JOURNAL OFVIROLOGY, May1992,P. 2846-2852 0022-538X/92/052846-07$02.00/0
Copyright ©1992,American SocietyforMicrobiology
Identification of Critical
cis
Elements
Involved in
Mediating
Epstein-Barr Virus Nuclear Antigen
2-Dependent
Activity
of
an
Enhancer
Located
Upstream
of the
Viral BamHI C Promoter
XIANW. JINAND SAMUEL H. SPECK*
Division ofTumorVirology, Dana-Farber CancerInstitute, andDepartment of Pathology, HarvardMedicalSchool, 44Binney Street, Boston, Massachusetts 02115
Received 27 November 1991/Accepted 11February 1992
The sixgenes encoding the Epstein-Barr virus nuclear antigens (EBNAs) aretranscribed fromoneof two promoters,BamHICpromoter(Cp)orBamHl Wpromoter(Wp), locatednearthe left end of the viralgenome.
During theestablishment of viral latency in B lymphocytes, Wp is used exclusively beforeaswitchtoCpusage.
We andothers havepreviously identifiedanenhancer in theregionupstreamofCp which requiresEBNA 2 for
activity(M. Woisetschlaeger, X. W. Jin, C. N. Yandava, L. A. Furmanski, J.L.Strominger, andS. H.Speck, Proc. Natl.Acad. Sci. USA88:3942-3946, 1991; N. S. Sung, S.Kenney, D. Gutsch, and J. S. Pagano, J.Virol. 65:2164-2169, 1991). Infection ofB lymphocytes with a mutant virus lacking the EBNA 2 gene results in prolonged usage ofWp and failure to switch to Cp usage, indicating that EBNA 2 transactivation of the
enhancer upstream ofCpmaybe critical forpromoterswitching.Inthis study,wehavedefinedthe minimal EBNA2-dependent enhancer by using a series ofdeletion mutants. The results ofsite-directed mutagenesis revealed that therearethree regions ofthe enhancer thatareimportant for activity,two of which appearto bind B-lymphocyte-specific factors.
Epstein-Barr virus (EBV) is a lymphotropic human
her-pesvirus which is the etiologic agentof infectious
mononu-cleosis, aself-limiting lymphoproliferative disorder. In
addi-tion, EBV is associated with two human cancers, African
Burkitt's lymphoma and nasopharyngeal carcinoma. Infec-tion of human B lymphocytes with EBV predominantly establishes a latent infection with littleor novirus produc-tion and concomitant growth transformation of the infected cells (immortalization).
In latently infected growth-transformed B lymphocytes, six EBV nuclear antigens (EBNAs) and three viral
mem-brane proteins (latent membrane proteins [LMPs]) are
known to be expressed (for reviews, see references 4, 12, and26). The viralgenesencoding these antigensare
distrib-utedthroughout the viralgenome. However, transcription of theseantigens is driven bypromotersthatareclusterednear
the terminal repeatsofthe viralgenome. Two viral promot-ers have been identified (BamHI C promoter [Cp] and BamHI W promoter [Wp]) that are involved in driving
transcription of the six EBNAgenes(Fig. 1). The activities
ofCpandWp are mutually exclusive in all clonal cell lines
which have been examined (34). Wp is exclusively used during the initial stagesofinfection and thenaswitchtoCp
usage occurs(35).
Severallines ofevidence indicate that EBNA 2 isessential for growth transformation of B lymphocytes and that it is involved in modulating the activity of several viral and cellular promoters (1, 5, 9, 11, 13, 21, 30, 31). EBNA 2 has been shown to transactivate Cp (28, 33) as well as the
promotersforthegenesencoding LMP 1, LMP 2a, andLMP
2b (1, 5, 32, 37). Characterization of the mechanisms by which EBNA 2 transactivates EBV latent promoters is essentialtounderstanding establishment and maintenance of latency and is likely to provide important insights into the
*Correspondingauthor.
roleof EBNA 2 plays in the processoflymphocyte immor-talization.
We and others have previously identified a region
up-stream of Cp (-429 to -245 bp) that contains an EBNA 2-dependent enhancer (28,33). Furthermore,wehaveshown that infection of B lymphocytes with a nonimmortalizing
strain of EBV lacking the EBNA 2 gene resulted in Wp activity andafailuretoswitchtoCp activity (33). The latter observationprovided evidence that EBNA 2 is required for viral promoter switching. In this study, we further dissect
the EBNA 2-dependent enhancer associated with Cp to evaluate the functional roles of various cis elementswithin thisregion thatare involved inmediating EBNA 2 transac-tivation.
MATERIALSANDMETHODS
Cellculture, transfection, and CATassays. DG75 cells (an
EBV-negative Burkitt's lymphoma cell line), Jurkat cells (a T-cell line), and HeLa cells (an epithelial cell line) were
grown at 37°C in RPMI 1640 medium supplemented with 10% fetal bovine serum as previously described(25).
DNA transfections were carried out by using DEAE-dextranasdescribedpreviously (16) with modifications. The cells (5 x 106 to 1 x
107)
were spun down at 1,000 x g,washed once with phosphate-buffered saline (PBS) and
resuspended in500,ulof RPMI 1640 medium withoutserum
(GIBCO). Cellswere then transferred to sterile tubes
con-taining2,ug of the relevantCsCl-purified plasmid DNA, and the final concentration of DEAE-dextran was 500 ,ug/ml.
