Replication Stress and Mitotic Dysfunction in Cells Expressing Simian
Virus 40 Large T Antigen
Liang Hu,cHarilaos Filippakis,aHaomin Huang,bTimothy J. Yen,bOle V. Gjoerupa
Molecular Oncology Research Institute, Tufts Medical Center, Boston, Massachusetts, USAa; Fox Chase Cancer Center, Philadelphia, Pennsylvania, USAb; Institute of
Reproductive and Stem Cell Engineering, Central South University, Changsha, People’s Republic of Chinac
We previously demonstrated that simian virus 40 (SV40) large T antigen (LT) binds to the Bub1 kinase, a key regulator of the spindle checkpoint and chromosome segregation. Bub1 mutations or altered expression patterns are linked to chromosome mis-segregation and are considered to be a driving force in some human cancers. Here we report that LT, dependent on Bub1 bind-ing, causes micronuclei, lagging chromatin, and anaphase bridges, which are hallmarks of chromosomal instability (CIN) and Bub1 insufficiency. Using time-lapse microscopy, we demonstrate that LT imposes a Bub1 binding-dependent delay in the meta-phase-to-anaphase transition. Kinetochore fibers reveal that LT, via Bub1 binding, causes aberrant kinetochore (KT)-microtu-bule (MT) attachments and a shortened interkinetochore distance, consistent with a lack of tension. Previously, we showed that LT also induces the DNA damage response (DDR) via Bub1 binding. Using inducible LT cell lines, we show that an activated DDR was observed before the appearance of anaphase bridges and micronuclei. Furthermore, LT induction in serum-starved cells demonstrated␥-H2AX accumulation in cells that had not yet entered mitosis. Thus, DDR activation can occur indepen-dently of chromosome segregation defects. Replication stress pathways may be responsible, because signatures of replication stress were observed, which were attenuated by exogenous supplementation with nucleosides. Our observations allow us to pro-pose a model that explains and integrates the diverse manifestations of genomic instability induced by LT.
S
imian virus 40 (SV40) naturally infects and replicates lytically in monkey cells (1,2). Large T antigen (LT) is the major early protein, which is capable of directing initiation of viral DNA rep-lication and oncogenic transformation in a wide variety of pri-mary or established cells. Fundamental insight into biological pro-cesses has come from the LT model system, for example, regarding initiation of DNA replication, nuclear translocation, transcrip-tional control, cellular immortalization, and malignant transfor-mation (2).To create a permissive cellular environment for viral replica-tion, LT extensively reprograms the host cell. This includes cell cycle deregulation, for example, LT-induced progression from quiescence into S phase, where both viral and cellular replication occurs (3). In part, oncogenic transformation results from this unscheduled cellular proliferation. LT is highly multifunctional and can be divided into modular domains (1). Almost all of its activities are linked to binding and alteration of host proteins, often via discrete, linear binding motifs on LT. Thus, LT interacts with pocket proteins, the retinoblastoma tumor suppressor (pRB), p107, and p130, via an LxCxE motif (1,2). Moreover, LT binds to the p53 tumor suppressor, whose gene is the most fre-quently mutated gene in human cancer (1,2). LT binding to pRB family members and p53 leads to their functional inactivation.
We have previously reported that LT additionally binds the mitotic spindle checkpoint kinase Bub1, and this requires LT res-idues 89 to 97 (4). This interaction is important for both Rat-1 transformation and viral replication activity (4) (data not shown). Furthermore, we have shown that LT via Bub1 induces tetraploidy and an activated DNA damage response (DDR) in normal BJ/tert human fibroblasts (5). DDR activation is manifested in nuclear foci of␥-H2AX and 53BP1, which are a hallmark of the DNA double-strand break (DSB) response (6). LT expression alone, in the absence of the viral origin of replication, induces cellular DNA damage, in part DSBs (7). Induction of an ataxia telangiectasia
mutated (ATM)- and ataxia telangiectasia- and Rad3-related (ATR)-dependent DDR promotes the viral replication program, partly by regulatory phosphorylation of LT on Ser120 and partly to maintain viral replication centers and repair replication-asso-ciated DNA damage (7–11).
Bub1 primarily acts at the spindle assembly checkpoint (SAC), which is a cellular genome protection mechanism that monitors tension and whether kinetochores achieve the correct bivalent at-tachment to spindle microtubules prior to anaphase onset (12, 13). Failure of the SAC undermines genome stability and is asso-ciated with cell death or oncogenic transformation when the checkpoint is weakened rather than fully inactivated (14). Bub1 also regulates chromosome segregation through correction of ab-errant kinetochore (KT)-microtubule (MT) attachments (12,15– 18). Importantly, alterations of Bub1 by mutation, or changes in the expression level either above or below normal, are associated with increased cancer incidence (19–22). Taken together, Bub1 is a key regulator of chromosomal stability, and interference with its function leads to genomic instability, gains and losses of whole chromosomes (aneuploidy), and, ultimately, tumorigenesis, which may be driven by a loss of heterozygosity of tumor suppres-sor genes (23).
LT has long been known to induce both structural and numer-ical chromosome instability, but the mechanisms have been elu-sive (24–29). Consequently, the link to Bub1 offers significant
Received7 August 2013 Accepted18 September 2013
Published ahead of print25 September 2013
Address correspondence to Ole V. Gjoerup, [email protected].
L.H. and H.F. contributed equally.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02224-13
on November 7, 2019 by guest
http://jvi.asm.org/
promise for an understanding of this process (4,5,30). Genomic instability is likely to contribute to long-term tumor formation induced by LT, given the accumulated evidence from numerous model systems that chromosomal instability (CIN) can be a driv-ing force in tumorigenesis (31). Here we report that LT induces several markers of CIN, such as micronuclei, lagging chromatin, and anaphase bridges. Moreover, LT perturbs mitotic progression by imposing a delay in metaphase-to-anaphase progression. LT also causes aberrant KT-MT attachments as well as loss of tension and cohesion between sister chromatids. All of these mitotic phe-notypes are dependent on LT binding to Bub1.
We aim to determine the molecular mechanisms underlying LT-induced genomic instability. The two main promoters of genomic instability are DNA damage and aberrant mitosis (32). Since LT induces both phenomena, we are interested in examining their potential relationship. Recent studies have concluded that DNA damage can cause chromosome missegregation and tetra-ploidy, but conversely, aberrant mitotic segregation can also lead to an activated DDR due to breakage of anaphase bridges or accu-mulation of DSBs in micronuclei (32–38). Using a LT doxycycline (Dox)-inducible BJ/tert cell line, our analysis reveals that␥-H2AX foci precede the formation of micronuclei and anaphase bridges, indicating that DNA lesions resulting from replication stress are early events that likely induce many, if not all, of the mitotic ab-normalities.
MATERIALS AND METHODS
Cell culture.BJ/tert cells stably expressing the empty vector, LT, or the dl89-97 mutant (BJ-V, BJ-LT, and BJ-dl89-97 cells, respectively) were derived by retroviral and blasticidin selection, as previously described (5). The BJ/tert cell lines were grown in 80% Dulbecco’s modified Eagle me-dium (DMEM) (Lonza) and 20% meme-dium 199 (Invitrogen) containing 10% fetal calf serum (FCS) (HyClone) and 1% penicillin-streptomycin. For Dox-inducible expression, special Tet-system-approved FCS was used (Clontech). BJ/tert cells infected with the pInducer20 lentiviral vec-tor (39) were selected with 750g/ml of G418 for 10 days and then tested for Dox-inducible expression by addition of 1g/ml Dox. All cell lines were maintained in a 37°C incubator with 5% CO2. 293FT cells
(Invitro-gen), used for lentiviral packaging, were grown in DMEM with 10% FCS and 1% penicillin-streptomycin. For the serum starvation experiment, cells were first plated overnight, followed by 3 days of incubation in DMEM without any FCS. Subsequently, cells were stimulated with DMEM and 10% FCS for 12 h in either the presence or absence of Dox.
Nucleoside supplementation.Uridine (Sigma) and cytidine (Sigma) were dissolved in milli-Q water to make a 10 mM stock, whereas adeno-sine (Sigma) and guanoadeno-sine (Sigma) were dissolved to make a 2 mM stock, and the suspension was briefly boiled to complete solubilization. Solu-tions were filter sterilized and added to complete medium until reaching a final concentration of 50M for 48 h (40).
