction sites for cloning purposes. The mutations are in boldface. H61K (59 -GAAGTTGACTTCAACTTT-GTTGCTGGTGTTTTG-39); C93S/C96S (59 -GCCCTGGAGCTCTCCC-CGCTGGGACTCCAG-39); L182R/L187R (59 -CTGTATGTACTGGAG-CGCTACTGCATCATTAGAGGACTTCCACCCTGT-39); mu2 was gen-erated by PCR using forward primer and 39downstream primer (59 -CGCGGGAATTCTCAATGGGGGTACCTTGGCCAAT-39); wild-type APOBEC-1 tagged with a 9-amino acid Haemophilus influenzae hemag-glutinin epitope (HA-1) preceded by a 3-alanine residue spacer (10) was generated by PCR using forward primer and 39downstream primer (59 -CGCGGGAATTCTCAAGCGTAATCTGGAACATCGTATGGGTAG-GCTGCGGCTTTCAACCCTGTAGCCCAAAGG-39). After PCR, the products were purified by electrophoresis on 1.5% agarose gels followed by extraction using a QIAquick gel extraction kit (Qiagen). The purified products were digested with BamHI and EcoRI and subcloned into the
BamHI/EcoRI sites of pBKCMV (Stratagene) or pBluescript KS. DNA
sequences were verified by double-stranded DNA sequencing.
In Vitro Translation—RNAs were transcribed from cDNAs subcloned
into pBKCMV and capped with T3 RNA polymerase using the mMES-SAGE mMACHINE kit (Ambion). They were translated in nuclease-treated rabbit reticulocyte lysate (Promega) in the presence of [3H]leucine (Amersham Corp.) for 2 h at 30 °C. Immunoprecipitation
was performed as described (10). The immunoprecipitated products were separated on an SDS-12% polyacrylamide gel. Gels were fixed, soaked in Amplify (Amersham Corp.), and dried before they were ex-posed to x-ray film at280 °C. The relative intensities of the fluoro-graphic images were determined with a photometric image scanner (BioImage).
In Vitro ApoB mRNA Editing Assay—The in vitro editing assay was
carried out as described previously (12) using 8 fmol of synthetic sub-strate in the presence of chicken enterocyte S-100 extracts. Primer extension products were fractionated on a 6% polyacrylamide, 8Murea gel and quantified by exposure to a phosphor screen (Molecular Dynamics).
Recombinant Adenoviral Vectors—The cDNA for mu1 was excised
from the KS vector by BamHI/ClaI digestion and subcloned into the
BglII/ClaI sites of pAvCvSv shuttle vector (13). The recombinant
ade-novirus was prepared by cotransfection of pAvCvSv containing mu1 cDNA and pJM17 (14) into 293 cells (15). Infection, propagation, screen-ing, and large scale production of high titer recombinant adenovirus were carried out as described (13, 16). AdLuc (17) contained luciferase cDNA instead of mu1 cDNA. Recombinant adenovirus stock was diluted with phosphate-buffered saline (PBS) to the appropriate concentra-tions, and 0.2 ml of diluted recombinant adenovirus (23109
plaque-forming units; particle/plaque-plaque-forming unit ratio;30) was injected into the external jugular vein of 3-month-old (;30 g) C57BL/6J mice that were maintained on laboratory chow (Teklad 4% mouse/rat diet 7001).
Northern Blot Analysis and Primary Extension Assay—Total cellular
RNA was prepared by using the Ultraspec RNA Isolation System (Bio-tecx Laboratory). 20mg of RNA was electrophoresed on a 1.0% agarose, 6% formaldehyde gel, transferred to a Hybond-N1membrane (Amer-sham Corp.), and hybridized to a 32P-labeled 0.7-kilobase pair mu1
cDNA probe. The same membrane was then hybridized to a mouse glyceraldehyde-3-phosphate dehydrogenase cDNA probe (Ambion). Di-rect primer extension was carried out as described (16). A32
P-end-labeled mouse antisense apoB DNA was annealed overnight at 45 °C with 20mg of total RNA. The annealed products were precipitated with ethanol and reconstituted in 10ml of first strand synthesis buffer (Life Technologies, Inc.) containing 0.5 mMeach dATP, dCTP, dTTP, 0.5 mM
ddGTP, 10 mMdithiothreitol, and 20 units of Superscript II reverse
transcriptase (Life Technologies, Inc.). The extension was carried out for 1 h at 45 °C. The extension products were resolved by 6% polyacryl-amide, 8Murea gel electrophoresis, and the radiolabeled products were quantitated by PhosphorImager (Molecular Dynamics).