Cellswere incubated at37°Cfor30 min and then 250 ,ulof 20% dimethyl sulfoxide (in RPMI 1640 medium without serum) was added for 2 min. After the dimethyl sulfoxide shock,cellsweretransferredtoa15-ml conical tube with 10
ml ofRPMI 1640 medium andcentrifuged at1,000 x gfor 5 min. The cellpelletwas thenresuspended in 10 ml of RPMI 2846
Vol. 66,No. 5
on November 9, 2019 by guest
http://jvi.asm.org/
EBNA4
C1C2 WOW1W2 W1W2 W1W2
IEBNA2 OTHER
EBNAs
W1W2
X
/2\V/T
40 60k(0
400 -380 -360
I
-340
MGATTATG_A_C GTCGAGTGCTAT CaAA
GTGcorm m
DNA" I proetonbd
REG0NSca _rDI
domI go
domah"
dom ain
TGCA 0.07
domain
CTGCA CGCG
0.12 0.26
domisnI
domainIl
domainIN'
doman o0go*
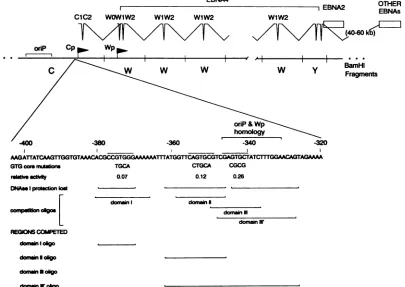
FIG. 1. Summary of transcription initiating from Cp and Wp, activities of the GTG core enhancer mutants and competition with
oligonucleotides containing specific domains within the EBNA 2-dependent enhancer. Thecommon5' leaderpresentin all the transcripts encodingthevarious EBNAs isillustrated in thetophalfof the figure. Alsoshown is thesequenceof the regionupstreamof Cp containing
anEBNA2-dependent enhancer. The specific nucleotide alterations introduced in the GTGcoremutantsareindicated,andthe regions which
lostprotection from DNase I digestion with these mutationsare indicated beloweachmutant. EBNA2 transactivation ofeach GTGcore
mutantisgivenrelativetotheunmutated enhanceractivity. In addition, the regions included in each competitor oligonucleotideareindicated
and the results of thebinding competitions shown in Fig. 5aresummarized.
1640 medium with 10% fetal bovine serum and cultured at 370C.
Transfectedcellswereharvested at72 h posttransfection,
spun down at 1,000 x g, washed once with PBS, and
resuspendedin 100 ,ulof0.25 M Trischloride (pH 7.5).The cell suspension was then lysed by three rounds of snap
freeze-thawing, and the debriswasremoved by
centrifuga-tion. The chloramphenicol acetyltransferase (CAT) assays wereperformed aspreviously described (8) and quantitated
by aBlotanalyzer (Betagen Corp., Waltham, Mass.).
Plasmid construction and site-directed mutagenesis. Plas-midscarryingdeletions in the EBNA2-dependent enhancer regionweregeneratedby cloning polymerasechain reaction
(PCR)-amplifiedenhancersequencesdownstream (XhoI and
Sacl sites) of a CAT reporter construct containing the enhancerless simian virus 40 (SV40) early promoter. The PCRs were carried out as described previously (18) with
slightmodifications. Pairs ofsingle-stranded synthetic oligo-nucleotides(30-mers) homologoustoregionsintheenhancer
were used as primers to generate each deletion by PCR amplification. The 5' primerscontained anXhoI restriction
site at their 5' ends, while the 3' primers contained aSacI
restriction site at their 3' ends. Twenty cycles of PCR amplificationwerecarried out.
The plasmids containing mutations within the EBNA 2-dependent enhancerwere generated by cloning the
PstI-Sacl fragment containing the SV40 promoter, CAT gene,
and the enhancer(from -415 to -321bp) intoaBlueScript vector(Stratagene), and then by generating single-stranded plasmid and performing site-directed mutagenesis as
previ-ously described (6). All mutations introducedwere
charac-terizedby DNAsequencing (22).
Preparation ofnuclearextracts and DNase I footprinting. Nuclear extracts were prepared essentially as previously
described (3). Briefly, 1 liter of cells were harvested and washed withPBS.All subsequentstepswerecarriedouton
ice. The cells were suspended in 5 volumes of prechilled
buffer A (10 mM HEPES [N-2-hydroxyethylpiperazine-N'-2-ethanesulfonicacid] [pH7.9],1.5 mMMgCl2, 10 mM
KCI,
0.5 mMdithiothreitol)andhomogenizedinaDounce homog-enizer, and the crude nuclear fraction was recovered by centrifugation at 10,000 x g for 10 min. The nuclei were
suspended in buffer C (20 mM HEPES [pH 7.9], 25% [vol/vol] glycerol, 0.42 M KCI, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol)andhomogenized again inaDounce homoge-nizer. The extractwas then stirredgentlyfor30min at4°C and clarifiedby centrifugationat25,000xgfor 30min.The supernatantwasdialyzed againsttwochangesof50 volumes of buffer D(20mM HEPES[pH 7.9],20%
[vol/voll
glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol) for 5 h and centrifuged at25,000 xgfor 20min.Thesupernatantwasrecovered,and
oriP
" . . .
w
BamHI
Fragments
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.105.510.79.366.2]2848 JIN AND SPECK
protein concentration was determined
by
Bio-Rad protein assay. Aliquotswere storedat -70°C.DNase Ifootprintingwas carried out asdescribed previ-ously
(14).
