Copyright©1976 AmericanSociety forMicrobiology Printed inU.SA.
N-Tropic Variants
Obtained
After Co-Infection with N- and
B-Tropic Murine Leukemia
Viruses
NANCY HOPKINS,* PAULATRAKTMAN, AND KATHLEEN WHALEN
Center for CancerResearch,Massachusetts InstituteofTechnology, Cambridge,Massachusetts02139 Received forpublication 22September1975
Sc-i cells co-infected with small XC plaque-forming N-tropic and large XC
plaque-forming B-tropicmurine leukemia virusesproduced, in addition to
pa-rentaltypes, progeny withthephenotype, largeXCplaquemorphology,and
N-tropism. Thisphenotyperemained stablethroughendpointtitration andplaque
purification on NIH/3T3 cells and growthon BALB/3T3 cells. These N-tropic
viruses (XLP-N virus) grow to unusually high titer and make very large XC plaques.
Co-infection with RNAtumorviruses
carry-ingappropriate genetic markerscanyield
prog-enywithnovelphenotypes.Inthe avian system
three typesof interactions betweentwo
exoge-nously infecting viruses orbetween an
exoge-nous and an endogenous virus have been
de-scribed: phenotypic mixing, heterozygosis, and
geneticrecombination(4, 11,23-25, 27).Studies
with mammalian RNA tumor viruses have
been less extensive than those with avian
vi-ruses, in part because of a lack of suitable
markers. However, evidence for phenotypic
mixing and for recombination between
exoge-nous viruses (21, 26) orbetween anexogenous
and an endogenous virus (20) has been re-ported.
In this paper we describe the isolation of
viruses with novelphenotype after co-infection
ofSc-icells with N- andB-tropic murine
leuke-mia virusesderived fromBALB/c mice. The
N-or B-tropism of ecotropic murine leukemia
vi-ruses provides a natural genetic marker that
allowsastrongselection for viruses with either
hostrange (7, 13,14, 16).Recently,Hartley and
Rowe (5)described the isolation ofa mousecell
line, Sc-i, on which N- and B-tropic murine
leukemia viruses grow equally well. Hopkins
and Jolicoeur (8) andStephensonand Aaronson
(19) have recentlydescribedtheisolation of N-tropic murine leukemia viruses, derived from
BALB/c mice, thatmake XCplaques with
al-tered morphology. TheXCplaqueassay
devel-oped byRowe etal. (18) isbasedonthe
observa-tion (12)that whenmouse cellsproducing cer-tainmurine leukemia viruses come in contact
with XC cells(22), a ratcell line transformedby
an avian sarcoma virus, the XC cells fuse to
form syncytia.
We describe here the isolation of large XC
plaque-forming N-tropic murine leukemia
vi-ruses(XLP-N virus)fromamong the progeny of
Sc-i cellsco-infected with the small XC
plaque-forming N-tropic (SP-N) virus ofHopkinsand
Jolicoeur (8) and large XC plaque forming
B-tropic (LP-B) virus. Infectionby SP-NorLP-B
alone didnotyieldXLP-N virus. MATERIALS AND METHODS
Cells. BALB/3T3 (clone A31) cells (1) were ob-tained from Robert Pollack, Cold Spring Harbor; NIH/3T3 cells (9) were from Stuart Aaronson, Na-tional Institutes of Health; and Sc-1 (5) and XCcells were from Janet Hartley and Wallace Rowe, Na-tional Institutes of Health. Medium was Dulbecco modified Eagle medium with 10% calf serum (10% inactivated fetal calf serum was used for Sc-1 cells) and 50 ,ugof penicillin and 50,gof streptomycin per ml.
Viruses. SP-N virus is a clonal isolate derived by Hopkins and Jolicoeur (8) from a stock of the LP-N virus (WN 1802N, pool 1898) derived from BALB/c by Hartleyetal. (6). The LP-B virus used is a clonal isolatederived by Paul Jolicoeur from a stock of the LP-B virus (WN 1802 B) derived from BALB/c by Hartley et al. (6, 10).
XC assays.NIH/3T3cells orBALB/3T3cells were plated at 105 cells/60-mmdish. Thenext day they were infected with 0.2 ml of appropriately diluted viruscontainingpolybrene (8 fig/ml). All dilutions were plated in triplicate. After 2 h ofadsorption with frequent rocking ofthe plates, medium was replaced. The next day the medium was changed. Theday before the cells reached confluence the me-dium waschanged. The next day the medium was removed, and the cells were irradiated and overlaid with 106 XC cells as described (18). Medium was changedthe following day, and on day 3 after the addition of XC cells the plates were fixed and stained, either with Harris hematoxylin or cresyl violet acetate (1% aqueous solution).