Expression constructs.The LT cDNA was cloned from pCMVNeo LT (41) into pENTR1A no ccdB (Addgene), using BamHI sites on both ends. Correct orientation was verified by sequencing. Gateway recombination into the destination vector pInducer20 (kindly provided by Stephen Elledge) (39) was performed with Clonase LR according to the manufac-turer’s instructions (Invitrogen). Recombinant clones were transformed into competentEscherichia coliStbl3 bacteria (Invitrogen) and grown at 30°C to minimize vector rearrangements. The pInducer20 vector was packaged into lentivirus as previously described (5).
Antibodies. Antibody to ␣-tubulin (Sigma), human centromere (CREST; Immunovision), the Bub1 N terminus (4), SgoI (42), H2ApT120 (43), BubR1 (BD Bioscience), PLK1 (Santa Cruz Biotechnology), Bloom syndrome helicase (BLM) (Ab2179; Abcam), and␥-H2AX (JBW301; Mil-lipore) were used for immunofluorescence analysis at a final
concen-tration of 0.5 to 1g/ml. Alexa 488-conjugated goat anti-mouse or anti-rabbit antibody (Invitrogen) and Cy3-conjugated goat anti-hu-man antibody (Jackson ImmunoResearch Labs) were used at 1g/ml. For Western blotting, LT mouse monoclonal antibodies PAb419 and PAb423 (4) were used, whereas rabbit polyclonal antibody was used for immunofluorescence experiments (v-300; Santa Cruz Biotechnol-ogy) to allow for costaining with␥-H2AX mouse monoclonal anti-body. Western blotting was performed by using standard procedures, as described previously (5).
Micronucleus and chromatin bridge analysis.V, LT, and BJ-dl89-97 cells were cultured on poly-L-lysine (Sigma)-coated glass cover-slips. Cells were stained by human anticentromere serum (CREST) and then counterstained with 1g/ml 4=,6-diamidino-2-phenylindole (DAPI). Fluorescence signals were then observed and captured by using an Olympus AX70 fluorescence microscope equipped with a Spot RT/KE charge-coupled-device (CCD) camera or by using a Nikon Eclipse 80i microscope. Note that anaphases in BJ/tert cell lines are rare, thus limiting total numbers that could be analyzed. On average, only 3 to 5 anaphases per coverslip could be found.
Time-lapse imaging.BJ-V, BJ-LT, and BJ-dl89-97 cells stably expressing histone H2B-green fluorescent protein (GFP) were grown on 40-mm poly-L-lysine-coated glass coverslips (Fisher) (44). Cells were synchronized with 2 mM thymidine for 16 h. After this, cells were washed twice with phosphate-buffered saline (PBS) and released into fresh medium for 7 h. The coverslips were then assembled with Focht chamber system 2 (Bioptechs) and observed under a fluorescence microscope (model IX81; Olympus). The camera, shut-ters, and filter wheel were controlled by Slidebook software (version 4.1; Olympus). Fluorescence and differential interference contrast (DIC) images were collected every 5 min, with 40-ms exposures.
Immunofluorescence microscopy.For KT/MT attachment analysis, cells were synchronized with 2 mM thymidine for 16 h and then released into fresh medium for 7 h (44). A proteasome inhibitor (MG-132; EMD Millipore) was added to cells at a final concentration of 20M for 2 h to allow correction of KT-MT attachment errors by provoking arrest in metaphase. Subsequently, cells were incubated in prechilled L15 buffer (Invitrogen) on ice for 10 min to depolymerize nonstable tubulin. Cells were then preextracted in microtubule-stabilizing buffer (MTSB) preex-traction buffer [4.0 M glycerol, 0.1 M piperazine-N,N= -bis(2-ethanesul-fonic acid) (PIPES) (pH 6.9), 1 mM EGTA, 0.25% Triton X-100] for 2 min. After that, cells were fixed in freshly prepared 4% paraformaldehyde for 7 min at room temperature and blocked in KB buffer (20 mM Tris [pH 7.5], 150 mM NaCl, and 0.1% bovine serum albumin [BSA]) at 4°C over-night. For regular immunostaining, cells were fixed in freshly prepared 4% paraformaldehyde for 7 min at room temperature, permeabilized with KB buffer containing 0.2% Triton X-100 for 5 min, and then blocked in KB buffer at 4°C overnight. Primary antibodies and secondary antibodies were diluted in KB buffer and added to coverslip for 30 min at room temperature. Images were visualized with a magnification of⫻ 100/nu-merical aperture of 1.4 objective on an inverted fluorescence microscope (model IX81; Olympus). Image stacks of 0.25 to 0.5 mm were captured by a Slidebook-controlled CCD camera (model Orca-ER; Hamamatsu), ex-ported, and then deconvolved with AutoQuant (Media Cybernetics). All maximal-intensity projection image files were reformatted to TIFF files and assembled by using Adobe Photoshop.
Chromatid cohesion analysis.Cells were trypsinized and hypotoni-cally treated with 0.075 M KCl. The swollen cells were fixed with metha-nol-glacial acetic acid at a ratio of 3:1 (Carnoy’s fixative). Metaphase spreads were stained by Giemsa staining solution (Sigma). Primary con-striction gaps (PCGs) were classified as previously described (45).
Statistical analysis.Statistical analyses were performed by using Mi-crosoft Excel to calculate standard errors of the means or statistical signif-icance based on the Studentttest. In most cases, differences were found to be highly significant (P⬍0.01).
on November 7, 2019 by guest
http://jvi.asm.org/
RESULTS
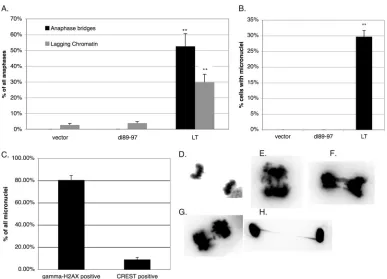
LT expression via Bub1 binding induces anaphase bridges and micronuclei, which are the hallmarks of chromosomal instabil-ity.We, as well as others, have previously demonstrated that ex-pression of LT, even in the absence of a viral origin of replication, destabilizes the cellular genome by inducing tetraploidy/aneu-ploidy and an activated DDR (5,7,24,29). We have found these responses to be mediated by Bub1 binding (5). Here, we seek to understand the full range of mitotic abnormalities that LT is ca-pable of inducing and how it relates to genomic damage. We found that LT expression in BJ/tert normal human fibroblasts (BJ-LT) causes the frequent appearance of anaphase bridges and lagging chromatin, which are the hallmarks of chromosomal in-stability (CIN) and are associated with poor cancer prognoses (Fig. 1AandEtoG) (46–50). Anaphase bridges were completely absent and lagging chromatin was very rare in BJ/tert cells express-ing an empty vector (BJ-V) or the Bub1 bindexpress-ing-deficient mutant (BJ-dl89-97) (Fig. 1AandD). Chromatin bridges frequently per-sisted and were also found in interphase cells (Fig. 1H).
Another important marker of genomic instability is micronu-clei. We next examined the presence of micronuclei, as it is an end product of chromosome missegregation or DNA damage (38,51, 52). Consistent with the high incidence of chromatin bridges, mi-cronuclei were highly abundant in BJ-LT but absent in BJ-V and
BJ-dl89-97 cells (Fig. 1B). Micronuclei that stain positive for cen-tromeres (centric) arise when a whole lagging chromosome fails to be segregated due to merotelic attachment, which happens when a single KT is attached to MTs emanating from both spindle poles (38). Conversely, micronuclei lacking centromere staining (acen-tric) are generated by chromosomal damage or breakage of chro-matin bridges (37,52). We found that the majority of micronuclei in BJ-LT cells were negative for centromere (CREST) staining, suggesting that they arose from DNA breakage (Fig. 1C) (53). Consistent with this interpretation, most of the micronuclei were positive by immunofluorescence staining for␥-H2AX, which is a marker for DNA breaks or, more specifically, DSBs (6).