Binding reactions wereperformed
in a solution containing 10 mM Tris(pH 7.9),
0.5 mM EDTA, 0.5 mMdithiothreitol,
2.5%glycerol,
2%polyvinylethanol
with 1 ,ug of poly(dI-dC) in the reaction buffer for 20 min at room temperature. DNaseI wasthenadded andincubatedatroom temperaturefor 30s.Digestion
wasstopped by
theaddition of 150 ,ul of stop buffer (8 M urea,0.5%
sodium dodecyl sulfate, 5 mM EDTA). The samples were extracted two times with phenol, two times withphenol-chloroform,
one time with chloroform; the samples were then precipitated with ethanol and run on a8% acrylamide denaturing gel.For thecompetition
studies,
thefollowing oligonucleotide
pairswereannealed and
ligated:
domain I, 5'-CTAGACACGCCGTGGGAAAAG-3',
5'-GATCCTll1TCCCACGGCGT
GT-3', domain II,
5'-CTAGAGGTTCAGTGCGTCGAG-3',
5'-GATCCTCGACGCACTGAACCT-3',
domain III, 5'-CT AGACGTCGAGTGCTATCTG-3',
5'-GATCCAGATAGC ACTCGACGT-3'; and domain III', 5'-CTAGATCGAGTGCTATCTTTGGAACAG-3',
5'-GATCCTGTITCCAAAGATAG
CACTCGAT-3'.
RESULTS
TheminimalEBNA2-dependent enhancer element mapsto theregion protectedfromDNase Idigestion byB-cellnuclear extracts. An EBNA 2-dependent enhancer has been previ-ously mapped to a184-bp region(-429to-245bp) upstream of Cp
(33).
To fine map the minimal sequence element(s) required for enhancer activity, a number of deletion con-structsweregenerated and cloned downstream of the CAT gene in a reporter construct driven by the minimal SV40 early promoter(Fig. 2). Characterization ofEBNA 2-depen-dent enhancer activity of the various deletion constructs revealed that theregion from -380to -331 bp is essential for enhanceractivity
[SVpCAT(-380/-331)
inFig.
2]. Thisregion
corresponds to the region protected from DNase I digestion by nuclear extracts prepared fromBlymphocytes(33).
It is important to note that our previous DNase Ifootprinting
results indicated that there was no striking difference in the protection pattern observed with extracts prepared from an EBV-negative Burkitt's lymphoma cell line and two EBV-positive Burkitt's lymphoma cell linesexpressing
EBNA 2(33).
This result suggested that EBNA 2 isnotdirectly interacting
with an enhancerelement.Inclusion of upstream sequences between -415 and -380
bp
resulted in an increase in enhancer activity, indicating the presence of a positive regulatory element(s) in this region. However, these upstream sequences alone did not exhibit any detectable EBNA 2-dependentenhancer activity[SVpCAT(-429/-370)
in Fig. 2]. In contrast, inclusion of downstream sequences to -245bp appeared tosignificantly inhibit enhancer activity [e.g., compare activities ofSVp-CAT(-429/-310)
andSVpCAT(-429/-245)
in Fig. 2]. In-terestingly, ourprevious DNase I footprinting studies dem-onstrated that nuclear extract prepared form the EBV-positive Burkitt'slymphoma
cell line clone 13 gave a protection pattern in the region from -330 to -310 bp that was distinct from that observed with nuclear extractspre-pared
from other B-cell lines (indicated in Fig. 2) (33).Activity
studies in clone 13 cells, whose resident EBV genome has a deletion that spans the EBNA 2 gene and therefore does not express EBNA 2, revealed that the enhancer(in the absence of cotransfection with an EBNA 2SVP7-
I CAT-380 -330 -310
'777i
b4p arFtp4 -245bp
*.___ -264bp
-286bp
-310bp
:.370bp
-415bp
. -321bp
A40 bp -264bp
-286bp
-310bp
-i80bp
-310bp:421
bp30bp j10bp
L...2.1bp
-34,bp -245bp
-283bp
SVpCATCONTRL
0 10 20 30 40 50
RELATIVE ACTIVITY
FIG. 2. Deletion analysis of the region
containing
the EBNA2-dependent
enhancer. CATconstructswerecotransfected into the DG75 cell line with either acontrol plasmid (solidbar)
orwith anEBNA 2
expression
vector(stippled bar).
Activities are given relativetotheactivity
observed with theparentSVpCAT
reporterplasmid
cotransfected with controlplasmid.