Plaquepurification ofvirus. Virus was diluted sufficiently so that a number ofplates received 1 PFU. An XC assay was performed as described above except that the plates were not irradiated beforebeingoverlaid with XC cells. Onday 3 after addition of XC cellsa plaquewaslocated and cir-324
on November 10, 2019 by guest
http://jvi.asm.org/
MLV PLAQUE VARIANTS 325
cled, and the cells from the circled area were re-moved with a cloning cylinder. The plate was then fixed and stained to confirm that no other plaques were present on the plate. The cells from the cloning were plated with 105 NIH/3T3 cells on a 60-mm dish. Four days later, culture fluids from this plate were collected and used to infect 105 NIH/3T3 cells on a 60-mm plate. When these cells had grown up their culture fluids were used as a source of plaque-puri-fied virus.
End point dilutions. NIH/3T3 cells (105) were plated on60-mm plates and infected the next day with 10-fold dilutions of virus. When the cells were confluenttheywere trypsinizedand transferredtoa 100-mmdish. At confluence they were split 1:5 and carried until reverse transcriptase assay (8) of the culture fluids showed a clear end point titer.
RESULTS
Properties of parental N- and B-tropic
vi-ruses. (i) Plaque morphology. XC plaques can
becharacterizedas tosize,sharpnessofoutline,
turbidity, and size and number of syncytia
withinthe plaque (18). For anyparticularvirus
these properties vary to some extent,depending
uponthecell lineemployedand the number of
cells present at the time of infection. Themore
cellspresent, the smallertheplaquesize. Iftoo
few cells are present at infection, the plaques
are large and diffuse and secondary plaques
appear.
Recently, we described the isolation ofan
N-tropic MLV that makes verysmallfuzzy-edged
plaques (SP-N virus) (8). This virus was
iso-lated from a stock of LP-N virusderived from
BALB/cbyHartley et al. (6) (WN 1802N, pool
1898). Aspreviouslydescribed, the XCplaques
made by SP-N are so tiny and indistinct on
NIH/3T3that theyare virtually impossible to
count accurately (Fig. 1 and 2). Furthermore, the endpoint titerof the virus is50 to100 times
higher than the (approximate) plaque titer.
Theplaque morphology ofSP-N appearstobe
stable since it has been retained through
re-peatedserial infectionsandendpointdilutions (8; unpublished data).
Thevisibility of SP-Nplaquesis sosensitive
to plating conditions that sometimes plaques
will bevisible on one but notanother
"dupli-cate" plate. Frequently plaques are seen on
someregionsofaplatebutnot
others,
presum-ably reflecting an uneven platingofthe NIH/
3T3 cells. SP-N plaques usually lack syncytia
altogether. Whensyncytiaarepresenttheyare
small andgenerallythereis not morethanone
perplaque. As we have shown previously (8),
SP-N virus is less infectious than LP-Nvirus.
Wedonotknowifthesmallplaquemorphology
ofSP-N is aresult ofitsreduced infectivityor
whether it is due to analteration inthe viral
proteins involved in the induction of XC syncy-tia.
The LP-B virus used in these experiments was a clonal isolate made by Paul Jolicoeur from the virus stock prepared by Hartley et al.
(6) from BALB/c mice (WN 1802 B). It forms
large plaques on BALB/3T3 cells (see Fig. 1). The syncytia in these plaques are larger and more numerous than those of SP-N or even of
LP-N plaques on NIH/3T3 cells. As stated
above, comparisons of plaque morphology on different cell lines must be made with reserva-tions.
(ii) Tropism. The preferential growth of
N-and B-tropic viruses on NIH N-and BALB cells, respectively, has been described extensively (7, 14, 16). The cellular restriction toward a virus
oftheopposite tropism isdetermined by a gene,
Fv-1, that acts internally to block viral growth (13, 14, 16). The stage of infection at which growth is blocked is not known. It is not known how many genetic markers on the virus
deter-mine its tropism, nor is it known if the viral
determinants of N- and B-tropism are allelic. Variations in the extent of restriction and in the ability of a virus to overcome the tropic restriction have been noted in different
labora-tories (3, 10, 15). However, different viruses
and cells have been employed in these studies.