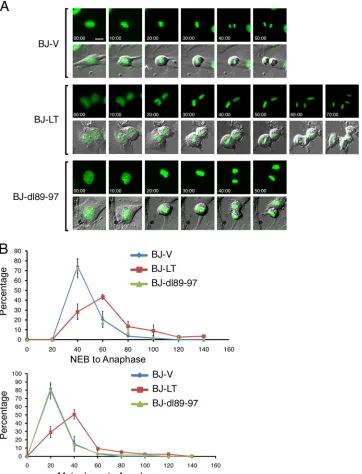
LT induces a delay in metaphase-to-anaphase transition de-pendent on Bub1 binding.Because LT induces anaphase bridges, which reflects mitotic dysfunction, and it binds the mitotic check-point kinase Bub1, we investigated whether LT affects mitotic pro-gression. BJ-LT, BJ-dl89-97, and BJ-V cells were engineered to express histone H2B-GFP for monitoring of mitotic progression by time-lapse microscopy (Fig. 2A) (44, 54). Fluorescence and DIC images were acquired every 10 min. Analysis of individual frames from time-lapse video microscopy allowed us to determine the time from nuclear envelope breakdown (NEB) until anaphase or from alignment on the metaphase plate until anaphase onset. Most BJ-V and BJ-dl89-97 cells (80%) entered anaphase on
aver-FIG 1Micronuclei and anaphase bridges are prevalent in LT-expressing cells. Error bars on the graphs indicate standard errors, whereas the double asterisks show that differences between LT and dl89-97 are considered highly significant. (A) Anaphases were evaluated by DAPI staining and microscopy for chromatin bridges and lagging chromatin. The frequency of anaphases with bridges or lagging chromatin is shown in the graph. Totals of 37, 26, and 57 anaphases were quantitated for BJ/tert cells expressing the vector, LT, and dl89-97, respectively. (B) The frequency of nuclei with an adjacent micronucleus was determined by DAPI staining and microscopy. Totals of 175, 134, and 538 nuclei were quantitated for BJ/tert cells expressing the vector, dl89-97, and LT, respectively. (C) For BJ/tert LT cells, the frequency of micronuclei containing clear␥-H2AX or CREST staining was determined based on 186 micronuclei. (D) Normal anaphase in BJ/tert cells expressing the vector. (E to G) Aberrant anaphases in BJ-LT cells exhibiting bridges and lagging chromatin. (H) Interphase chromatin bridge in BJ-LT cells. In panels D to H, using Adobe Photoshop, the acquired DAPI images were converted to grayscale, the color was inverted, and contrast was maximized in order to most clearly depict the bridges.
SV40 T Antigen-Mediated Genome Destabilization
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.99.486.63.342.2]age 40 min after NEB. In contrast, only 30% of BJ-LT cells entered anaphase 40 min after NEB. While most of BJ-LT cells required 60 min for the progression from NEB to anaphase, some took even longer than 100 min (Fig. 2B). Similarly, after metaphase, most BJ-V and BJ-dl89-97 cells entered anaphase after 20 min, while 70% of BJ-LT cells required 40 min or longer for the progression from metaphase to anaphase (Fig. 2B). When we studied⬎30 cells and plotted the times required for each cell to complete the
NEB-to-anaphase or metaphase-NEB-to-anaphase transition, it became clear that BJ-LT cells are consistently delayed by at least 20 min at the metaphase-to-anaphase boundary (Fig. 2B). BJ-V and BJ-dl89-97 cells behaved identically with regard to their mitotic progression. All of the cell lines required about 30 min to align the chromo-somes on the metaphase plate. Collectively, live-cell imaging re-vealed a metaphase delay induced by LT depending on the Bub1 binding site.
FIG 2LT transiently delays the metaphase-to-anaphase transition in a Bub1 binding-dependent manner. (A) BJ-V (empty vector), BJ-LT, and BJ-dl89-97 cells stably expressing histone H2B-enhanced GFP were released from a thymidine-induced G1/S block. After 8 h, cells were monitored by time-lapse microscopy
(minutes elapsed are indicated). Selected frames of both FITC and FITC/DIC merged images are shown. (B) Time-lapse videos were analyzed frame by frame to discern times of nuclear envelope breakdown (NEB), metaphase alignment, and anaphase onset. The percentage of all cells that completed the transition (from NEB to anaphase or from metaphase to anaphase) for each time point is depicted in the graphs. Data from two independent experiments are included. Analysis was based on 30 cells for BJ-V and BJ-dl89-97 and 50 cells for BJ-LT. The values are means⫾standard deviations (error bars).
on November 7, 2019 by guest
http://jvi.asm.org/
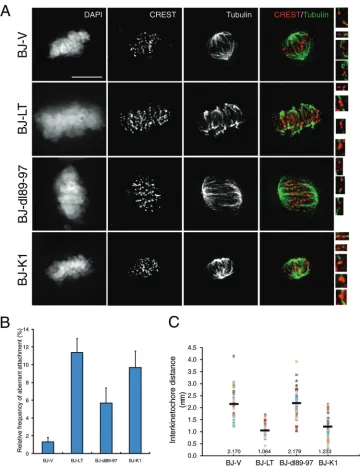
[image:4.585.113.475.68.542.2]LT compromises kinetochore-microtubule attachments and attenuates sister chromatid tension.Since LT imposes a meta-phase delay, we reasoned that this might be caused by aberrant KT-MT attachments. Indeed, Bub1 has been shown to participate in the error correction machinery (17,18,22). Therefore, we di-rectly visualized the KT-MT attachments by immunofluorescence staining of centromeres (CREST) and tubulin (Fig. 3A) (44). BJ-V and BJ-dl89-97 cells exhibited mostly normal end-on attach-ments, whereas BJ-LT cell attachments were highly aberrant, in-cluding lateral, monotelic, and syntelic abnormalities (Fig. 3A,
insets) (44). Because pRB inactivation has sometimes also been implicated in genome destabilization phenotypes, we included the K1 mutant of LT, which is pRB binding defective, as a control (55, 56). The K1 mutant behaved similarly to wild-type LT, excluding pRB binding from playing a major role (Fig. 3AandB). We quan-titated the frequency of aberrant attachments and observed an ⬃6-times-higher frequency in BJ-LT cells (Fig. 3B). The SAC monitors both lack of tension and attachment of MTs to KTs (20,57). To determine the tension across sister chromatids, we measured the interkinetochore distance between two CREST
FIG 3LT affects kinetochore tension and error correction of KT-MT attachments. (A) Thymidine-synchronized BJ-V, BJ-LT, BJ-dl89-97, and BJ-K1 cells were released for 8 h and treated with MG-132 for 3 h. Kinetochore fibers were visualized by chilling cells on ice for 10 min, followed by extraction, fixation, and staining for CREST, tubulin, and DAPI. Signals of tubulin and CREST were deconvolved, and individual bioriented kinetochores with microtubule attachment are shown in the insets. (B) The frequency of aberrant attachments is depicted (n⬎150). Error bars show the ranges of three different data sets. (C) Interkinetochore distances were determined by measuring the distances between two CREST signals with end-on attachment. The graph depicts individual values as well as the means, which are indicated by black bars (n⫽50).
SV40 T Antigen-Mediated Genome Destabilization
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.112.472.64.530.2]signals with end-on attachment (12,44). Consistent with at-tachment defects, the mean interkinetochore distance was sig-nificantly reduced in BJ-LT (1.064m) and BJ-K1 (1.233m) cells compared with BJ-V (2.17 m) and BJ-dl89-97 (2.179 m) cells, thus indicating a lack of tension (Fig. 3C). The lack of tension should be sensed by the SAC, which might account for the delay in the metaphase-to-anaphase transition. On the other hand, we have previously demonstrated that LT, by bind-ing to Bub1, is able to partially overcome the SAC, which might explain why the delay is rather minimal and the cells continue proliferating (4).
