Vol.67,No. 1 JOURNAL VIROLOGY, Jan. 1993, 476-488
0022-538X/93/010476-13$02.00/0
Copyright ©C1993,AmericanSocietyforMicrobiology
Identification of Specific
Adenovirus
ElA
N-Terminal Residues
Critical
to
the
Binding of Cellular Proteins and
to
the Control of Cell Growth
HWEI-GENEHEIDIWANG, YASUJIRIKITAKE,t MARY CORRIGANCARTER,4 PETERYACIUK, SUSHMAE.ABRAHAM,§ BRADZERLER,II AND ELIZABETHMORAN*
ColdSpring Harbor Laboratory, Cold Spring Harbor, New York 11724 Received 17June1992/Accepted 23 September 1992
Adenovirus early region IA (EIA) oncogene-encoded sequences essential for transformation- and cell
growth-regulating activities are localized atthe N terminus and in regions ofhighly conserved amino acid sequencedesignatedconservedregions1and 2. Theseregions interacttoformthebindingsites fortwoclasses ofcellular proteins: those, suchasthe retinoblastomageneproduct,whoseassociation with theE1A products
isspecificallydependent onregion 2, and another class whichsofar is knowntoincludeonlyalargecellular
DNA-binding protein, p300, whose association with the ElA products is specifically dependent on the N-terminalregion. Association betweenthe ElAproductsand either class of cellularproteinscanbedisrupted bymutations in conservedregion 1.While region2 hasbeen studied intensively, verylittle is known sofar concerning the nature of the essential residues in the N-terminal region, or about the manner in which
conserved region 1 participates in the binding of two distinct sets ofcellular proteins. A combination of
site-directedpoint mutagenesis andmonoclonalantibody competition experiments reportedhere suggests that
p300 binding is dependent on specific, conserved residues in the N terminus, including positively charged residues atpositions 2 and 3 oftheE1A proteins, and that p300 andpRBbindto distinct, nonoverlapping subregions within conserved region 1. Theavailability ofprecise point mutations disrupting p300 binding
supportspreviousdatalinking p300withcellcyclecontrol and enhancer function.
The adenovirus (Ad) early region 1A (ElA) gene is re-quired for normal transcriptional activation of the remainder of the viral genes (4, 42, 46, 55) and is sufficientto induce extended proliferation in quiescent rodent cells such as
primary baby rat kidney (BRK)epithelial cells (36). TheElA gene encodesthree regions of highly conserved amino acid sequenceinexon1(72),which also appearstocontain three
independent active sites (for reviews, see references 6 and
51); the sequence of theElAprotein isillustrated schemat-ically in Fig. 1.Conserved region 3,specifically required for efficient viral gene transactivation (8, 26, 70), can be re-moved selectively by differential splicing (11, 56). Exon 1 sequences in theabsence ofregion 3aresufficient for the cell growth control activities of the ElAproducts (48, 50, 83).
However,thetwoactive sites in thisregionare notlimitedto
sequences withinconservedregions 1 and 2; growth control
activity requires the relatively nonconserved region at the
extreme Nterminusaswell(41, 66, 69, 79). The active site
linkedwith conservedregion 2 is supported by sequences in
conserved region 1 (17, 79) (Fig. 1). These noncontiguous sequences form an apparently direct binding site for the product of the retinoblastoma (RB) tumor susceptibility gene
(15, 17, 79). ApRB-related gene product, designated p107
(20), likewise binds via region 2 (17, 79) and is itself
complexed with cyclinAandp33cdk2(19, 21, 37). Binding of
* Corresponding author.
t Present address:Medical Environment Co.,Tokyo, Japan. tPresentaddress: Oncogene Science, Uniondale,NY11553. §Present address: Department of Microbiology, Washington StateUniversity,Pullman, WA 99164-4233.
11Present address: Miles Research Center, West Haven, CT 06516.
a less well-characterized cellular product, p130, is also
dependenton sequencesin region2(25).
In addition to supporting the binding ofregion
2-associ-atedproducts,conservedregion1participatesin the
forma-tion ofasecond activesite,onethat requiresnoncontiguous sequences at the extreme Nterminus of the ElA proteins. The active site linked with the N terminus is involved,
directlyorindirectly, inbindinga300-kDa cellularproduct,
p300(17, 66,79).
Binding of these various cellular products isclosely linked withthe cell growth control functions of the ElAproducts.