Inour studies, we have used only NIH/3T3 cells
derived by Jainchill et al. (9) and BALB/3T3 (clone A31) cells derived by Stuart Aaronson
andGeorge Todaro (1). When XC plaque assays
are performed asdescribed above andplaques arescoredmacroscopically, then the N-and
B-tropic viruses derived from BALB/c mice
be-have as follows. N-tropic viruses form plaques onBALB/3T3cellsthatarecomparable insize
and morphology to those formed on NIH/3T3
cells;however, the titer of the virus is approxi-mately 100-fold lower on BALB/3T3 cells than
on NIH/3T3 cells. Tenfold dilutions of the
N-tropic virus result in 10-fold reductions in the
number of plaques observed on BALB/3T3 cells. These observations apply to LP-N virus (6, 8). It
is difficult to determine accurately the ratio
(PFU on NIH/PFU on BALB) for SP-N virus
and difficult to determine ifSP-N titrates on
BALB/c cells with a one- or two-hit pattern,
sinceSP-Nplaquesare sodifficultto countand
since their visibility and appearance varies
from assaytoassay andevenfromplatetoplate
within asingle assay. However, there is a
re-ductioninthe number ofSP-Nplaques
appear-ing on BALB/3T3 cells relative toNIH/3T3 (see
alsoHopkins and Jolicoeur [8]), andin assays
in which plaque visibility is optimal, this
re-duction appears to be 50- to 100-fold. The
B-tropic virus showsamuch greaterrestriction in
VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
326 HOPKINS, TRAKTMAN, AND WHALEN
on
NIH / 3T3
In
SP-N
+
us
I"'
A)
eJ
I
I
'I 'I B
I
I
sP-:B
Ad I
.SP-N
and
C-B
c.k;x
-M'"X.-f
Si
r
MALts/
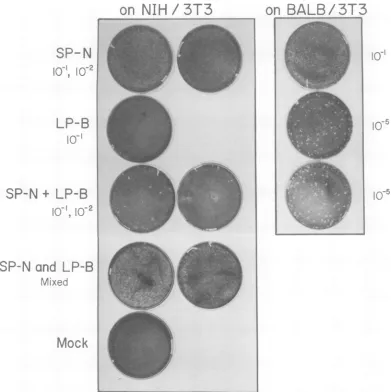
IFIG. 1. XCplaquesonNIH13T3andBALB/3T3ofprogenyofSc-i cellsfromcross1singlyorco-infected with SP-N and LP-B. SP-N and LP-B mixed is acontrol showing thatifSP-N and LP-B aremixed in amountscomparabletothat emergingfromco-infectedSc-i cells(10-1 SP-Nplus10-1LP-B,first plate; 10'2 SP-Nplus10-1LP-B, secondplate) and the mixture is titeredonNIH/3T3, onedoes notobservethelarge plaques thatone seesamongthe progenyfromSc-icellsco-infectedwith SP-N and LP-B.Mock-infectedplates received 0.2 ml of medium containing polybrene (8y/ml).Todetermine the titer(PFUlml) ofvirusthe number ofplaques on aplateshould be counted andmultiplied bythe dilutionshown.
plaque formationonNIH/3T3 cells than does
N-tropic virus on BALB cells. B-tropic virus
stocks withatiter of 107PFU/ml show less than
10PFU/ml onNIH/3T3 cells. Ifoneplates the
virusonNIH/3T3atatotal dilutionof 101, only
anoccasionalsmall plaque isseen.
Co-infection of Sc-i cells with SP-N and LP-B viruses. (i) Plan of the experiment.
Plates (60mm)wereseeded with2x 105, 105, 5 X 104, 2.5 x 104, 104,5 x 103, and2.5 x 103Sc-i
cells in triplicate for infection the following day. The maximum multiplicity of infection ob-tainable was limited by the titer of the SP-N
stockemployed, whichwas5 x 104 (>5 x 104,
<105)infectious units/mlbyendpointtitration.
The LP-B virus was diluted to 105 PFU/ml.
Both viruses werethenbroughtto 8My of
poly-brenepermlby the addition of concentrated(80
y/ml) polybrene,andaplateofSc-i cellsateach
celldensitywastheninfected with SP-Nor
LP-B virus (0.1 ml of virus plus 0.1 ml of 8 y
polybreneperml)orco-infected with SP-Nand
LP-B(0.1mlofSP-Nplus0.1mlofLP-Bvirus).
Cellsplated at2 x 105arehenceforth referred
to as cross1, 105as cross2, etc.
No attempt was made to prevent further
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.509.61.452.69.461.2]MLV PLAQUE VARIANTS 327
on
NIH/3T3
on
BALB/3T3
SP-NW''
LP-B
Soj 10-2
SP-N
+LP-N
N-1-5
IU1
0Ad
FIG. 2. XCplaques onNIHI3T3andBALB/3T3 cellsofprogenyfromthesecondNIHI3T3infection for
cross1.
rounds ofinfection. Rather, the cells were
al-lowedtogrow for varyingperiods oftimebefore
culture fluids were collected. This was
neces-sarytoaccumulateenoughcellstoproduce
rea-sonable quantities of virus. Further, we
as-sumed that once all the cells had become
in-fected there would benofurtherchangeinthe
amountand type of virusbeing produced. This
wasconfirmed forcross1, inwhichanalysis of
culturefluids taken3daysand 24days
postin-fectionyielded similar results.