Plasma Lipoprotein and ApoB Analysis—Animals were fasted for 6 h
and blood was collected into tubes containing EDTA. Plasma (0.2 ml) collected from individual mice was fractionated by fast protein liquid chromatography (FPLC) using two Superose 6 columns (Pharmacia Biotech, Inc.) connected in series (17). Fifty 0.5-ml fractions were col-lected using the eluent 1 mMEDTA, 154 mMNaCl, and 0.02% NaN3(pH
8.2) (18). Cholesterol and triglyceride contents of each FPLC fraction and of plasma were determined enzymatically (Sigma kits 352-50 and 336-10). For analysis of plasma apoBs, lipoproteins of d,1.21 prepared by ultracentrifugation from plasma pooled from four individual animals were fractionated on 4 –15% SDS gel and stained by Coomassie Blue. The apoB-100 and apoB-48 bands were quantified by densitometer measurements (13).
RESULTS AND DISCUSSION
Heterodimer Formation between Mutant and Wild-type APO-BEC-1—We examined the capacity of various APOBEC-1 mu-tants to dimerize with epitope-tagged wild-type APOBEC-1 (10). mu1 (H61K/C93S/C96S) efficiently formed a heterodimer with epitope-tagged wild-type APOBEC-1 (Fig. 1, lane 4). How-ever, a mutant that lacks the leucine-rich region (mu2) or one with mutations in this region (mu3) showed a markedly de-creased ability to dimerize with epitope-tagged wild-type APO-BEC-1 (Fig. 1, lanes 6 and 8). These results indicate that the leucine-rich region is important for dimer formation.
Capacity of Mutant APOBEC-1s to Inhibit the Editing Activ-ity of Wild-type APOBEC-1s—In order to study the effect of heterodimer formation on apoB mRNA editing activity, indi-vidual mutant and wild-type APOBEC-1s were cotranslated in vitro and the editing activity of the translation products was determined. By itself, mu1, in which the 3 zinc-coordinating residues were mutated, displayed no detectable editing activity in vitro. Increasing the ratio of mu1 mRNA in the presence of a constant amount of wild-type APOBEC-1 mRNA produced an APOBEC-1 product that had lost.95% of its activity in the in
FIG. 1. Immunoprecipitation of APOBEC-1 and mutants
co-translatedin vitro. Translation products before and after
immuno-precipitation with anti-HA-1 antibody were analyzed as described un-der “Materials and Methods.” wt, wild-type APOBEC-1; wt*, HA-1 epitope-tagged wild-type APOBEC-1; mu1, (H61K/C93S/C96S) mutant;
mu2, APOBEC-1 1–172; mu3, (L182R/L187R) APOBEC-1. In each lane,
the upper band represents the epitope-tagged APOBEC-1 and the lower
band represents the untagged wild-type or mutant APOBEC-1. Since
the antibody is only against the HA-1 epitope, the coprecipitation of the untagged smaller APOBEC-1 band indicates that the tagged and un-tagged proteins existed as heterodimers (10).
by guest on November 7, 2019
http://www.jbc.org/
vitro editing assay (Fig. 2). Both mu2 and mu3 also had essen-tially no editing activity by themselves (,5% editing activity compared to that of wild type). The presence of increasing amounts of mu2 or mu3 translation products did not affect the editing activity of wild-type APOBEC-1 (Fig. 2). These results suggest that only a mutant that can efficiently dimerize with the wild-type APOBEC-1 is able to inhibit the editing activity of the latter. This supports the hypothesis that active APO-BEC-1 functions as a dimer.