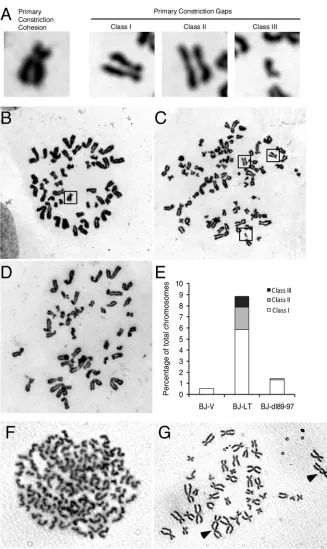
LT reduces sister chromatid cohesion via Bub1 binding and promotes dicentric chromosome formation.Because Bub1 was previously implicated in regulation of sister chromatid cohesion, either directly via recruitment of SgoI (shugoshin) (42, 58) or indirectly via SAC regulation (13,16,22), we investigated the ef-fect of LT on cohesion. After metaphase chromosome preparation and Giemsa staining, we observed a significant proportion of chromosomes with a premature loss of sister chromatid cohesion when LT was expressed (Fig. 4AandC) but not when an empty vector or dl89-97 was expressed (Fig. 4BandD). One hundred Giemsa-stained mitotic spreads were evaluated to estimate the frequency of sister chromatid cohesion loss (Fig. 4E). Occasion-ally, a cell exhibited a complete loss of sister chromatid cohesion, although this was quite rare (Fig. 4F). The loss of sister chromatid cohesion could be a contributing cause of CIN or at least exacer-bate the phenotype (45). Finally, the chromosome spreads also revealed dicentric chromosomes, which is consistent with previ-ous reports of LT (Fig. 4G) (24,26). Dicentric chromosomes are often caused by telomeric fusion after telomere erosion, but since telomerase is expressed, the underlying mechanism may be differ-ent.
LT induces prometaphase DNA damage and micronu-cleation that possibly arises from breakage of chromatin bridges.Although LT clearly induces mitotic dysfunction in the form of anaphase bridges (Fig. 1), metaphase delay (Fig. 2), aber-rant KT-MT attachments (Fig. 3A), loss of tension (Fig. 3B), and reduced sister chromatid cohesion (Fig. 4), LT has also been shown to induce DNA damage (7). Additionally, the micronuclei lack centromere staining, consistent with an origin from acentric chromosome fragments (Fig. 1C). Despite the general consensus in the past that CIN arises from mitotic dysfunction (31,59), for example, through loss of SAC activity, reduced cohesion, or mero-telic attachments, recent evidence from colorectal cancers indi-cates that replication stress and the resulting DNA damage can also be a driver of CIN and lead to chromosome missegregation (33). With that concept in mind, we explored the relationship between DNA damage and mitotic abnormalities.
We noted that the micronuclei in BJ-LT cells stained strongly for␥-H2AX and were positioned as if they originated from break-age of an anaphase bridge (Fig. 5A) (37,60). Indeed, it has been demonstrated that one of the origins of micronuclei is through severing of chromatin bridges at cytokinesis (37). Some of the anaphase bridges were also decorated by␥-H2AX (Fig. 5B), and the breakpoints or extremities of the bridge often exhibited bright ␥-H2AX foci (Fig. 5BandC) (61). Furthermore, prometaphase BJ-LT cells showed multiple␥-H2AX foci (Fig. 5D). When com-paring the average number of␥-H2AX foci per prometaphase cell, the LT-expressing cells had a much greater number than the vec-tor-expressing cells (Fig. 5E).
LT expression promotes formation of BLM-decorated ultra-fine bridges that are indicative of replication stress.The appear-ance of anaphase bridges in BJ-LT cells prompted us to investigate the presence of another type of bridge, known as “ultrafine bridges” (UFBs) (62–65). UFBs cannot be stained by conventional DNA dyes, although fluorescencein situhybridization and bro-modeoxyuridine (BrdU) incorporation indicate that they contain DNA. Replication stress causes an increased number of UFBs that are coated with the Bloom syndrome helicase (BLM) and the Plk1-interacting checkpoint helicase (PICH) (63). UFBs often connect centromeres or FancD2 foci and result from unresolved DNA rep-lication structures or decatenation failure (63,64). We found mul-tiple UFBs in BJ-LT cell anaphases after staining by immunofluo-rescence for the BLM protein (Fig. 6AtoC). Sometimes, the UFBs coincided with DAPI-stained conventional chromatin bridges, but most often, they did not. When quantitating the number of UFBs, we found that LT-expressing cells had a greatly increased number compared to cells expressing the vector control, which likely reflects LT-mediated replication stress from unresolved rep-lication intermediates (Fig. 6D) (33,64,65).
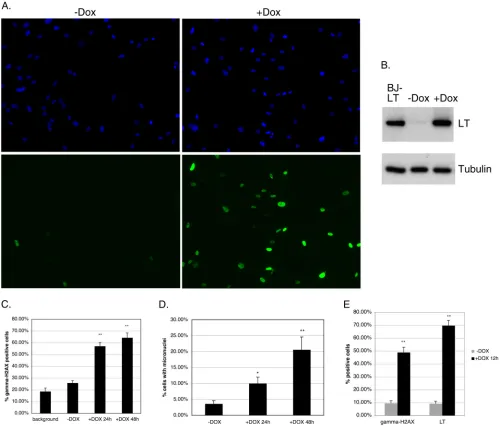
A conditional LT expression system demonstrates that the DDR is induced acutely and precedes the majority of micronu-cleus formation.Given signatures of both replication stress and mitotic dysfunction in BJ-LT cells, we wondered if the replication stress resulted from mitotic dysfunction or whether it was a sepa-rate phenomenon. To approach this question, we sought to deter-mine their temporal relationship, if any. We used a doxycycline (Dox)-regulated system that allowed us to rapidly induce LT ex-pression in BJ/tert cells (39). In the absence of Dox, only a small percentage of the cells showed LT expression by immunofluores-cence (Fig. 7A). Addition of Dox for 24 h led to induction of LT in ⬎80% of the cells (Fig. 7A). Western blotting also showed strong Dox-dependent induction of LT expression and minimal expres-sion without Dox (Fig. 7B). Expression after Dox induction was approximately equivalent to stable LT expression in BJ-LT cells (Fig. 7B). Thus, this expression system provides a robust means of inducibly expressing LT with minimal background.
At 24 h or 48 h after addition of Dox, cells were stained by immunofluorescence for both LT and␥-H2AX, and the frequen-cies of␥-H2AX-positive cells were calculated. The dual staining for LT also allowed us to determine the background, which we define as the population of cells that are␥-H2AX positive but LT negative (Fig. 7C). As shown inFig. 7C, LT induction for 24 h caused a dramatic increase in the proportion of␥-H2AX-positive cells compared with the background. Similar results were ob-tained in 3 independent experiments. Induction for 48 h only slightly increased the␥-H2AX frequency compared with induc-tion for 24 h, presumably because of a moderate increase in the number of LT-expressing cells. Note that the value without Dox is slightly higher than the background, because it includes the minor population of cells that express LT in the absence of Dox induc-tion. We also quantitated the frequency of micronuclei, which reflect errors in chromosome segregation following DNA break-age. As shown inFig. 7D, the frequency of micronuclei began to increase at 24 h, but increased much further at 48 h, post-LT induction. Taken together, this argues strongly that DNA damage occurs acutely within 24 h of LT expression and is followed later, likely after mitosis, by micronucleus formation. A separate exper-iment demonstrated that the majority of anaphase bridges occur prior to micronucleus formation, which is suggestive of a
on November 7, 2019 by guest
http://jvi.asm.org/
ral relationship between these (data not shown; also see Discus-sion).
To corroborate that LT can induce DNA damage in the absence of mitotic missegregation events, we performed a serum
starva-tion experiment with Dox-inducible cells. As shown inFig. 7E, when comparing cells subsequently serum stimulated in the ab-sence of Dox to those in the preab-sence of Dox for 12 h, we observed a significant increase in the number of␥-H2AX foci when LT was
FIG 4LT attenuates sister chromatid cohesion. (A) Representative images of normal primary constriction cohesion and various classes of primary cohesion gaps: PCGI (mild), PCGII (moderate), and PCGIII (severe). (B to D) BJ-V (B), BJ-LT (C), and BJ-dl89-97 (D) cells were trypsinized, hypotonically treated, and fixed with methanol-glacial acetic acid (3:1). Metaphase spreads were prepared and stained with Giemsa. (E) Frequency of cohesion defects. (F) Metaphase spread showing complete loss of sister chromatid cohesion in a BJ-LT cell. (G) Two examples of dicentric chromosomes in a BJ-LT cell, indicated by arrowheads.