ElAregion2isadiscretegenetic element that is shared by
theproducts of the transforming proteinsof several classes of small DNAtumorviruses (22, 49, 57,65) andbycellular
pRB-binding proteins as well (13). These all contain the
conserved amino acid motif(D)-L-X-C-X-E, and the integ-rity of this motif is essential for the transforming and pRB-binding functions of the ElA, simian virus 40 (SV40)
largeTantigen, and human papillomavirus(HPV)E7
trans-forming gene products (3, 12, 49). Most of the region
2-associated proteins have also been identified as
compo-nentsof the cellulartranscription factorE2F(2, 7, 9, 14,63). The N terminus of theElA products is likewise strictly
requiredforElAtransforming functions(41, 66,69,79). In
addition,the N-terminalfunction has been linkedspecifically
with ElA-mediated repression of viral enhancers (41) and
tissue-specific gene expression (66). Moreover, p300 has
been identified as a nucleus-localized phosphoprotein (81)
withsequence-specificDNA-bindingactivity andaffinityfor enhancer element-related DNA motifs (59). Nevertheless,
little is known about the exact amino acid sequences in-volved in ElA N-terminal function. Since the extreme
N-terminal sequence is not strongly conserved among the ElAproducts of the various Ad serotypes (Fig. 2), there are 476
on November 9, 2019 by guest
http://jvi.asm.org/
CELLULAR PROTEIN ASSOCIATIONS AT THE ElA N TERMINUS 477
25 37 80 121 139
p300 cell cycle control
differentiationstate p1O5RB
enhancerrepression p107-cyclinA-p33 p130
[image:2.612.63.302.75.154.2]cellcycle control
FIG. 1. Schematic diagram of ElA active sites and associated proteins. The ElA regions required for cell growth-regulating func-tions (shownasunshadedregions in this diagram) arethebinding
sites for several cellular proteins. These regions constitute two
independent active sites that can be inactivated selectively by mutations confined toeither the extremeN terminus orregion 2.
The activity of either site is sufficienttoinduce the G1 and S phases in resting cells, although both sites are required for the ElA immortalization function (38, 77, 84). While both sitescan induce
cellcycle functions, the abilityof ElA torepress enhancers
con-trolling tissue-specificgeneexpression is linked withthe N-terminal
active site and the binding of p300 (41,66). A.A., amino acids.
fewcluesto thenatureof thepotentially essential residues. However, since thesesequences are implicated in the ElA functionstargeting control of cell growth and differentiation,
it is ofsignificantinteresttodetermine whetherspecificElA
residues areessential forN-terminal function, especiallyas thesesequences could conceivablycontain motifs homolo-gous toimportant cellular proteins.
MATERIALSAND METHODS
Cells and viruses. Monolayer cultures ofHeLa, 293 (29),
and COS (27) cells were grown in Dulbecco's modified Eagle'smedium supplementedwith10% calfserum, 100,ug
of penicillin per ml, and 100 ,ug of streptomycin per ml. Primary baby rat kidney (BRK) cells were prepared from 6-day-old Fisher rats as described previously (50) and in-fected 2to3days afterplating.
Ad2 ElA
Ad5
Ad4
Ad7
Adl2
Ad40
Ad4l
SA7
SV40 TAg
M HII-CHGGVITEEMAASL
MRHII-CHGGVITEEMAASL
MRHLRDLPDEEIIIASGSEI
MRHLRFLPQEIISSETGIEI
M.RTEMTPL--VLSYQEADDI
MRKLPDFFTGNWDDMFQ-GL LPDFFTGNWDDMFQ-GL
-ALEM--ISELLD-LG
MDKVLNREE--SLQLMDLL9
X LIE
LE
LE
LE
LE
LE
LE
LE
2LIE VVN
VVN
HLVD
rEYV
kEHP
rIDG
RSAW FIG. 2. Comparison of N-terminal ElAproteinsequencesfrom variousprimateAds. The first 25 amino acid residues of Ad5 ElA
are compared with the N-terminal sequences of ElA from other serotypes. The sequences and alignments of Ad serotypes Ad5, Ad7,Adl2, andSA7arepresentedasreportedbyKimelmanetal.
(43); dashes indicategaps introduced tomaximizehomology. The
Ad2,Ad4,and Ad4Oand Ad4lsequencesaretaken fromTokunaga
etal.(71),Ishinoetal.(40),and Allard and Wadell(1), respectively.
Conserved featuresasnotedbyKimelmanetal.(43)areboxed. The first 24 residues ofSV40 largeTantigen(TAg),taken fromSeifetal.
(62), arealso shown forcomparison.