(ii) Progeny ofSc-icells. The progeny of
Sc-1 cells infected with SP-N,LP-B, or SP-N plus
LP-Bwere titered on NIH/3T3 and BALB/3T3
cells todetermine theirtiterand tropism.
Fig-ure 1 shows theplaques produced on NIH/3T3
andBALB/3T3 by the progeny of
Sc-i
cells fromcross 1. Table 1A shows the titer on NIH/3T3 and BALB/3T3 cells of the progeny of singly
andco-infected cells from all seven crosses. As
expected, inallseven crosses
Sc-i
cellsinfectedwith SP-N alone produced only SP-N virus,
whereas cells infected with LP-B alone
pro-ducedonlyLP-B virus.However, the seven
co-infections all yielded SP-N, LP-B, and also
vi-rusesthat madelargeXC plaques onNIH/3T3
cells. Toanswerthe question of whether these
plaquesweremadeby viruses derivedfrom
co-infection of
Sc-i
cells by SP-N and LP-B orwhethertheyaroseas aresult of co-infection of
the NIH/3T3 cellswithSP-N and LP-B atthe
timeof XC assay, SP-NandLP-Bprogenyfrom
Sc-i cells of cross 1 were mixed in amounts
comparable to those emerging from the
co-in-fected cells, and the mixture was titered on
NIH/3T3. Nolarge XC plaqueswere observed,
suggesting thatthe large plaques seen among
the progeny of the co-infected Sc-1 cells arose
from viruses present priortotheXCassay(Fig.
1). Henceforth, virusesresponsiblefor thelarge
XCplaquesonNIH/3T3cellsarereferredto as
XLP-N virus.
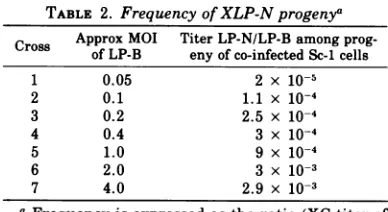
Inallseven crossesthetiterofvirusproduced
bythe co-infected Sc-1 cellswaslow relativeto
the titer of parental B-tropic virus produced
(see Tables 1A and2).However, the proportion
of XLP-N virus to LP-Bemerging from the
co-infected cells increased from2x 10-5 to 3 x 10-3
with increasing multiplicity of infection of the
Sc-i cells (Table 2). This frequency is not an
accurate measure of thefrequencywithwhich
XLP-Nviruses arisesince, asstatedabove, no
attempt was madeto prevent multiple rounds
of reinfection and since we have not yet
at-temptedto determine the actual percentage of
co-infected cells in each cross. Note that
be-cause of the low titer of SP-N virus it was
necessary to alterthe multiplicity of infection
of Sc-i cells by reducing the number of cells
inoculated rather thandilutingthe virus. It is
not clear towhatextentthe effective titer ofa virusdependsonthecelldensityatthetimeof VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.509.63.458.67.324.2]J. VIROL. 328 HOPKINS, TRAKTMAN, AND WHALEN
ck
0
p24
04 V0-cicC
._C)004
X X
Vcm CD
ce XX
z4c
E-~~~~~~~-~~~ ~ ~ ~ ~ ~ ~ ~ ~
=~~~~~~~~~~~C
m"0 x00 4 ~~~00 - 00
.w~~¢~~ X X
oX
Xet XX XX
1o E-v J Jc p
,~0p4
(124 - 0 04 x=~~~~~~~~~~~~Xooo X
Xc X- C>- - . -4*''-C
.v - ) x x
X x
C2cq -1
0~~~ 0
-_-P-o
cC)
CC P4 P. P,Pc P P.
rX4 cxp4x-C1P.ci Ci
-C0 °-~C~_OC o-'I4 ~-°°
C-78 Xtwq + C; X X
E-4 Pc p4 P4 o + - P V 4 o PV V
Ci)~ ~~~4 p4 0+ 1
- XX -4x x Ci) x Ci w ¢mXO X x x
C) .> C
Z-
C-
O
oOX
,, ~ U-4LOMC- .- r- c
i~~~~~+:x° x
VZ x 4 M q
U~~~~~~-
coX4
C)0-4 _-14C)M
¢; + + X x
X X
e~~~~~~~~
Xo-u:0 X oD X X C)° Leo o =°CD qV4 u : V
0
m
+
-zCAz 0-z.zoU; co
u
+
C4zm z
Q
m
+ 04
M zm0z
00W04C
I.. ) C
04
m
+ 04
10zmz
0a. a.a
2°C, J4U
V
m
+
04
CD Z00Z
0W4
M,
oo1 c
Cu
04
X
0
LO
Un
0 0
o0
C>
x
LO
0 0 X
Cd Q 0
0o
u
Ci.