SV40 T Antigen-Mediated Genome Destabilization
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.124.451.61.610.2]induced (49%), relative to the noninduced control (9.5%). Ap-proximately 70% of the cells expressed LT after 12 h of induction (Fig. 7E). Importantly, after 12 h of serum stimulation, the cells had not yet reached mitosis. Taken together, we conclude that LT can induce DNA damage without passage of the cells through mitosis, possibly as a result of replication stress.
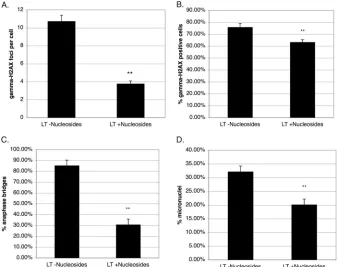
Nucleoside supplementation reduces DNA damage markers, anaphase bridges, and micronuclei, consistent with a replica-tion stress response.Replication stress may result from aberrant origin firing or replication fork progression. Recent evidence in-dicates that exogenous nucleoside supplementation can attenuate the replication stress response, because it is often fueled by deple-tion of the nucleotide pool due to insufficient nucleotide synthesis (33,40). Therefore, we supplemented BJ-LT cells with 50 M nucleosides for 48 h and quantitated␥-H2AX formation, ana-phase bridges, and micronuclei (40). As shown inFig. 8A, the average number of␥-H2AX foci per cell was greatly decreased by the addition of nucleosides. The overall frequency of␥ -H2AX-positive cells was also reduced upon nucleoside addition, although this effect was less pronounced than the effect on the number of foci per cell (Fig. 8B). Strikingly, the frequency of anaphase bridges was markedly reduced (Fig. 8C), and the proportion of micronuclei was also clearly reduced (Fig. 8D). Perhaps the reason for the effect on micronuclei being less pronounced than the effect on anaphase bridges relates to the micronuclei already being pres-ent to begin with in BJ-LT cells and only gradually being lost. Overall, the changes occurring upon nucleoside supplementation are of the same magnitude as those previously reported (33,40). Taken together, the observation that nucleoside addition attenu-ates the genome instability phenotypes is consistent with a caus-ative role of replication stress (33,40).
FIG 5LT induces␥-H2AX foci in mitotic cells. (A to D) Merged immunofluorescence images of BJ-LT cells stained for␥-H2AX (green) and DAPI (blue). Telophase cells (A and C) show remnants of a chromatin bridge, which was converted into micronuclei.␥-H2AX staining decorates micronuclei (A), the presumed breaking point of the bridge (C), as well as the anaphase bridge itself (B). Prometaphase cells frequently exhibit multiple␥-H2AX foci (D). (E) Average numbers of␥-H2AX foci per prometaphase cell compared between BJ-V cells (22 cells) and BJ-LT cells (17 cells). Error bars depict standard errors, whereas the double asterisk indicates that the difference between LT and the vector is considered highly significant.
FIG 6LT expression promotes formation of BLM ultrafine anaphase bridges. (A to C). Merged immunofluorescence images of BJ-LT anaphase cells stained for BLM (green) and DAPI (blue). (D) Frequency of UFB formation in BJ-V (43 anaphases) and BJ-LT (24 anaphases) cells. Error bars depict standard errors, whereas the double asterisk indicates that the difference between LT and the vector is considered highly significant.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.114.473.63.313.2] [image:8.585.44.285.414.665.2]DISCUSSION
The two main promoters of genomic instability are DNA damage and aberrant mitosis (32). We have previously demonstrated that LT is capable of inducing both DNA damage as well as a compromised SAC and tetraploidy in BJ/tert normal human fibroblasts (4,5). Here we significantly extend the genome destabilization phenotypes to in-clude anaphase bridges (both DAPI-stainable bridges and UFBs),
acentric micronuclei, metaphase delay, shortened interkinetochore distance, aberrant KT/MT attachments, reduced chromatid cohe-sion, DNA damage in mitosis, and replication stress. While aberrant mitotic segregation can cause DNA damage, the DNA damage aris-ing, for example, from replication stress can also induce mitotic ab-normalities (32–38). Here we present evidence of their temporal re-lationship when LT is expressed.
FIG 7Inducible expression of LT acutely elicits␥-H2AX foci indicative of replication stress prior to micronucleus formation. Error bars on the graphs indicate standard errors. (A) BJ/tert cells stably expressing pInducer20 LT were examined by immunofluorescence with Dox (⫹Dox) or without Dox (⫺Dox) for 48 h. Nuclei are visualized with DAPI in blue, whereas LT staining of the same field is shown in green. (B) Western blot analysis of LT expression in BJ/tert pInducer20 LT cells grown with or without Dox for 48 h as well as in BJ-LT cells, which were included as a control. The same samples were also blotted for tubulin to verify even loading. (C) BJ/tert pInducer20 LT cells were grown with or without Dox for 24 h or 48 h, respectively. Cells were subsequently stained by immunofluo-rescence for␥-H2AX and LT. The percentage of all cells that were positive for␥-H2AX staining was calculated and is depicted in the graph. “Background” refers to the population of cells that are␥-H2AX positive but LT negative. Total numbers of cells counted were 251, 280, and 194 for cells grown without Dox, cells grown with Dox for 24 h, and cells grown with Dox for 48 h, respectively. The double asterisk indicates that differences between cells grown without Dox and those grown with Dox (24 h and 48 h, respectively) are highly statistically significant. (D) Same analysis as outlined above for panel C except that micronuclei were quantitated and are depicted. The single asterisk indicates that for growth with Dox for 24 h, the difference is significant, whereas the double asterisk indicates that for growth with Dox for 48 h, the difference is highly significant compared to growth without Dox. (E) LT-inducible cells were serum starved for 3 days, followed by serum stimulation without Dox or with Dox for 12 h. Cells were then stained for␥-H2AX and LT. The frequency of␥-H2AX- or LT-positive cells was calculated based on 252 cells (without Dox) or 245 cells (with Dox), respectively. The double asterisk shows that differences between growth without Dox and growth with Dox for 12 h are highly significant.
SV40 T Antigen-Mediated Genome Destabilization
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.42.544.67.493.2]Our studies are significant for two principal reasons. First, they define the nature of mitotic and DNA damage defects caused by LT that affect genome stability. There is evidence to support a role of genomic instability in LT-induced tumorigenesis. When LT was inducibly expressed in the submandibular gland of transgenic mice, hyperplasia could be reversed after 4 months but not after 7 months of age, suggesting that CIN may drive the cells to ulti-mately become independent of LT (66). Moreover, experiments with SV40 expression in human keratinocytes demonstrated that these cells become tumorigenic only after extensive passage in culture, again suggesting the importance of secondary genetic changes (67). Second, LT provides an excellent model system to gain insight into the links between replication stress, DNA dam-age, and mitotic abnormalities. Currently, there is much focus on improving our understanding of these responses, because cellular oncogenes are thought to induce replication stress, leading to an activated DDR. DDR activation is observed at the earliest stages of tumorigenesis, where it serves as a barrier to full oncogenic trans-formation (68,69).
Collectively, our results presented here allow us to introduce a model that integrates potential links between the various genomic instability phenotypes that LT provokes (Fig. 9). The inducible LT expression system suggests that replication stress and the associ-ated DNA damage from stalled or collapsed forks might be the earliest events, although we cannot exclude that LT binding to Bub1 also contributes to overall genomic instability by perturbing mitosis directly. Strikingly, replication stress was recently shown to be a major cause of CIN in colorectal cancers, where it had been
previously assumed that SAC dysfunction or merotelic attach-ments were responsible for CIN (31,33,59). Anaphase bridges and lagging chromatin are hallmarks of CIN and are correlated with poor prognoses (46–50). Replication stress can lead to both visible and ultrafine anaphase bridges, but these bridges can also be caused by aberrant DNA repair, telomere fusion, or decatena-tion failure (33,64,65,70). Abnormalities arising from telomere erosion are likely to be minimal, since telomerase is expressed. Anaphase bridges can break, leading to DNA fragments, which are then incorporated into micronuclei upon chromosome segrega-tion (37). Because cells expressing the Bub1 binding mutant lack both anaphase bridges and micronuclei, it strengthens the argu-ment for a link between these cellular phenomena. Observations from our LT-inducible system are indicative of anaphase bridges arising before micronuclei. The arrangement of␥-H2AX foci in telophase, or interphase after mitosis, is also suggestive of a broken bridge. DNA contained in micronuclei is subject to further break-age, since DNA replication is defective or asynchronous and repair is dysfunctional (38). Indeed, we found most of the micronuclei to be␥-H2AX positive and to contain acentric fragments.