All viruses were propagated on 293 monolayers. The 12S.WT (50, 83) and 12S.928 (53) viruses have been de-scribedpreviously. The deletion mutant viruses used for the monoclonal binding site mapping experiments are 12S vari-antsof constructsdescribedpreviously, as indicated: 12S.2-36, 12S.15-35, 12S.51-116, 12S.81-120 (66); 12S.86-120, for-merly called 12S.NCdl (53); 12S.38-67 (58). The mutant
deleted for residues 1 to 14 isthe virus d1309-68 (32a), which encodes an altered initiation codon such that translation begins at the residue 15 methionine. The single-residue substitution mutants pl2S.RG2, pl2S.HN3, pl2S.LS20, pl2S.DA21, pl2S.YG47, pl2S.YH47, pl2S.LP49, and pl2S.EV55 were constructed by standard oligonucleotide-directed mutagenesis techniques. Subsequently, restriction fragments containing the point mutations were subcloned into a 12S.WT parent pUC plasmid, and the presence of the mutations directing the single-residue substitutions was ver-ified by dideoxy sequencing through the entire restriction fragment.
The 12S double mutants 12S.RG2/928 and 12S.YH47/928 wereconstructed by first isolating a 773-bp EcoRI (bp 1 of Ad type 2
[Ad2J)-to-StyI
(bp 875 of Ad2) fragment from genomic constructs pElA.RG2 and pElA.YH47, containing thesubstitution, andligating each to the larger EcoRI-to-StyI fragment ofp12S.928 containing thevectorsequences. The structures of the recombinant plasmidswere confirmed by restriction digest analysis and selective hybridization withtheoligonucleotides used togenerate themutations.
Plasmid pl2S.T:1-24/E:25-243 is a 12S variant of pT.1-24/ E.25-289 described previously (80). It was constructed by ligating equimolar amounts of the following four gel-purified restrictionfragments and a synthetic double-stranded DNA oligomer: a 146-bp SflI-EcoRI (SV40 nucleotide positions 5240 to 5094) restrictionfragment from pT.1-100/E.121-289 (80); a 13-bp synthetic oligonucleotide with
EcoRI-RsaI-complementaryends,
CCTGGGAAGAGGT CTTCTCCA,
a 239-bp RsaI-StyI (ElA nucleotide positions 638 to 877) restrictionfragment from pl2S.WT(53); a208-bpStyI-XbaI (ElA nucleotide positions 877to1336, minus the 12S intron) restrictionfragment from pl2S.WT; anda4,420-bpXbaI-SflI (ElA nucleotideposition 1336 to SV40 nucleotideposition
5240)restrictionfragmentfrompT.1-100/E.121-289.
Plasmidpl2S.T:1-14/E:20-243 was constructedby ligating equimolaramountsofthefollowing threegel-purified
restric-tionfragments: the923-bpEcoRI-AvaI restrictionfragment
from pT.1-100/E.121-289 and the 257-bp AvaII-Sty and
3,319-bpStyI-EcoRI restriction fragmentsfrompl2S.WT.
The structures of the recombinant plasmids were
con-firmedby restriction digestanalysisandsequencing through
mutated regions. Mutant and chimeric ElA regions were
rebuilt into virusconstructsbythe method ofStow (68) as
described in detailpreviously(50).
Immunoprecipitations. Immunoprecipitations were per-formed as described previously (77). HeLa or BRK cells
were infected at multiplicities of 10 to 50 PFU per cell.
Electrophoretically separated immunocomplexcomponents
were visualized by autoradiography. ElA-specific mouse
monoclonal antibodies M73, Ml, M29, and M37 (31) and
pRB-specificmouse monoclonal antibody XZ133
(39)
weregenerously provided by Ed Harlow and
Qianjin
Hu. Theresultspresentedarerepresentativeof
experiments
repeated atleast three times.VOL. 67,1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.71.282.478.619.2]478 WANG ET AL.
Virus immortalization assay. In all experiments, primary BRKcells were infected at a multiplicityof 10 PFUper cell. After adsorption at 37°C for 1 h, nonadsorbed virus and old medium were removed and replaced with fresh
antibiotic-supplemented Dulbecco's modified Eagle's medium contain-ing 5% fetal bovine serum. The infected cells were incubated at37°Cfor4weeks with weekly feedings. At the end of this time, the monolayers were fixed with methanol-acetone (50:50) and stained with 20% Giemsainphosphate-buffered saline (PBS) solution. Immortalization activity was deter-mined by counting the number of established colonies of Giemsa-staining cells. For each experiment, two to seven
plates were averaged pervirus isolate.