Allactivitiesrepresent the averages ofat least fiveindependent
experiments. CI-13 Ftpt, clone 13footprint.
expression
vector)
significantly
suppressed
activity
of the minimal SV40 minimalearly
promoter
(33).
This result alsosuggested
the presence cis elements in thisregion
of the viral genome that may bind cellular factors that function to suppress enhancer function.Three domains within the
protected
region
areimportant
for EBNA2-dependent
enhanceractivity.
Since theregion
protected
by
nuclear extractsprepared
fromB-lymphocyte
cell lines was
large,
wesuspected
that several cellular factorswere involved inbinding
to theenhancer.Thus,
toidentify
functional domainsbinding
cellulartranscription
factors within the minimal enhancer
element,
a series of site-directed mutationswereintroduced into theregion
from -415 to -321 bp andassayed
for EBNA2-dependent
enhancer
activity (Fig.
3).
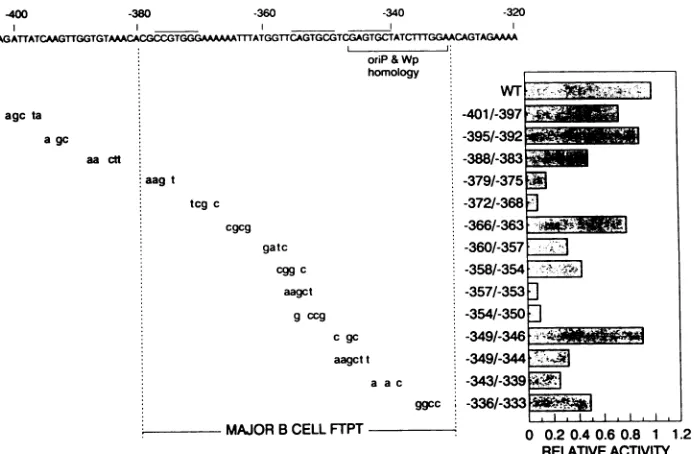
Each mutation introduced substi-tutions of 3 to 6 nucleotides. Two domainsappeared
to be critical for enhanceractivity.
Mutations in theregion
from -379to -368bp
and mutations in theregion
approximately
-357to-350
bp dramatically
reducedactivity
(see
activities of-3701-375, -3721-368, -3571-353,
and-3541-350
mu-tants inFig.
3).
In addition, the-343/-339
mutant alsosignificantly
reduced EBNA 2-dependent enhanceractivity
to
approximately
20% ofthe unmutated enhancer.J.VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.330.565.73.397.2]400 -380 -360 -340 -320
AAGATTATCAAGTTGGTGTAAMCACGCCGTGGGMAAAAATXTATGGTTCAGTGCGTCGAGTGCTATCITTGGAAAGTAGAAA
agcta
agc
aa ctt
oriP &Wp
homology
aagt
tcgc
cgcg
gatc
cggc
aagct
g ccg
cgc
aagctt a ac
ggcc
MAJOR B CELL FTPT
WT ;e', >ist! |
-401/-3977 -395/-392
-Mic
-388/-383 -379/-375 i -372l-368 -366/-363 -.
-360/-357 ; -358/-54A E,,
-357l-3533 -354/-350 3
-349,-46-..",1.
-349/-344 <# Fg
-343,-339 z
-336/-333 l
0 0.2 0.40.6 0.8 1 1.2 RELATIVEACTIVITY
FIG. 3. Mutational analysis of the EBNA 2-dependent enhancer. Site-directed mutations were introduced into an SVpCAT reporter constructcontaining the EBNA 2-dependent enhancer (from -415 to -321 bp) cloned downstream of the CAT gene. EBNA 2-dependent enhancer activities aregiven relative to that of the parent construct. The activities are expressed relative to the unmutated promoter and represent theaverages of three independent experiments. WT, wild type; FTPT, footprint.