Anaphase bridges may act as trapped chromatin, which can cause regression of the cleavage furrow, failed cytokinesis, and formation of tetraploid cells (71). Conversely, DNA damage, aris-ing from telomere dysfunction or from other sources, can also cause tetraploidy (34,72). The mechanistic basis for tetraploidiza-tion is not fully understood. According to a recent study, LT causes tetraploidy mainly via endoreduplication or mitotic failure (73). Tetraploid cells exhibit CIN, may constitute an intermediate
FIG 8Nucleoside supplementation reduces␥-H2AX foci, anaphase bridges, and micronuclei, consistent with a replication stress response. Error bars on the graphs depict standard errors, whereas the double asterisk indicates that differences between LT effects without nucleosides and those with nucleosides are considered highly significant. (A) BJ-LT cells were grown with or without 50M nucleoside supplement for 48 h and subsequently stained for␥-H2AX. The average number of␥-H2AX foci per cell is outlined. Total numbers of cells counted were 171 and 314 without and with nucleoside supplementation, respectively. (B) Same analysis as in panel A except that the frequency of␥-H2AX-positive cells is depicted. (C) Same analysis as in panel A except that the frequency of anaphase bridges is depicted. (D) Same analysis as in panel A except that the frequency of micronuclei is shown.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.123.462.67.334.2]to aneuploidy, and are predisposed to tumorigenesis in a p53-deficient background (74–76). Tetraploid cells are known to gen-erate merotelic attachments, which in turn promote micronucleus formation (76). DNA damage activates the DDR and p53, which should block tetraploidy and tumorigenesis, but LT overcomes this block by inactivating p53, thus propagating CIN and DNA damage (74,77–80). Finally, given the presence of dicentric chro-mosomes, it is also likely that LT fuels genetic instability via break-age-fusion-bridge (BFB) cycles, where breakage of chromosomes or loss of telomeres leads to fusion events, the resulting dicentric chromosomes form a chromatin bridge in anaphase, and finally, breakage of the bridge occurs, starting a new cycle (50).
The chromosome segregation defects that we observed are reminiscent of Bub1 insufficiency and an attenuated SAC (12,13, 21). Thus, the aberrant mitotic phenotypes that we observed in LT-expressing cells, such as micronuclei, lagging chromosomes, chromatin bridges, and loss of sister chromatid cohesion, were noted previously for cells with reduced Bub1 expression levels (12, 13,21,42,58). Bub1 is an essential protein, even in somatic cells (13). As expected, LT does not interfere with the crucial scaffold-ing role of Bub1 in assembly of the kinetochore (Bub1, BubR1, Aurora B, and PLK1 remain at the kinetochore [data not shown]) (15,81). There is controversy as to whether Bub1 directly regu-lates cohesion via phosphorylation of histone H2A on T120 and recruitment of shugoshin (SgoI) or whether the effect is indirect through Bub1’s role in the SAC (13,16,22,42,58). Since we saw no consistent effects of LT on phosphohistone H2A T120 or SgoI localization in mitotic cells, we conclude that the observed reduc-tion in cohesion likely results from SAC interference (data not shown). Since Bub1 is normally involved in KT-MT error correc-tion, defective attachments may cause the loss of tension (17,18). The mitotic delay may be caused by DNA damage, aberrant at-tachments, or tetraploid DNA content (82).
LT is not localized at the kinetochore, suggesting that it targets
the non-kinetochore-bound Bub1 population (4). Indeed, Bub1 is also known to play an important kinetochore-independent role in chromosome segregation (16). We speculate that specific protein complexes of Bub1 may be disrupted, or the kinase activity may be redirected to new substrates. As a potential caveat, we note that our conclusions are based on a single Bub1 binding mutant (dl89-97). We cannot formally exclude that another yet-to-be-identified binding partner for this region exists and is responsible for the phenotypes, but this seems unlikely. Furthermore, we have evi-dence that dl89-97 retains the ability to disrupt pRB and p53 path-ways like wild-type LT, thus arguing against a grossly perturbed structure of the mutant (data not shown). Point mutations in the region from residues 89 to 97 have not been as helpful as the deletion because of their residual Bub1 binding (4).
Since DNA damage induced by LT depends on Bub1 binding, it suggests a role of Bub1 in DNA replication or repair (5,43,83). This is consistent with our unpublished observations that LT binding to Bub1 is a prerequisite forin vivoviral replication. Bub1 has been shown to play a role in DDR and repair pathways. Thus, Bub1 is phosphorylated on S314 by ATM and colocalizes with ␥-H2AX foci following DNA damage, confirming that nonmitotic functions of Bub1 exist (83,84). Exactly how Bub1 regulates DNA replication and repair is a captivating subject of future studies.
There is considerable evidence of replication stress in BJ-LT cells, given the high frequencies of BLM UFBs (Fig. 6), 53BP1 foci in G1phase (7), prometaphase␥-H2AX foci (Fig. 5), and inter-phase FancD2 foci (7), taken together with clear phenotypic ef-fects of nucleoside supplementation (Fig. 8). BLM-decorated UFBs often connect fragile sites (64). Indeed, fragile sites and telo-meres may be the preferential breakpoints in LT-expressing cells, as replication stress preferentially causes DSBs in hard-to-repli-cate regions (85). The ATR/Chk1 pathway is activated by LT to cope with replication stress (7,9,11,86), which highlights a ther-apeutic opportunity that targeted inhibition of this pathway could
FIG 9Comprehensive model for genomic instability phenotypes induced by LT. The model integrates all of the genome destabilization phenotypes observed in LT-expressing cells and attempts to link these temporally. Note that in some cases, the relationship between two phenotypes can be bidirectional, which is illustrated by black and red arrows, respectively. For example, DNA damage can cause anaphase bridges, but breakage of the bridges can also induce DNA damage. A detailed overview is included in Discussion. Note that LT, as indicated, causes Bub1 dysfunction while simultaneously inactivating p53, thus blocking apoptosis and senescence programs.
SV40 T Antigen-Mediated Genome Destabilization
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.128.461.64.276.2]be relevant, as it would increase DNA damage and cell death (87). Replication stress may result from aberrant origin firing or repli-cation fork progression. The exact nature of the replirepli-cation defect remains to be shown. It was recently shown that human papillo-mavirus (HPV) E6/E7 expression causes replication stress due to depletion of the nucleotide pool (40). Consistent with a role of replication stress, we also found reduced DDR activation and numbers of micronuclei and anaphase bridges when exogenous nucleosides were supplemented (33,40). Moreover, the related viral initiator of DNA replication, HPV E1, also induces the DDR (88, 89). In the case of E1, origin-specific DNA binding and ATPase activity are required, and the mechanism has been pro-posed to involve nonspecific unwinding of host DNA or depletion of cellular replication factors. Replication stress and the resulting lesions in S phase can be carried over to the subsequent M phase, resulting in missegregation events, cytokinesis failure, and tetra-ploidy (34,65). It is not entirely clear why intra-S and G2/M check-points are not preventing this from happening, but it appears that lesions below a certain threshold can bypass the checkpoint.
Why induce DNA breaks and the accompanying DDR? For SV40, the answer seems to be that activated DDR promotes effi-cient viral replication by maintaining replication fork integrity (11). Induction of DSBs could also enhance integration events. In the case of LT expression alone, it may be viewed more as a case of “collateral damage.” Even in the absence of an origin, LT may be coopting and perturbing the cellular replication machinery. It is worth noting that the induction of genomic instability may be inherently beneficial to the virus also by driving cell heterogeneity and allowing adaptation to an ever-changing environment. Un-fortunately, if a tumor is created, CIN allows resistance to thera-peutic drugs and correlates with unfavorable prognoses (90). Our findings and model reported here are likely to apply more broadly to other viruses. For example, mitotic abnormalities and DNA damage have been observed in the context of the HPV E6/E7 on-coproteins, and the HIV Vpr protein also causes replication stress, micronuclei, and polyploidy (91–94).