Analysis of DNA synthesis. Infected BRK cells were la-beled with[3H]thymidine (ICN) (25 ,Ci/ml of medium) and assayed for trichloracetic acid-precipitable counts, as de-scribedpreviously (84).
Enhancer repression assay. For transfection with HeLa
cells, 5 x 105 cells were plated on tissue culture plates
(100-mm diameter) 24 h before transfection. Cells were
transfected by the calcium phosphate coprecipitation method described by Graham and van der Eb (30). The humanimmunodeficiency virus (HIV) long terminal repeat
(LTR)-chloramphenicol acetyltransferase (CAT) reporter
plasmid, pBennCAT, has beendescribed previously (24), as wasthe NF-KB element deletion plasmid, pNFA-CAT(45). The total plasmid DNA concentration was adjusted with pUC18 to 20 ,g, and the total DNA concentration was
adjusted to 40 ,ug with salmon sperm DNA as the carrier. Thecalcium phosphate precipitate remained on the cells for 6h,atwhichpoint the cellsweretreated with15% glycerol-PBSfor2minand then fed with fresh medium and incubated foranadditional 30 h.
CATactivitywas assayed asdescribed by Gorman etal.
(28) and quantified with an AMBIS Beta Scanner. The percent conversion of
[14C]chloramphenicol
to acetylated formswascalculated and normalized to the amountof total cellprotein in the lysate.RESULTS
Requirement for specific sequences at the extremeN termi-nus.Asafirst approachtoidentifying the essential N-termi-nal sequences, we explored the general requirement for
specific N-terminal residues in the p300 binding function.
Required N-terminal sequences, exclusive of conserved
region 1, are known to belimited to the first 25 residues,as adeletion of residues 26to35doesnotsignificantly alter the
p300binding ability ofElA, orany of its knownbiological
activities (41). Mutants encoding deletions of residues4 to
25, 15to35,or2to36make fairly stable proteins but fail to bindp300or totransformprimary cells (17, 41,66).Theonly single N-terminal residue so far demonstrated to have an essentialrole is Arg-2,which, when changed to Gly, results inElAproducts defective for transforming activity andp300 binding (79).
Since the N-terminal 25 residues are not well conserved amongtheElAproducts ofdifferent Ad serotypes (Fig. 2),
we first tested whether there is an overall requirement for specific residues in this region. To do this, we replaced Ad sequences encoding the first 20 N-terminal ElA residues withSV40 sequences encoding the N-terminal 14 residues of large Tantigen. This chimeric virus construct (described in detail in Materials and Methods) also exchanges the ElA
promoter forthe SV40 early promoter. The chimeric virus is
designated 12S.T:1-14/E:21-243. A second, similar,
con-Ocr:
LOU t)L\
0 CM)N
200
-PC rr)
C\j
4-:
ucmn
AI Nj
CQ C\i
W- -P300
-p130
92 _ p105-RB
68
-doEIA
2 3 4 5
FIG. 3. AssociationofcellularproteinswithN-terminal chimeric
constructs. Coprecipitation was assayed by immunoprecipitating
Tran-35S-labeledinfected COS celllysateswithElA-specific
mon-oclonalantibodyM73(31).Thepositionsofp300,theElAproteins, and theregion 2-associated groupofproteinsareindicated atthe right. Thepositionsofmolecularsizemarkers(in kilodaltons)are
indicated at theleft.
struct, 12S.T:1-24/E:25-243, replacesElA residues 1 to 24
withT-antigen residues1 to24. WecomparedtheT-antigen
N-terminal sequence with theElA N-terminal sequences in Fig. 2. The ability of these chimeric virusproteins to bind
p300wasassayedbycoimmunoprecipitationin severaltypes
of infected cells, including HeLa and primary BRK cells.