._.
*C 0
0C-
ci-)0*-
1--P.i b0
z-
@.^00
45) 00o,
cr m=W Y
0 0
Oho
0 0
Co)
E-4 E-- E-E-E m
Cl
C.)
0z C.
Cl
P.
c;)
ci)
ci)
ci) % C.
*P.
Cl)
on November 10, 2019 by guest
http://jvi.asm.org/
MLV PLAQUE VARIANTS 329 TABLE 2. Frequency of XLP-N progenya
Cross Approx MOI Titer LP-N/LP-Bamong
prog-of LP-B eny of co-infectedSc-1cells
1 0.05 2 x 10-5
2 0.1 1.1x 10-4
3 0.2 2.5x 10-4
4 0.4 3 x 10-4
5 1.0 9x10-4
6 2.0 3 x 103
7 4.0 2.9x 10-3
a Frequency is expressed as the ratio (XC titer of
large plaques on NIH/XC titer of large plaques on BALB) for the progeny of Sc-1 cells co-infectedwith SP-N and LP-B. MOI, Multiplicity of infection. infection. Therefore, the calculated multiplicity
ofinfection, and thus the calculated number of
co-infected Sc-1 cells, particularly for crosses
performed at very low cell densities, may be quite unreliable.
Two critical questionsnow arose concerning
the nature of the viruses responsible for the
large XC plaques appearing on NIH/3T3 cells.
(i) Would they breed true? (ii) Whatwastheir
tropism?To answer these questionsitwas
nec-essaryto trytoisolate XLP-N viruses.
Selection of XLP-N viruses. Wewere
reluc-tanttoattempttopurify theXLP-Nvirusesby
directly selecting the large plaques on NIH
cells,sincetherewasalargeexcessof SP-N and
LP-Bviruspresent atthe time ofinfection and
since plaque purification would have required
co-cultivation withXC cells. The lowfrequency
of XLP-N viruses made selection bymicrotiter
plating prohibitive. Thus, we sought to enrich
the titer of XLP-N virus relative to SP-N and LP-B.
Aspreviously reported (8), SP-N virusisonly
moderately infectious on NIH cells relative to
the LP-N virus ofHartleyet al. (6). As stated
above, B-tropic viruses are severely restricted
in growth on NIH cells. Since XLP-N viruses
made verylarge XCplaques onNIH/3T3 cells,
it seemed possible that ifthese viruses were
genetically stable they would have a growth
advantage over both the SP-N and LP-B
par-ents inNIHcells. In this case, serial passage of
the progeny of co-infected Sc-i cells through
NIHcells should enrich the proportion of
XLP-Nvirus.
NIHcells (5 x 105)wereplatedon each of 21 100-mmplates and infectedthenextdaywith 0.5 ml (diluted by 10% with concentrated
poly-brenetomake the solution8y ofpolybreneper
ml) oftheprogeny fromsingly andco-infected
Sc-1 cellsfrom the sevencrosses. Medium was
changed every24hand collectedonday3after
infection. Theseculture fluids were then used
for asecond infection ofNIH cells, which was
performed in the same way as the first
infec-tion. On day 3 postinfection culture fluids from
all 21 infections of the second NIH selection
were then titered on NIH/3T3 and BALB/3T3
cells(Table 1C). In addition, the culture fluids
from the first NIHselection of cross 1 (2 x 105
Sc-i cells) were titered (Table 1B). The XC
assay of progenyfrom the second NIHinfection
for cross 1 is shown in Fig. 2.
In all seven crosses, XLP-N virus was
re-coveredonly fromNIH-passaged progeny of the
co-infected Sc-i cells. The titer of this virus
increased rapidly with passage in NIH cells
(Table 1Band C). For example, in cross 1 in the
first NIHselection, the cells were infected with
approximately 50 PFU of large NIH
plaque-forming virus. Within three days the titer of
this virus had increased to 1.2 x 105 PFU/ml,
whereas the titer of parental LP-B had dropped
to 2.3 x 104PFU/ml. After the second infection
ofNIH/3T3 cells, the titer of XLP-N virus had
risen to 107i, whereas the titer of LP-B had
dropped sufficiently to allow one to observe the N-tropism of the XLP-N virus.
Sc-1 cells that had been infected with SP-N
alone yielded virus that retained the small
plaque morphology through NIH passage.