Effect of mu1 Gene Transfer—Normal C57BL/6J mice were treated with an intravenous injection of Admu1, an adenoviral vector containing mu1, or AdLuc, an adenovirus containing luciferase cDNA. Liver and blood samples were obtained at days 4 and 9 after treatment. The highest level of mu1 mRNA was detected in the liver on day 4. It decreased substantially but was still readily detectable on day 9 (Fig. 3A). In contrast, mu1 mRNA was not detectable in the small intestine either on day 4 or day 9. This agrees with our previous experience (13, 16) and that of others (reviewed in Ref. 19) that intravenous administration of adenoviral vectors in mice in vivo targets the transgene to the liver almost exclusively. To determine whether the Admu1 effectively inhibited endogenous apoB mRNA editing, we measured the relative amounts of apoB-100 and apoB-48 mRNAs by primer extension (Fig. 3B). On day 4 following adenovirus administration, the hepatic apoB mRNA from Admu1 animals contained 23.365.0% (n54) and that from AdLuc animals contained 65.3 6 11% (n 5 4) edited mRNA. On day 9, the proportions of edited mRNA were 36.86 5.7% for Admu1 animals (n53) and 71.365.2% for AdLuc animals (n54). The ratio for PBS-treated animals was 69.36 4.3% (n54). Therefore, Admu1 but not AdLuc administration markedly reduced the amount of edited apoB mRNA in the liver. The amount of edited apoB mRNA in the small intestine in Admu1-treated animals was 89.563.5% (n54) and 88.46 2.2% (n 54) for days 4 and 9, respectively, compared with AdLuc values of 92.762.2% (n54) and 94.760.48% (n54) for these two days; it was 93.860.7% (n54) for PBS-injected controls. Thus, adenovirus-mediated transfer of a dominant negative mutant APOBEC-1 in vivo selectively inhibited apoB mRNA editing in the liver. Furthermore, this maneuver re-sulted in a significant increase in apoB-100 level (15.264.0 mg/dl in Admu1, 2.84 60.5 mg/dl in Adluc, and 3.63 62.1 mg/dl in PBS animals, p,0.0005, on day 9) in Admu1-treated animals.
Effect of Inhibiting Hepatic ApoB mRNA Editing on Plasma ApoB-100 and ApoB-48 and Plasma Lipids—The ratio of
apoB-100 and apoB-48 protein was examined in mouse plasma by SDS gel analysis (Fig. 3C). Pooled plasma from four AdLuc-treated mice on days 4 and 9 contained 78 and 72%, respec-tively, of the total apoB as 100. The proportion of apoB-100 increased to 95% at day 4 and remained at 93% at day 9 in Admu1-treated animals.
There were significant differences in plasma lipids among the three groups following adenovirus treatment. In Admu1 animals, plasma cholesterol (day 9) and triglyceride (both day 4 and day 9) were almost double the values in the other two groups (Table I).
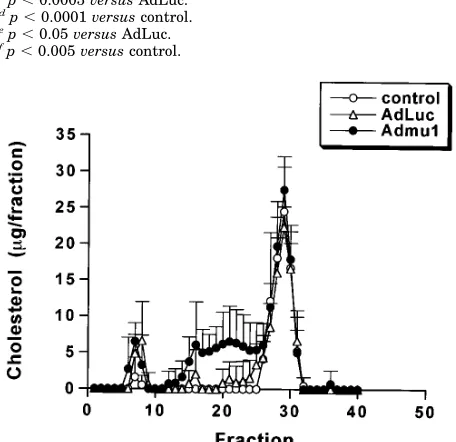
In order to determine which lipoprotein fractions were af-fected by Admu1 injection, plasma lipoproteins from day 9 samples were fractionated by FPLC and their cholesterol con-tent determined (Fig. 4). It is evident that there is no signifi-cant difference in the very low density or high density lipopro-tein fractions between Admu1-treated and the AdLuc- or PBS-treated control samples. However, compared with controls the
FIG. 2. Effect of the presence of excess mutant APOBEC-1s on
editing activity of wild-type APOBEC-1. Capped wild-type
APO-BEC-1 cRNA and mutant cRNAs were cotranslated in vitro using a rabbit reticulocyte lysate. Translation products were analyzed for apoB mRNA editing activity in vitro. ApoB RNA editing activity of wild-type APOBEC-1 in the absence of mutant APOBEC-1 was 7464% (mean6
S.D., n54) of input RNA in this assay and this activity is set to 100%. All assays were done at a constant amount of wild-type APOBEC-1 in the presence of increasing amounts of mutant APOBEC-1s.