Comparison of the nucleotide sequences in these regions of the enhancerrevealedalowlevel of homology(indicated by overbars inFig. 1 and 3). Each domaincontains a GTG coresequence andsubsequent mutations targetingthe GTG core in each of these domains also significantly reduced enhanceractivity (see GTGcore mutants inFig. 1), indicat-ing that the region of homology between these sites are important forfactorbinding. It ispossiblethatthese domains bind the same or related cellular transcription factors and that these factors mediate EBNA 2 transactivation. How-ever,itshould be noted thatourprevious DNaseI footprint-ing (33) indicated that the region from approximately -365to -380bp (domain I) binds afactor(s)presentin bothBandT
lymphocytes,
while the other two regions (domains II and III)were not protected byextractfromaT-lymphocyte cell line (Jurkat). We have now extended this observation to extractsprepared from theHeLacell line which alsodidnot protectdomain II or III but didprotect domain I (datanot shown).Cellularfactors present inan EBV-negative Burkitt's lym-phoma cell line bind independently to the domains in the EBNA 2-dependent enhancer. To determine whether muta-tions thatsignificantly diminishedenhanceractivityresulted inonlyalocalperturbationoffactor bindingorwhether they disrupted bindingtoother sites within theenhancer, DNase Ifootprintingwascarriedoutwiththe mutantsthattargeted the conserved GTG core sequence (refer to Fig. 1 for structuresofGTG core mutants). Nuclearextractprepared from the DG75 cell line (an EBV-negative Burkitt's
lym-phomacellline)wasutilizedinthe
protection
assay employ-ingthewild-type and mutant EBNA2-dependentenhancers (Fig. 4). In addition, as a control, the-366/-363
mutant which targeted theA/T-rich
region between domain I and domainII(andhadlittle effectonenhanceractivity)
wasalso analyzed.Analysis of the protection pattern exhibited by these
mutants demonstrated that there was a direct correlation between mutations that cause asignificant loss in enhancer activity and loss of binding of a cellular factor(s). The
-366/-363
mutation, which had little effect on EBNA 2-dependent enhancer activity, did not appear to signifi-cantly affect the binding of the cellular factors to the en-hancer, although it slightly altered the DNAse cleavage patternboth in the absenceandpresenceofextract.DNase I footprinting of three GTG core mutants (-375/-372,-354/-350,
and-344/-341
mutants), all of which exhibit significantly reduced EBNA2-dependent enhanceractivity, revealed loss of protection in the region of -380/-370,-362/-345,
and-343/-326,
respectively (Fig. 4; results summarized inFig. 1). The 3' boundary of domain I,aswell as the 5' boundary of domain II, could not be accuratelydetermined,
because there are no DNAse I sites within theA/T-rich region
located between these two domains. Nota-bly, with each of the three GTGcore mutantsthere wasonly loss of binding of the cellular factor(s) to the mutated domain, indicating thatbinding of cellular factorstoeach of these domains is independent of binding to the other do-mains within the enhancer. Thus, the loss ofactivity
ob-served with each mutant is due tospecific
loss ofbinding onlyatthat siteintheenhancer,demonstrating
thatefficient transactivationbyEBNA 2 appears torequire thebinding
of all these cellular factors totheenhancer.Multipledomains within the enhancer binddistinct cellular factors. To assess the possibility that common or closely relatedtranscription factorsbindtothe
regions
ofhomology within the enhancer, DNase Ifootprinting
inconjunction
with binding competition was carried out employing syn-thetic double-stranded
oligonucleotides
specific
foreach of the domains (Fig. 5; refer toFig.
1 foroligonucleotide
structuresandsummaryof
results).
Addition ofanunlabeled multimerized double-strandedoligonucleotide
containing
ei-therdomain Iordomain IIeffectively
competed
forbinding
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.139.486.75.302.2]2850 JIN AND SPECK
co =
E c c c
r
E E E
-- -- r- -- D.
+- + + - + DG75 EXTRACT
i i~~
0 0co 0
0=0 = 0=_
-o0 0 'OX -OX
r
(D - + -4 + ++ + + DG75EXTRACT + + +
-330 -340 -350 -360
4:
:.:.30
0*!w
i.~~~~~z.~
-370 - -
---380
-*a
*as--390 ---e _
d -3W-- .
FIG. 4. DNase I footprinting analysis of wild-type and mutant
EBNA 2-dependent enhancers. The DNA fragments used in the protectionassaycontained theupstreamregion oftheCppromoter
from -415 to -321 bp. Footprinting was performed with crude
nuclear extracts prepared from DG75 (an EBV-negative Burkitt's lymphoma cell line). Distinct domains revealed by loss ofprotection with the specific domain mutantsare bracketed on the right-hand
sideof thegel, and the relative positions of the protected region with
respect totranscription initiation from Cp are indicated. The
mu-tants employed are indicated at the top of the gel by the region mutated.The A/Tdomainmutantis the-366/-363mutantshownin Fig. 3. The domain I, lI, and IIImutantsemployedarethe GTGcore
mutantsshown inFig. 1.w.t.,wildtype. +,withDG75extract; -,
in the absence of DG75extract.
of theDG75cellular factor(s)totherespective domain. This result indicatesthat distinct cellular factors bindtodomains I and II, since these oligonucleotides did not efficiently
compete forbinding tothe other sites.
Whenanoligonucleotide spanning the homology region in domain III (domain III oligo [Fig. 1]) was employed as a
competitor in the DNase I footprinting assay, no loss of protectionwas observed. Carefulexamination of the loss of DNase I protection exhibited by the -344/-341 domain III mutant (Fig. 4), revealed that unlike the domain I and domain IImutations the lossofprotection wasasymmetric
withrespect tothe mutation (results summarized in Fig. 1). The domain III competitor oligonucleotide only contained
sequences extending tothecenterof the loss of protection. Therefore, it may not contain the entire sequence required
forbinding the appropriate cellularfactor(s). Indeed, extend-ing the competitor oligonucleotide to contain the down-stream sequences (domain III' oligo [Fig. 1]) resulted in effective competition for binding to domain III (Fig. 5). In addition, this oligonucleotide also effectively competed for binding to domain II but did not compete for binding to
[image:5.612.353.528.76.350.2]domain I. This suggests that a common or closely related
FIG. 5. Competition for binding of cellularfactorsto individual
domains within the EBNA 2-dependent enhancer. DG75 nuclear
extractwas preincubated with either 160 or320 ng(left and right lanes, respectively, of each oligonucleotide competition) of the
indicatedoligonucleotides for 10 minat roomtemperature,before
labeled fragments were added and DNase I digestion was
per-formed. Thestructuresof thecompetition oligonucleotidesaregiven
in Materialsand Methods andareshownschematicallyinFig. 1. +,
with DG75extract; -, in the absenceof DG75extract.
factormaybind todomainsIIand III. However, the failure
of either the domain IIoligonucleotide orthe short domain
III oligonucleotide to effectively compete for domain III binding raises the possibility that more than one factor is
involved in binding to domain III and thatbinding of these factors to domain III is cooperative. This model would explain the asymmetric loss of protection exhibited by the -344/-341 mutant and the ability of the longer domain III oligonucleotide (domain III' oligo [Fig. 1]) to compete for binding of factors toboth domains II and III.