In four out of seven cases, after passage
through NIH/3T3 cells the progeny from Sc-i cells infected with LP-B alone acquired the
abilitytoplaqueonNIHcellsatthesametiter
as onBALBcells (see Table 1C andFig.2). The
gradual conversion of B-tropic murine
leuke-mia virus to NB-tropism by serial passage in
NIHcells inculturehas been observed
repeat-edly by Janet Hartley (see reference 12) and
presumably accounts for the results obtained. Interestingly, the plaques of this presumed
NB-tropicvirus aresmalleronNIHthanon BALB
cells and smalleronNIHcells than theplaques
made by the XLP-N viruses (Fig. 2). The
pre-sumed NB-tropic viruseswere unable to grow
rapidly on NIH cells, and after three serial
passages on NIH/3T3 never achieved titers
higher than5 x 103 (seeTable 1C).
Properties ofXLP-Nviruses. (i)Stability of
plaque morphology and tropism. To further
test the stability of the large plaque
morphol-ogyandN-tropism of the XLP-N viruses, virus
obtained from the secondNIHselection ofcross
1 (2 x 105 Sc-i cells) was passed once more
through NIH/3T3 cells andalsothrough BALB/
3T3 cells by the procedure described above.
Bothplaque morphology and tropismwere
re-tainedby passageineither hostcell(Table iD).
The culture fluids from thesecondNIH
selec-tionof the co-infected cellsfrom thiscrosswere
then titered by end point dilution. The end
pointtiter was 107(>107, <108) infectiousunits/
VOL.18,1976
on November 10, 2019 by guest
http://jvi.asm.org/
330 HOPKINS, TRAKTMAN, AND WHALEN
ml. Virus from this end point dilution (10-i)
remained large XC plaque forming and
N-tropic. This virus was plaque purified. The
plaque-purified virus formed large XC plaques andwas N-tropic. To furthertest thestability
of thesemarkers, theplaque-purifiedviruswas
passedthrough BALB/3T3 cells. A virus stock
withatiterof2x 107PFU/mlwasusedtoinfect
BALB/3T3 cellsat10-folddilutions from10-lto
10-4. Virus emerging from these infections
re-mained N-tropic and produced large XC
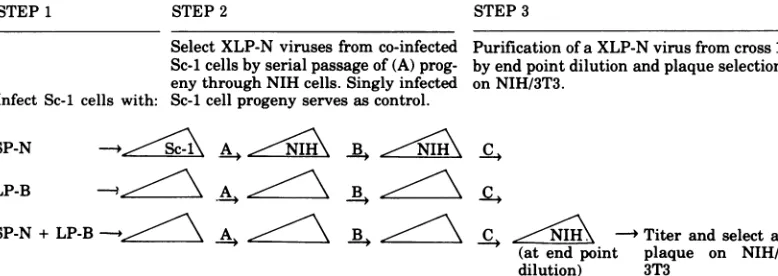
plaques.Figure3showsaschematicdiagramof the entire experiment from co-infection of Sc-1
cells through plaque purification ofa XLP-N
virus.
(ii) Plaquemorphology and titer. The
XLP-Nviruses obtainedfromthesestudiesgrow to
unusually high titers, at least 5- to 10-fold
higher than thoseobtainedwiththe LP-Nvirus
ofHartleyetal. (6).LP-Balsogrows tohigher
titersthan the LP-N of Hartleyetal. (6),andit
ispossible thatthis property was acquired by
the XLP-N viruses.
The plaquesmade by the XLP-Nviruses are
unusually large, and the syncytia within the
plaques are more numerous and considerably
larger than those observed for other N-tropic
variantderived from BALB/c mice.
DISCUSSION
Co-infection ofSc-icells withSP-Nand LP-B
viruses yields progeny XLP-N virus with the phenotype large XC plaque morphology and
N-tropism.Infection ofSc-icells withSP-Nor
LP-B alone does not yield XLP-N viruses. The
phenotype of XLP-N is stable through many
cycles ofgrowthon bothNIH/3T3 and BALB/
3T3 cells. XLP-Nviruses make largerplaques
andgrow tohighertitersthananyof the LP-N
STEP 1 STEP2
virusesderivedfrom BALB/c micethatwehave
studied previously(8).
A number of explanations for the origin of
XLP-Nviruses canbeconsidered. Probably the
simplest explanation for the results
summa-rized above is genetic recombination between
plaquemorphologyand tropism markers of
SP-Nand LP-Bviruses. Other explanationsmight
bephenotypic mixingorheterozygosis between
SP-NandLP-B. Wedonotknow whether these
mechanisms could generate viruses with the
XLP-N phenotype. Although they are not
ex-cluded as explanations, they seem somewhat
unlikely because ofthe stabilityof the XLP-N
phenotype andfailure tosegregateLP-B virus
aftergrowthof XLP-NonBALB/3T3 cells.