FIG. 3. Admu1 gene transfer. A, northern blot analysis of liver and intestinal RNA using32P-labeled APOBEC-1 cDNA probe on days 4 and
9 of treatment; the asterisk indicates mu1 mRNA. B, primer extension assay of liver and intestine RNA. The edited (UAA) and unedited (CAA) products are as marked. C, pooled day-9 plasma apoBs from PBS-, AdLuc-, or Admu1-treated mice fractionated on 4 –15% SDS gels.
by guest on November 7, 2019
http://www.jbc.org/
IDL/LDL fractions were markedly elevated in the animals treated with the dominant negative mutant APOBEC-1 vector. SDS-polyacrylamide gel analysis indicates that the apoB-100 band was substantially higher in the Admu1 compared with the AdLuc1 samples (data not shown).
The experiments above lead us to the following conclusions. (i) Active APOBEC-1 works as a dimer and can be inhibited by a dominant negative mutant. Here we show that a mutant APOBEC-1 (H61C/C93S/C96S) with its zinc-coordinating resi-dues mutated is totally inactive. This is consistent with previ-ous observations (8, 20, 21) and with the catalytic role of the homologous residues in E. coli cytidine deaminase (11). The other two mutants, mu2 and mu3, which had mutations in the leucine-rich region, were also enzymatically inactive. The loss of catalytic activity in these mutants is probably related to their inability to efficiently form dimers, the active form of the enzyme. The fact that the mutations in these dimerization-incompetent APOBEC-1s involve residues in the leucine-rich domain indicates that this region is important for dimer for-mation. In any case, only mu1, which is competent in dimer-ization, acts as a dominant negative mutant when coexpressed with wild-type APOBEC-1 (Fig. 2). Therefore, these experi-ments support the contention that the active form of APO-BEC-1 is a homodimer (10).
(ii) Liver-specific inhibition of APOBEC-1 action leads to elevated IDL/LDL cholesterol. We hypothesized that the low IDL/LDL cholesterol observed in mice compared to humans is partly the result of the diversion of apoB mRNA to the produc-tion of predominantly apoB-48, severely limiting the amount of
apoB-100, which is essential for the formation of IDL/LDL. Here we showed that hepatic APOBEC-1 action in mice was inhibited by adenovirus-mediated transfer of a dominant neg-ative mutant. As a result of the decrease in apoB mRNA edit-ing, the plasma apoB-100/apoB-48 ratio was greatly increased and the plasma IDL/LDL cholesterol was much higher in these animals than in the luciferase- or PBS-treated controls. We have repeated the adenovirus experiments and consistently observed the IDL/LDL elevation (data not shown). We conclude that the availability of apoB-100 (indirectly determined by apoB mRNA editing efficiency in the liver) limits the amount of IDL/LDL that can be formed in mice. It is interesting that in APOBEC-1 knockout mice that do not edit apoB mRNA either in the liver or small intestine there is little (22) or no change (23, 24) in plasma IDL/LDL. The difference in phenotype be-tween the animals reported in this study and the complete knockout mice may be related to the fact that 1) the liver-specific nature of APOBEC-1 inactivation and the normal ed-iting in the small intestine may in some way contribute to enhanced very low density lipoprotein and subsequently IDL/ LDL production, and/or more likely 2) the acute inhibition of APOBEC-1 action by adenovirus-mediated gene transfer does not allow sufficient time for the animals to marshal a compen-satory response that may play a role in minimizing the lipopro-tein changes in the APOBEC-1 gene knockout animals. In any case, future experiments aimed at “humanizing” the mouse should include the permanent liver-specific inactivation of apoB mRNA editing, which would greatly enhance the value of this animal as a model for human lipoprotein metabolism and atherosclerosis.
Acknowledgments—We thank Dr. R. D. Gerard for providing AdLuc,
and Linda Phillips and Irene A. Harrison for excellent secretarial assistance.