DISCUSSION
We and othershavepreviously shown thata184-bp region
(-429to -254bp)upstreamofCp functionsas anenhancer in EBNA 2-positive cell lines, butnotin EBNA 2-negative cell lines(28, 33). Recent studies (5, 29, 32, 37) have shown thatupstreamregions of the LMPpromoterscontain EBNA 2-dependent enhancers. Like the EBNA 2-dependent Cp enhancer, a region upstream of the LMP 1 promoter has
been shown tofunction asan EBNA2-dependent enhancer
when cloned ineither orientationupstreamofaheterologous
promoter. Similarly, Wang et al. (31) recently reported a
186-bp fragment (-275to-89bp) from theupstreamregion ofthe cellularCD23promoterthatexhibits EBNA 2-depen-dent enhancer activity which is largely orientation and distance independent.
domainlII
domain11
-330
-340
-350
domain
domairnIII
-360
domairn 11 a i
-370
-380 *
-390_....
-390 f
^*m ,_ft _. -
-domain
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.119.246.77.353.2]In this study, we have mapped the minimal EBNA 2-de-pendent enhancer associated with Cp to a 50-bp region (from -380 to -331 bp). However, it is clear from the deletion analysis that surrounding sequences influence enhancer ac-tivity (Fig. 2). Sequences upstream of the minimal active enhancer (-415 to -380 bp) had a positive effect on en-hancer activity, while downstream sequences (-330 to -245 bp) had a negative impact on activity. This indicates the presence of both positive and negative regulatory elements in the region surrounding the minimal EBNA 2-responsive enhancer fragment.
Within the minimal Cp EBNA 2-dependent enhancer we have identified three important cis elements involved in binding cellular factors (domains I to III). These domains contain a region of low-level homology centered around a GTG core sequence. Mutations targeting the GTG core sequences all resulted in significant reduction in enhancer activity, directly implicating the homology regions in en-hancer function. DNase I footprinting analyses revealed that mutations in these domains abrogated binding of cellular factors to these sites. Furthermore, binding of cellular fac-tors to each domain appeared to be independent of binding to the other domains.
Competition analyses employing specific oligonucleotides provided evidence that the factor(s) which binds to domain I is distinct from the factors binding to domains II and III, since an oligonucleotide containing domain I did not com-pete for binding to any of the other domains and, conversely, oligonucleotides containing either domain II or domain III did not compete for binding to domain I. A common factor may be involved in binding to both domainII and domainIII, since an oligonucleotide containing domain III (domain III' oligo [Fig. 1])competed effectively for the factor(s) binding to domain II. However, an oligonucleotide containing do-main II did not effectively compete for binding of the factor(s) bound at domain III. The latter result, in conjunc-tion with the observaconjunc-tion that a mutaconjunc-tion indomainIII inthe region of homology between domainsII and III resulted in an asymmetric loss in protection, supports a modelin which two factors bind cooperatively to domain III. One of these factors binding to domain III is predicted to be identical or closely related to the factor binding to domainII.This model predicts that the common factor, in conjunction with the other domain III-binding factor, exhibits a significantly higher affinity for domain III than domain II.
Previously, extensive analysis of the SV40 enhancer has identified two domains, A and B, which areimportantforfull enhancer activity (10). Mutational analysis of domain B revealed the presence of a repeated sequence motif (GTG TGGAAAG) which is important for enhancer activity (36). Similar motifs have also been found in the enhancers of a variety of other viruses. Comparisonofthesesequences has identified a viral enhancer core consensus sequence
TGTGG(A/T)(A/T)AG.
Domain I within the Cp enhancer matches the core consensus sequence at seven of nine positions and may bind the same or a related factor(s). It should be noted, however, that the polyomavirus enhancer contains several sequence motifs resembling those ofSV40 enhancer, yet thepolyomavirus enhancer doesnot compete efficiently with theSV40 enhancer in cell-free transcription, electrophoretic mobility shift, and DNase I footprinting assays (20, 23).At the present time, the mechanism by which EBNA 2 functions to activate transcription is not clear. Our present studies implicate at least some of the cellular factors in-volved inbindingto the minimal enhancer. Currently, there
is no evidence that EBNA 2 functions by directly binding DNA. Indeed,ourpreviouslyreported DNase I footprinting data employing extracts prepared from EBV-negative and -positive B-cell lines did not reveal any consistent differ-encesin theobservedprotection pattern when EBNA 2 was present or absent (33).