It isalsoconceivable that duringco-infection
LP-Bcausedageneticalterationinthegrowth
and plaque-forming properties of SP-N by a
mechanismotherthan recombinationor
heter-ozygosis.Hopkins and Jolicoeur (8) and Rappet
al. (17) have described the "conversion" of
weakly infectious XC N-tropic virus to LP-N
virus. Although infection ofSc-i or NIH cells
with SP-N under a variety of conditions has
never yielded either LP-N or XLP-N viruses,
andeventhoughnoneofourotherstudies with
N-tropic plaque variants derived from BALB/c
mice (8) or induced fromBALB/c cells in
cul-ture(unpublished data) hasyielded XLP-N
vi-ruses, we can not exclude the possibility that
co-infection with SP-N and LP-B facilitated
conversion of SP-N to this novel N-tropic
var-iant.
Nor we can exclude the possibility that the
XLP-N viruses we obtained resulted from
ge-neticexchange betweentheexogenously
infect-ing viruses and an endogenous virus of
Sc-i
cells. However,we cansay that if such an event
occurred, itapparently requires co-infection of
STEP3
Select XLP-N viruses from co-infected Purification of a XLP-N virus from cross 1 Sc-icellsby serialpassageof (A) prog- by end point dilution and plaque selection enythrough NIH cells. Singly infected onNIH/3T3.
Infect Sc-1 cells with: Sc-icell progeny serves as control.
SP-N -* Sc-i A. NIH B1\NIH C,
LP-B
SP-N + LP-B --.ii6 \ A 2\ .
.-..IH;
-+ Titer and select a(at end point plaque on NIH/ dilution) 3T3
FIG. 3. Schematic diagram of the experiment from infection ofSc-i cells toplaque purification ofaXLP-N virus. XC assays of (A) progeny are shown inTable 1A and Fig. 1; (B) progeny are shown in Table 1B; (C) progeny are shown inTable 1C and Fig. 2.
----4 A B
C
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.509.63.452.490.628.2]VOL. 18, 1976
theSc-1 cells with both N- and B-tropic viruses
toproduce XLP-N.
Hopefully, further analysis of the XLP-N
vi-ruses will aid in determining the genotypic
basis for theirphenotype. Recently Buchhagen
et al. (2) succeeded in distinguishing the
pro-teins of N- and B-tropic viruses derived from
BALB/c mice. P. O'Donnell and E. Fleissner
(personalcommunication) have observed a
dif-ference intheability of these viruses to induce
G1X antigen upon infection.
(Gx
is anantigenicdeterminant found on the envelope
glycopro-tein gp 69/71of some murine leukemia viruses.)
Analysis of the proteins and of the ability to
induce
Gx
of the parental and phenotypicallyrecombinant viruses from our studies may
provehelpful indetermining whether these
vi-ruses are geneticrecombinantsbetween N- and
B-tropic viruses. Acollection of such
recombi-nants might prove useful in defining the
pro-teinsand regions of the genomeinvolved in XC
plaque morphology, induction of
Gx
antigen,and N-and B-tropism.
ACKNOWLEDGMENTS
Wethank Terry Grodzicker and Rex Risser for interest-ingdiscussions and David Botstein for acriticalreading of the manuscript.
Thiswork wassupportedby grantnumber 76-03 from the Health SciencesFundandPublic Health Service grant CA 14051from the National Cancer Institute.
LITERATURE CITED
1. Aaronson,S.A.,andG. J. Todaro. 1968.Development of3T3-like lines from BALB/c mouse embryo cul-tures:transformationsusceptibilitytoSV40. J.Cell Physiol. 72:141-148.
2. Buchhagen,D.L.,0. Stutman,and E. Fleissner. 1975. Chromatographicseparation andantigenicanalysis ofproteins of the oncornaviruses. IV. Biochemical typing ofmurine viral proteins. J. Virol. 15:1148-1157.
3. Declsve,A.,0.Niwa, E.Gelmann, andH.S.Kaplan. 1975.Replication kineticsof N-and B-tropicmurine leukemia viruses on permissive and non-permissive cellsinvitro.Virology 65:320-332.
4. Hanafusa, H., and T. Hanafusa. 1966. Determining factorinthecapacity of Roussarcomavirustoinduce tumorsin mammals. Proc. Natl. Acad. Sci. U.S.A. 55:532-538.
5. Hartley,J.W.,and W. P. Rowe.1975.Clonal cell lines from a fetal mouse embryo which lackhost-range restrictionsfor murine leukemia viruses. Virology 65:128-134.