REFERENCES
1. Paigen, B., Plump, A. S., and Rubin, E. M. (1994) Curr. Opin. Lipidol. 5, 258 –264
2. Chien, K. (1996) J. Clin. Invest. 97, 901–909
3. Powell, L. M., Wallis, S. C., Pease, R. J., Edwards, Y. H., Knott, T. J., and Scott, J. (1987) Cell 50, 831– 840
4. Chen, S.-H., Habib, G., Yang, C.-Y., Gu, Z.-W., Lee, B. R., Weng, S.-a., Silberman, S. R., Cai, S. J., Deslypere, J. P., Rosseneu, M., Gotto, A. M., Jr., Li, W.-H., and Chan, L. (1987) Science 238, 363–366
5. Chan, L., and Seeburg, P. (1995) Science & Medicine 2, 68 –77
6. Teng, B., Burant, C. F., and Davidson, N. O. (1993) Science 260, 1816 –1819 7. Nakamuta, M., Oka, K., Krushkal, J., Kobayashi, K., Yamamoto, M., Li, W.-H.,
and Chan, L. (1995) J. Biol. Chem. 270, 13042–13056
8. Yamanaka, S., Poksay, K. S., Balestra, M. E., Zeng, G.-Q., and Innerarity, T. L. (1994) J. Biol. Chem. 269, 21725–21734
9. Hadjiagapiou, C., Giannoni, F., Funahashi, T., Skarosi, S. F., and Davidson, N. O. (1994) Nucleic Acids Res. 22, 1874 –1879
10. Lau, P. P., Zhu, H.-J., Baldini, A., Charnsangavej, C., and Chan, L. (1994) Proc. Natl. Acad. Sci. U. S. A. 91, 8522– 8526
11. Betts, L., Xiang, S., Short, S. A., Wolfenden, R., and Carter, C. W., Jr. (1994) J. Mol. Biol. 235, 635– 656
12. Teng, B., and Davidson, N. O. (1992) J. Biol. Chem. 267, 21265–21272 13. Kobayashi, K., Oka, K., Forte, T., Ishida, B., Teng, B., Ishimura-Oka, K.,
Nakamuta, M., and Chan, L. (1996) J. Biol. Chem. 271, 6852– 6860 14. McGrory, W. J., Bautista, D. S., and Graham, F. L. (1988) Virology 163,
614 – 617 p,0.05 versus AdLuc.
f
p,0.005 versus control.
FIG. 4. FPLC profile of mouse plasma. 9 days after administration of PBS, AdLuc, or Admu1, 0.2 ml of plasma from individual animals was fractionated by FPLC. Cholesterol content of each fraction was meas-ured and expressed as mean6S.D. of six individual mice for each group. Fractions 6 – 8, very low density lipoprotein; fractions 14 –25, IDL/LDL; fractions 26 –31, high density lipoprotein; control, PBS-treated.
by guest on November 7, 2019
http://www.jbc.org/
[image:4.605.63.291.160.381.2]15. Graham, F. L., Smiley, J., Russell, W. C., and Norin, R. (1977) J. Gen. Virol. 36, 59 –72
16. Teng, B., Blumenthal, S., Forte, T., Navaratnam, N., Scott, J., Gotto, A. M., Jr., and Chan, L. (1994) J. Biol. Chem. 269, 29395–29404
17. Herz, J., and Gerard, R. D. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 2812–2816 18. Cole, T., Kitchens, R., Daugherty, A., and Schonfeld, G. (1988) FPLC
BioCom-munique 4, 4 – 6
19. Gerard, R. D., and Chan, L. (1996) Curr. Opin. Lipidol. 7, 105–111 20. Driscoll, D. M., and Zhang, Q. (1994) J. Biol. Chem. 269, 19843–19847
21. MacGinnitie, A. J., Anant, S., and Davidson, N. O. (1995) J. Biol. Chem. 270, 14768 –14775
22. Morrison, J. R., Paszity, C., Stevens, M. E., Hughes, S. D., Forte, T., Scott, J., and Rubin, E. M. (1996) Proc. Natl. Acad. Sci. U. S. A. 93, 7154 –7159 23. Hirano, K.-I., Young, S. G., Farese, R. V., Jr., Ng, J., Sande, E., Warburton, C.,
Powell-Braxton, L. M., and Davidson, N. O. (1996) J. Biol. Chem. 271, 9887–9890
24. Nakamuta, M., Chang, B. H.-J., Zsigmond, E., Kobayashi, K., Lei, H., Ishida, B., Oka, K., Li, E., and Chan, L. (1996) J. Biol. Chem. 271, 25981–25988
by guest on November 7, 2019
http://www.jbc.org/
http://www.jbc.org/content/272/3/1456
Access the most updated version of this article at
Alerts:
When a correction for this article is posted
•
When this article is cited
•
to choose from all of JBC's e-mail alerts
Click here
http://www.jbc.org/content/272/3/1456.full.html#ref-list-1
This article cites 24 references, 14 of which can be accessed free at
by guest on November 7, 2019
http://www.jbc.org/