ACKNOWLEDGMENTS
We thank Stephen Elledge for providing us with the pInducer20 vector, Assaf Bester and Batsheva Kerem for advice on nucleoside addition, and Ian Hickson for recommendations on visualization of BLM-coated UFBs. We also thank Hongtao Yu and Yoshinori Watanabe for providing anti-bodies to SgoI and phosphohistone H2A T120, respectively. We are in-debted to Brian Schaffhausen for reading the manuscript.
We gratefully acknowledge financial support from the NIH (grant R01 AI078926 to O.V.G. and grant CA169706 and core grant CA06927 to T.J.Y.) and the DOD (grant OC100172 to T.J.Y.) and an appropriation from the Commonwealth of Pennsylvania.
REFERENCES
1.Gjoerup O, Chang Y.2010. Update on human polyomaviruses and can-cer. Adv. Cancer Res.106:1–51.
2.Ahuja D, Saenz-Robles MT, Pipas JM.2005. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene24:
7729 –7745.
3.Dickmanns A, Zeitvogel A, Simmersbach F, Weber R, Arthur AK, Dehde S, Wildeman AG, Fanning E.1994. The kinetics of simian virus 40-induced progression of quiescent cells into S phase depend on four independent functions of large T antigen. J. Virol.68:5496 –5508. 4.Cotsiki M, Lock RL, Cheng Y, Williams GL, Zhao J, Perera D, Freire R,
Entwistle A, Golemis EA, Roberts TM, Jat PS, Gjoerup OV. 2004. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc. Natl. Acad. Sci. U. S. A.101:947–952.
5.Hein J, Boichuk S, Wu J, Cheng Y, Freire R, Jat PS, Roberts TM, Gjoerup OV. 2009. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J. Virol.
83:117–127.
6.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM.1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem.273:5858 –5868.
7.Boichuk S, Hu L, Hein J, Gjoerup OV.2010. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T an-tigen. J. Virol.84:8007– 8020.
8.Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS. 2005. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem.280:40195– 40200.
9.Rohaly G, Korf K, Dehde S, Dornreiter I.2010. Simian virus 40 activates ATR-delta p53 signaling to override cell cycle and DNA replication con-trol. J. Virol.84:10727–10747.
10. Zhao X, Madden-Fuentes RJ, Lou BX, Pipas JM, Gerhardt J, Rigell CJ, Fanning E.2008. Ataxia telangiectasia-mutated damage-signaling kinase-and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in simian virus 40-infected primate cells. J. Virol.82:5316 –5328. 11. Sowd GA, Li NY, Fanning E.2013. ATM and ATR activities maintain
replication fork integrity during SV40 chromatin replication. PLoS Pat-hog.9:e1003283. doi:10.1371/journal.ppat.1003283.
12. Meraldi P, Sorger PK.2005. A dual role for Bub1 in the spindle check-point and chromosome congression. EMBO J.24:1621–1633.
13. Perera D, Tilston V, Hopwood JA, Barchi M, Boot-Handford RP, Taylor SS.2007. Bub1 maintains centromeric cohesion by activation of the spindle checkpoint. Dev. Cell13:566 –579.
14. Holland AJ, Cleveland DW.2009. Boveri revisited: chromosomal insta-bility, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol.10:478 – 487.
15. Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS.2004. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci.117:1577–1589. 16. Klebig C, Korinth D, Meraldi P. 2009. Bub1 regulates chromosome
segregation in a kinetochore-independent manner. J. Cell Biol.185:841– 858.
17. Logarinho E, Bousbaa H.2008. Kinetochore-microtubule interactions “in check” by Bub1, Bub3 and BubR1: the dual task of attaching and signalling. Cell Cycle7:1763–1768.
18. Elowe S.2011. Bub1 and BubR1: at the interface between chromosome attachment and the spindle checkpoint. Mol. Cell. Biol.31:3085–3093. 19. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD,
Kinzler KW, Vogelstein B.1998. Mutations of mitotic checkpoint genes in human cancers. Nature392:300 –303.
20. Schliekelman M, Cowley DO, O’Quinn R, Oliver TG, Lu L, Salmon ED, Van Dyke T.2009. Impaired Bub1 function in vivo compromises tension-dependent checkpoint function leading to aneuploidy and tumorigenesis. Cancer Res.69:45–54.
21. Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM.
2007. Bub1 mediates cell death in response to chromosome missegrega-tion and acts to suppress spontaneous tumorigenesis. J. Cell Biol.179:255– 267.
22. Ricke RM, Jeganathan KB, van Deursen JM.2011. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyper-activation. J. Cell Biol.193:1049 –1064.
23. Baker DJ, Jin F, Jeganathan KB, van Deursen JM.2009. Whole chro-mosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell16:
475– 486.
24. Chang TH, Ray FA, Thompson DA, Schlegel R.1997. Disregulation of mitotic checkpoints and regulatory proteins following acute expression of SV40 large T antigen in diploid human cells. Oncogene14:2383–2393. 25. Friedrich TD, Laffin J, Lehman JM.1992. Simian virus 40 large T-antigen
function is required for induction of tetraploid DNA content during lytic infection. J. Virol.66:4576 – 4579.
26. Ray FA, Meyne J, Kraemer PM.1992. SV40 T antigen induced chromo-somal changes reflect a process that is both clastogenic and aneuploido-genic and is ongoing throughout neoplastic progression of human fibro-blasts. Mutat. Res.284:265–273.
27. Ray FA, Peabody DS, Cooper JL, Cram LS, Kraemer PM.1990. SV40 T antigen alone drives karyotype instability that precedes neoplastic trans-formation of human diploid fibroblasts. J. Cell. Biochem.42:13–31.
on November 7, 2019 by guest
http://jvi.asm.org/
28. Stewart N, Bacchetti S.1991. Expression of SV40 large T antigen, but not small t antigen, is required for the induction of chromosomal aberrations in transformed human cells. Virology180:49 –57.
29. Woods C, LeFeuvre C, Stewart N, Bacchetti S. 1994. Induction of genomic instability in SV40 transformed human cells: sufficiency of the N-terminal 147 amino acids of large T antigen and role of pRB and p53. Oncogene9:2943–2950.
30. Gjoerup OV, Wu J, Chandler-Militello D, Williams GL, Zhao J, Schaff-hausen B, Jat PS, Roberts TM.2007. Surveillance mechanism linking Bub1 loss to the p53 pathway. Proc. Natl. Acad. Sci. U. S. A.104:8334 – 8339.
31. Schvartzman JM, Sotillo R, Benezra R. 2010. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev. Cancer10:102–115.
32. Ganem NJ, Pellman D.2012. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol.199:871– 881.
33. Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, Chew SK, Rowan AJ, Schenk A, Sheffer M, Howell M, Kschischo M, Behrens A, Helleday T, Bartek J, Tomlinson IP, Swanton C.2013. Replication stress links structural and numerical cancer chromosomal instability. Nature494:
492– 496.
34. Ichijima Y, Yoshioka K, Yoshioka Y, Shinohe K, Fujimori H, Unno J, Takagi M, Goto H, Inagaki M, Mizutani S, Teraoka H.2010. DNA lesions induced by replication stress trigger mitotic aberration and tetra-ploidy development. PLoS One 5:e8821. doi:10.1371/journal.pone .0008821.
35. Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH.2011. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science333:1895–1898.
36. Hayashi MT, Karlseder J.2013. DNA damage associated with mitosis and cytokinesis failure. Oncogene32:4593– 4601.
37. Hoffelder DR, Luo L, Burke NA, Watkins SC, Gollin SM, Saunders WS.
2004. Resolution of anaphase bridges in cancer cells. Chromosoma112:
389 –397.
38. Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D.2012. DNA breaks and chromosome pulverization from errors in mitosis. Nature482:53–58. 39. Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Hersch-kowitz JI, Burrows AE, Ciccia A, Sun T, Schmitt EM, Bernardi RJ, Fu X, Bland CS, Cooper TA, Schiff R, Rosen JM, Westbrook TF, Elledge SJ.
2011. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A.108:3665–3670. 40. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon
A, Zamir G, Shewach DS, Kerem B.2011. Nucleotide deficiency pro-motes genomic instability in early stages of cancer development. Cell145:
435– 446.