In the absence of specific binding of EBNA 2 to DNA, there are two distinct mechanisms which may explain how EBNA 2 functions: (i)EBNA 2mayindirectly interactwith the enhancer bybindingtoacellularfactor(s) boundto acis element(s)withintheenhancer;or (ii) EBNA 2 may act at a distance by affecting complexes formed between a cellular factor(s) involved in binding to the enhancer and some cellular control protein(s), thereby activating the transcrip-tion factor. There is evidence for both mechanisms among viraltransactivators. Theherpessimplexvirus VP16 protein targetsviralpromotersbydirectinteraction with the cellular transcription factor Oct-1 (7, 27). Similarly, it has been shown that adenovirus Ela protein stimulates transcription oftheviralE4promoter by interacting with promoter-bound ATF-2 (15). Human T-cell leukemia virus type I Tax and hepatitis Bvirus X protein appear to function viaATFand AP-2, respectively (19, 24). Alternatively, it has
recently
been shown that adenovirus Ela protein transactivates the adenovirus E2 promoter in some cells by
liberating
the cellular transcription factorE2Ffrom a complexcontaining
either the retinoblastoma gene product or cyclin A
(2,
17).
Thus, for adenovirus Ela protein both mechanisms appear to be important.
ACKNOWLEDGMENTS
We thankmembers of S.H.S. laboratoryfor critical readingof the manuscript.
This work was supported by grant CA43143 from the National Institutes of Health (to S.H.S.), a Leukemia Society of America Scholar Award (toS.H.S.), and a Postdoctoral Fellowshipfrom the Lady TataMemorial Trust (toX.W.J).
REFERENCES
1. Abbot, S. D., M. Rowe,K.Cadwallader,A.Ricksten, J.Gordon, F. Wang, L. Rymo, and A. B. Rickinson. 1990. Epstein-Barr virusnuclear antigen2 inducesexpression of the virus-encoded latentmembrane protein. J. Virol. 64:2126-2134.
2. Chellappan, S. P., S. Hiebert, M.Mudryj,J. M. Horowitz, and J. R. Nevins. 1991. The E2F transcription factor is a cellular target for the RB protein. Cell 65:1053-1061.
3. Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a
soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489.
4. Epstein, M. A., and B. G. Achong. 1979.Introduction:discovery andgeneral biology of thevirus, p. 1-22. In M. A. Epstein and B. G. Achong (ed.), The Epstein-Barr virus. Springer-Verlag, NewYork.
5. Fahraeus, R., A. Jansson, A. Ricksten, A. Sjoblom, and L.
Rymo. 1990. Epstein-Barr virus-encoded nuclear antigen 2
ac-tivates thevirallatentmembrane proteinpromoterby modulat-ing the activity of a negative regulatory element. Proc. Natl. Acad. Sci. USA87:7390-7394.
6. Foss, K., and W. H. McClain. 1987. Rapid site-specific
muta-genesis in plasmids. Gene 59:285-290.
7. Gerster, T., and R. G. Roeder. 1988. A herpesvirus
trans-activating protein interacts withtranscription factor OTF-1 and other cellular proteins. Proc. Natl. Acad. Sci. USA 85:6447-6351.
8. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982.
Recombinant genomes which express chloramphenicol
acetyl-transferase in mammalian cells. Mol. Cell. Biol. 9:1044-1051. 9. Hammerschmidt, W., andB.Sugden. 1989. Genetic analysisof
on November 9, 2019 by guest
http://jvi.asm.org/
2852 JIN AND SPECK
immortalizingfunctions of
Epstein-Barr
virus in Blymphocytes. Nature(London)340:393-397.10. Herr, W., and Y. Glutzman. 1985. Duplications of a mutated simianvirus 40 enhancer restore itsactivity. Nature(London) 313:711-713.
11. Jeang, K.-T., and S. D. Hayward. 1983. Organization of the Epstein-Barr virus DNA molecule. III. Location of the P3HR-1 deletionjunctionand characterization of the NotI repeats units that form part of the template for an abundant 12-O-tetrade-canoylphorbol-13-acetate-induced mRNA transcript. J. Virol. 48:135-148.
12. Kieff, E., and D. Liebovitz. 1990. Epstein-Barr virus and its replication, p. 1889-1920. In B.Fields,D.Knipe, R.Chanock, M.Hirsh,J.Melnick,T.Monath,and B. Roizman(ed.),Field's virology.RavenPress, New York.
13. Knutson, J. C. 1990. The level ofc-fgrRNA isincreased by EBNA-2, anEpstein-Barr virus gene required for B-cell immor-talization. J. Virol. 64:2530-2536.
14. Lee, W., P. Mitchell, and R. Tjian. 1987.Purifiedtranscription factor AP-1 interacts with TPA-inducible enhancer elements. Cell 49:741-752.
15. Liu, F., and M. Green. 1990. Aspecific member of the ATF transcriptionfactor family can mediate transcription activation bytheadenovirusElaprotein.Cell 61:1217-1224.