6. Hartley, J. W.,W. P. Rowe, W. I.Capps, and R. J. Huebner. 1969. Isolation ofnaturally occurring
vi-rusesof the murine leukemia virusgroupintissue culture. J. Virol. 3:126-132.
7. Hartley,J.W.,W. P.Rowe,and R. J.Huebner.1970.
Host-range restrictions ofmurine leukemiaviruses inmouseembryocell cultures. J. Virol. 5:221-225.
8. Hopkins, N., and P. Jolicoeur. 1975. Variants of N-tropic leukemia virusderived fromBALB/cmice. J.
MLV PLAQUE VARIANTS 331 Virol. 16:991-999.
9. Jainchill,J. L., S. A. Aaronson, and G. J. Todaro. 1969. Murinesarcomaand leukemia viruses: assay using clonal lines ofcontact-inhibited mouse cells. J. Virol. 4:549-553.
10. Jolicoeur, P., and D. Baltimore, 1975. Effect of the Fv-1 locus on the titration of murine leukemia viruses. J. Virol. 16:1593-1598.
11. Kawai, S., and H. Hanafusa. 1971. Genetic recombina-tionwith avian tumor viruses. Virology 49-.37-44. 12. Klement, V., W. P. Rowe, J. W. Hartley, and W. E.
Pugh. 1969. Mixed culture cytopathogenicity: a new testfor growth ofmurine lukemia viruses in tissue culture. Proc. Natl. Acad. Sci. U.S.A. 63:753-758. 13. Lilly, F., and T. Pincus. 1973. Genetic control of
mu-rineviral leukemogenesis. Adv. Cancer Res. 17:231-277.
14. Pincus, T., J. W. Hartley, and W. P. Rowe. 1971. A major geneticlocus affecting resistance to infection with murine leukemia viruses. I. Tissue culture stud-ies of naturally occurring viruses. J. Exp. Med. 133:1219-1233.
15. Pincus, T., J. W. Hartley, and W. P. Rowe. 1975. A major geneticlocus affectingresistance toinfection withmurineleukemia viruses. IV. Dose-response re-lationships in Fv-1 sensitive and resistant cell cul-tures.Virology65:333-342.
16. Pincus, T., W. P. Rowe, and F. Lilly. 1971. A major geneticlocus affecting resistance to infection with murine leukemia viruses. II. Apparentidentityto a major locusdescribed for resistance toFriend murine leukemia virus. J. Exp. Med. 133:1234-1241. 17. Rapp, U. R., R. C.Nowinski, C. A. Reznikoff, and C.
Heidelberger. 1975. Endogenous oncornaviruses in chemically induced transformation. I. Transforma-tion independent of virus production. Virology 65:392-409.
18. Rowe, W. P., W. E. Pugh, and J. W. Hartley. 1970. Plaque assay techniques for murine leukemia vi-ruses.Virology42:1136-1139.
19. Stephenson, J. R., and S. A. Aaronson. 1972. Genetic factors influencing C-typeRNA virus induction. J. Exp.Med. 136:175-184.
20. Stephenson, J. R.,G.R.Anderson,S.R.Tronick,and S. A. Aaronson. 1974. Evidence for genetic recombi-nation between endogenous and exogenous mouse RNAtype-Cviruses.Cell 2:87-94.
21. Stephenson, J. R., S. R. Tronick, and S. A. Aaronson. 1974. Temperature-sensitivemutantsof murine leu-kemia virus. IV. Further physiological characteriza-tionand evidence for genetic recombination. J. Virol. 14:918-923.
22. Svoboda, J. 1960. Presence of chicken tumor virus in the
sarcomaof theadultratinnoculated afterbirth with Roussarcomavirus.Nature(London)186:980-981. 23. Vogt, P. K. 1967. Phenotypic mixing in the avian tumor
virusgroup. Virology32:708-717.
24. Vogt, P. K. 1971. Genetically stablereassortmentof markers during mixed infection with avian tumor
viruses.Virology46:947.
25. Weiss, R. A., W. S. Mason, and P. K. Vogt. 1973.
Genetic recombination betweenendogenous and
ex-ogenous avianRNAtumorviruses.Virology
52:525-535.
26. Wong, P. K. Y., and J. A. McCarter. 1973. Genetic studies oftemperature-sensitivemutantsof Moloney-murineleukemiavirus.Virology53:319-326.
27. Wyke, J. A., J. G. Bell, and J. A. Beamand. 1974.
Genetic recombination amongtemperature-sensitive mutantsof Rous sarcoma virus. ColdSpringHarbor Symp. Quant.Biol.39:897-905.
on November 10, 2019 by guest
http://jvi.asm.org/