41. Campbell KS, Mullane KP, Aksoy IA, Stubdal H, Zalvide J, Pipas JM, Silver PA, Roberts TM, Schaffhausen BS, DeCaprio JA.1997. DnaJ/ hsp40 chaperone domain of SV40 large T antigen promotes efficient viral DNA replication. Genes Dev.11:1098 –1110.
42. Tang Z, Sun Y, Harley SE, Zou H, Yu H.2004. Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mito-sis. Proc. Natl. Acad. Sci. U. S. A.101:18012–18017.
43. Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, Watanabe Y.2010. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science327:172–177.
44. Huang H, Hittle J, Zappacosta F, Annan RS, Hershko A, Yen TJ.2008. Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J. Cell Biol.183:667– 680.
45. Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C, Hieter P. 2008. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U. S. A.105:3443–3448.
46. Thompson SL, Compton DA.2008. Examining the link between chro-mosomal instability and aneuploidy in human cells. J. Cell Biol.180:665– 672.
47. Bakhoum SF, Danilova OV, Kaur P, Levy NB, Compton DA.2011. Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin. Cancer Res.17:7704 –7711. 48. Montgomery E, Wilentz RE, Argani P, Fisher C, Hruban RH, Kern SE,
Lengauer C.2003. Analysis of anaphase figures in routine histologic
sec-tions distinguishes chromosomally unstable from chromosomally stable malignancies. Cancer Biol. Ther.2:248 –252.
49. Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA.
2000. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature406:641– 645.
50. Gisselsson D, Pettersson L, Hoglund M, Heidenblad M, Gorunova L, Wiegant J, Mertens F, Dal Cin P, Mitelman F, Mandahl N. 2000. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc. Natl. Acad. Sci. U. S. A.97:5357–5362.
51. Shimura M, Tanaka Y, Nakamura S, Minemoto Y, Yamashita K, Hatake K, Takaku F, Ishizaka Y.1999. Micronuclei formation and aneuploidy induced by Vpr, an accessory gene of human immunodeficiency virus type 1. FASEB J.13:621– 637.
52. Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD, Thomas P.2011. Mo-lecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis26:125–132. 53. Majone F, Semmes OJ, Jeang KT.1993. Induction of micronuclei by
HTLV-I Tax: a cellular assay for function. Virology193:456 – 459. 54. Meraldi P, Draviam VM, Sorger PK.2004. Timing and checkpoints in
the regulation of mitotic progression. Dev. Cell7:45– 60.
55. Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R.2011. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell19:701–714.
56. Kalderon D, Smith AE.1984. In vitro mutagenesis of a putative DNA binding domain of SV40 large-T. Virology139:109 –137.
57. Musacchio A, Salmon ED.2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol.8:379 –393.
58. Kitajima TS, Hauf S, Ohsugi M, Yamamoto T, Watanabe Y. 2005. Human Bub1 defines the persistent cohesion site along the mitotic chro-mosome by affecting Shugoshin localization. Curr. Biol.15:353–359. 59. Thompson SL, Bakhoum SF, Compton DA.2010. Mechanisms of
chro-mosomal instability. Curr. Biol.20:R285–R295. doi:10.1016/j.cub.2010 .01.034.
60. Medvedeva NG, Panyutin IV, Panyutin IG, Neumann RD.2007. Phos-phorylation of histone H2AX in radiation-induced micronuclei. Radiat. Res.168:493– 498.
61. Suzuki M, Suzuki K, Kodama S, Watanabe M.2006. Phosphorylated histone H2AX foci persist on rejoined mitotic chromosomes in normal human diploid cells exposed to ionizing radiation. Radiat. Res.165:269 – 276.
62. Chan KL, Hickson ID.2011. New insights into the formation and reso-lution of ultra-fine anaphase bridges. Semin. Cell Dev. Biol.22:906 –912. 63. Chan KL, North PS, Hickson ID.2007. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J.26:3397–3409.
64. Chan KL, Palmai-Pallag T, Ying S, Hickson ID.2009. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol.11:753–760.
65. Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, Lukas J.2011. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol.13:243–253. 66. Ewald D, Li M, Efrat S, Auer G, Wall RJ, Furth PA, Hennighausen L.
1996. Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science273:1384 –1386.
67. Brown KW, Gallimore PH.1987. Malignant progression of an SV40-transformed human epidermal keratinocyte cell line. Br. J. Cancer56:545– 554.
68. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J.2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature434:864 – 870.
69. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F.2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature444:638 – 642.
70. Laulier C, Cheng A, Stark JM.2011. The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome sep-aration. Nucleic Acids Res.39:5935–5944.
71. Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar
SV40 T Antigen-Mediated Genome Destabilization
on November 7, 2019 by guest
http://jvi.asm.org/
S, Gerlich DW.2009. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell136:473– 484.
72. Davoli T, Denchi EL, de Lange T.2010. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell141:81–93.
73. Davoli T, de Lange T.2012. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell21:765–776.
74. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D.
2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature437:1043–1047.
75. Ganem NJ, Storchova Z, Pellman D.2007. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev.17:157–162.
76. Ganem NJ, Godinho SA, Pellman D.2009. A mechanism linking extra centrosomes to chromosomal instability. Nature460:278 –282. 77. Lanni JS, Jacks T.1998. Characterization of the p53-dependent
postmi-totic checkpoint following spindle disruption. Mol. Cell. Biol.18:1055– 1064.
78. Thompson SL, Compton DA.2010. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol.188:369 –381. 79. Aylon Y, Oren M.2011. p53: guardian of ploidy. Mol. Oncol.5:315–323. 80. Bargonetti J, Reynisdottir I, Friedman PN, Prives C.1992. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev.6:1886 –1898.
81. Boyarchuk Y, Salic A, Dasso M, Arnaoutov A.2007. Bub1 is essential for assembly of the functional inner centromere. J. Cell Biol.176:919 –928. 82. Mikhailov A, Cole RW, Rieder CL.2002. DNA damage during mitosis in
human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol.12:1797–1806.
83. Yang C, Wang H, Xu Y, Brinkman KL, Ishiyama H, Wong ST, Xu B.
2011. The kinetochore protein Bub1 participates in the DNA damage re-sponse. DNA Repair (Amst.)11:185–191.
84. Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, Wong ST, Fu L, Xu B.2011. Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol. Cell
44:597– 608.
85. Durkin SG, Glover TW.2007. Chromosome fragile sites. Annu. Rev. Genet.41:169 –192.
86. Jiang M, Zhao L, Gamez M, Imperiale MJ.2012. Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus in-fection. PLoS Pathog.8:e1002898. doi:10.1371/journal.ppat.1002898. 87. Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S,
Oyar-zabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O.2011. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for can-cer-associated mutations. Nat. Struct. Mol. Biol.18:721–727.
88. Sakakibara N, Mitra R, McBride AA.2011. The papillomavirus E1 heli-case activates a cellular DNA damage response in viral replication foci. J. Virol.85:8981– 8995.
89. Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J.2011. Nuclear accumulation of the papillo-mavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol.85:8996 –9012.
90. Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, Kschischo M, Swan-ton C.2011. Chromosomal instability confers intrinsic multidrug resis-tance. Cancer Res.71:1858 –1870.
91. Duensing S, Munger K.2002. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromo-some instability. Cancer Res.62:7075–7082.
92. Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF, Duensing S.2007. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi anemia (FA) pathway and causes accelerated chro-mosomal instability in FA cells. J. Virol.81:13265–13270.
93. Zimmerman ES, Sherman MP, Blackett JL, Neidleman JA, Kreis C, Mundt P, Williams SA, Warmerdam M, Kahn J, Hecht FM, Grant RM, de Noronha CM, Weyrich AS, Greene WC, Planelles V.2006. Human immunodeficiency virus type 1 Vpr induces DNA replication stress in vitro and in vivo. J. Virol.80:10407–10418.
94. Shimura M, Onozuka Y, Yamaguchi T, Hatake K, Takaku F, Ishizaka Y.
1999. Micronuclei formation with chromosome breaks and gene amplifi-cation caused by Vpr, an accessory gene of human immunodeficiency virus. Cancer Res.59:2259 –2264.