16. McCutchan, J. H., and J. S. Pagano. 1968. Enhancement of the infectivityof simian virus 40deoxyribonucleicacid with diethyl-aminoethyl-dextran.J. Natl. Cancer Inst.41:351-357. 17. Mudryi,M., S. H. Devoto, S. W. Hiebert, T. Hunter, J. Pines,
and J. R. Nevins. 1991. Cell cycle regulation of the E2F transcription factor involvesaninteraction with cyclinA. Cell 65:1243-1253.
18. Mullis, K. B., and F. A. Faloona. 1987. Specific syn.hesis of DNAin vitro via a polymerase chain reaction. Methods En-zymol. 155:335-350.
19. Park, R. E., W. A. Haseltine, and C. A. Rosen. 1988. A nuclear factoris required for transactivation ofHTLV-1gene expres-sion. Oncogene3:275-280.
20. Piette, J., M. H. Kryszke, and M. Yaniv. 1985. Specific interac-tionofcellular factors with the B enhancer of polyoma virus. EMBO J. 4:2675-2685.
21. Rabson, M., L. Gradoville, L. Heston, and G. Miller. 1982. Non-immortalizing P3J-HR-1 Epstein-Barr virus: a deletion
mutantof its transformingparent,Jijoye. J.Virol.44:834-844. 22. Sanger, F., A. R. Coulson, B. G. Barrel, A. J. H. Smith, and
B. A.Roe.1980.Cloning insingle-stranded bacteriophageas an aid torapidDNAsequencing. J. Mol.Biol. 143:161-178. 23. Sassone-Corsi, P., A. Wilemann, and P. Chambon. 1985. A
trans-acting factor is responsible for the simian virus 40 en-hanceractivityin vitro. Nature(London)313:456-458. 24. Seto, E., P. J. Mitchell, and T. S. B. Yen. 1990. Transactivation
bythehepatitis Bvirus Xprotein depends on Ap-2 and other transcription factors. Nature(London)344:72-74.
25. Speck, S. H., and J. L. Strominger. 1985. Analysis of the transcript encoding the latent Epstein-Barr virus nuclearantigen I:a potentially polycistronic message generatedbylong-range splicingof severalexons.Proc. Natl.Acad. Sci. USA 82:8305-8309.
26. Speck, S. H., and J. L. Strominger. 1989. Transcription of Epstein-Barr virus in latently infected, growth-transformed lym-phocytes. Adv. Viral Oncol. 8:133-148.
27. Stern, S., M. Tanaka, and W. Herr. 1989. The Oct-I homeo-domain directs formation ofamultiprotein-DNA complex with theHSV transactivator VP16. Nature(London) 341:624-630. 28. Sung, N. S., S. Kenney, D. Gutsch, and J. S. Pagano. 1991.
EBNA-2 transactivates a lymphoid-specific enhancer in the BamHI C promoter of Epstein-Barr virus. J. Virol. 65:2164-2169.
29. Tsang, S. F., F. Wang, K. M. Izumi, and E. Kieff. 1991. Delineation of the cis-acting element mediating EBNA-2 trans-activation of latentinfection membrane protein expression. J. Virol. 65:6765-6771.
30. Wang,F., C. D. Gregory, M. Rowe, A. B. Rickinson, D. Wang, M. Birkenbach, H. Kikutani, T. Kishimoto, and E. Kieff. 1990. Epstein-Barr virus nuclear antigen 2 specifically induces expres-sion ofthe B-cell activation antigen CD23. Proc. Natl. Acad. Sci. USA 84:3452-3456.
31. Wang, F., H. Kikutani, S. F. Tsang, T. Kishimoto, and E. Kieff. 1991. Epstein-Barr virus nuclear antigen transactivates a cis-acting CD23 DNA element. J. Virol. 65:4101-4106.
32. Wang,F., S. F. Tsang, M. G. Kurilla, J. I. Cohen, and E. Kieff. 1990.Epstein-Barr virus nuclear antigen 2 transactivated latent membrane protein LMP1. J. Virol. 64:3407-3416.
33. Woisetschlaeger, M., X.W.Jin, C. N. Yandava, L. A. Furman-ski, J. L. Strominger, and S. H. Speck. 1991. Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages ofinfection. Proc. Natl. Acad. Sci. USA 88:3942-3946.
34. Woisetschlaeger, M., J.L.Strominger, andS.H. Speck. 1989. Mutuallyexclusive use of viral promoters in Epstein-Barr virus latently infected lymphocytes. Proc. Natl. Acad. Sci. USA 86:6498-6502.
35. Woisetschlaeger, M., C. N.Yandava, L. A. Furmanski, J. L. Strominger, and S. H. Speck. 1990. Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc. Natl.Acad. Sci. USA 87:1725-1729. 36. Zenke, M., T. Grundstrom, H. Matthes, M. Wintzerith, C.
Schatz,A.Wileman,and P.Chambon. 1986.Multiplesequence motifs are involved in SV40 enhancer function. EMBO J. 5:387-397.
37. Zimber-Strobl, U., K. 0. Suentzewich, G. Laux, D. Erik, M. Cordier, A. Calender, M. Billaud, G. Lenoir, and G. W. Bornkamm. 1991.Epstein-Barrvirus nuclear antigen 2 activates transcription of the terminal protein gene. J. Virol. 65:415-423. J. VIROL.