Thechimericproteinsshowednoevidence ofbinding p300in
these cells(datanotshown).Association withp300wasalso assayed in COS cells, in whichexpression of the chimeric
constructs is optimal as a consequence of theirexpression
from the SV40 earlypromoter. This promoter is autoregu-latedbyTantigen,whichisconstitutivelyexpressedin COS cells (27). The chimeric proteinswere immunoprecipitated
with anElA-specificmonoclonal antibody(series M73; see
reference31)thatrecognizesanepitopenearthe Cterminus
of the ElAproducts. Even at a high expression level, no
p300 bindingwasdetectedwiththechimeras(Fig.3, lanes4
and5). Inthesameconditions, the products of 12Swild-type
ElA and the pRBbinding-defective region2 mutant,12S.928
(witha single substitution changing Cys-124to Gly), retain
theabilitytobindp300efficiently (Fig.3, lanes2and3),even
though they are expressed at lower levels relative to the
chimeric products. Significantly, the chimeras retain the
abilitytobind theregion2-associatedsetofproteins,
includ-ing pRB, verifying that substitution with the heterologous sequences does notgrossly distort the protein structure of the chimeras. These results suggest that there is a specific
sequenceorstructuralrequirementwithin theElA N-termi-nal sequences for binding p300 that cannot be satisfied
simply by substitutingN-terminal sequences froma
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.377.495.76.333.2]CELLULAR PROTEIN ASSOCIATIONS AT THE ElA N TERMINUS 479
0
-OJ ff) N\1 N
(5 Z Lo
-v 3:
ot:
ILj
Q 5l -i (n E C CM CM NI )w _
-* 2Jw
200
p!30
j_-*W _Wl _-i07
92- - 056
68-
-45
A1E
1254-2 3 4 5 6
FIG. 4. Association of cellular proteins with N-terminal point
mutant constructs. Coprecipitationwasassayed by immunoprecip-itatingTran-35S-labeledinfectedHeLa celllysates with ElA-specific
monoclonal antibody M73 (31). The positions of p300, the ElA proteins,and theregion2-associatedgroupofproteinsareindicated
attheright.Thepositionsof molecular size markers(in kilodaltons)
areindicatedatthe left. Theexperimentsshown inFig.4through 8
arerepresentativeof results observedmultipletimes inindependent experiments.
ogousprotein.Note also that Tantigenencodesabiological function that cancomplementthe ElA N-terminal function
(80), althoughit is not known whetheranystructural
homol-ogy underlies thisactivity orwhere, precisely, in Tantigen
the complementing function lies. The chimeric construct results, do not, of course, rule out the possibility of a p300-bindingsite somewhere in T antigen.
Pointmutantanalysisindicatesarequirementinstablep300 binding for specific residues in the N terminus, including positively charged residues at positions 2 and 3. As noted
above,adeletionfrom residues 26to35previouslydefineda regionbetween the Nterminus and conservedregion1 that is notrequired forp300 binding (41). This deletion demon-strated thatp300-bindingsequencesin the ElAproteinsare noncontiguous and indicated that there is some degree of
flexibility in the spacing acceptable between the binding
sequencesatthe N terminus and those in conservedregion 1. Another deletion, larger by 10 residues, extending the deletedregionNterminallyto include residue 15,results in anElAproteinwithnodetectablep300-binding activity (66). Thisresultdid notdistinguishwhether the 15-to-35 deletion loses activity because the region from residues 15 to 25 contains specific essential residues, or more indirectly be-causethe spacingbetweenrequiredN-terminal residues and conservedregion1wasreduced toalevelincompatiblewith p300 binding. We addressed this question by introducing specific pointmutations in theregionfrom residues 15to25. The only conserved feature that has been noted in this stretch (43) (Fig. 2) is the appearance of a leucine residue immediatelyfollowedbyanegatively charged residue,either glutamicoraspartic acid. We used oligonucleotide-directed site-specific mutagenesis to change each of these residues selectively. Leucine 20was changedto serine in a mutant
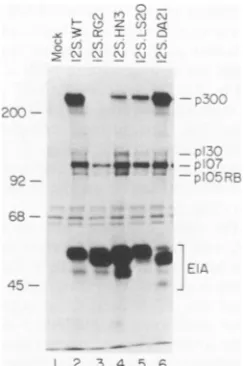
designated 12S.LS20. Aspartic acid 21 was changed to alanine in a mutant designated 12S.DA21. Both of these
constructs remained ableto bindp300 in HeLa celllysates
(Fig. 4, lanes 5 and 6); however, while 12S.DA21 showed an essentially wild-type level of p300-binding activity, 12S.LS20 consistently showed a much reduced level,
sug-gesting that stable p300 association does involve specific
residues in the small stretch from positions 15 to 25. Although there is little sequence conservation at the
extremeNterminus ofthe ElA products, it is notable that all
serotypicallydistinct ElA forms so far sequenced have an
arginine at position 2, and several encode a histidine at
position 3 (1, 40, 43, 71, 72) (Fig. 2). It has already been
noted that changing arginine 2 to glycine disrupts
p300-bindingand-transformingactivity(79).The conservation of
thesepositively chargedresidues is intriguing, especiallyas
the occurrence ofpositively charged residues in the highly acidicElA.12Sproductis ratherrare(sequencereviewed in reference 51). To determine whether the histidine residue plays a role in p300 binding in Ad5 ElA, we changed the histidinetotheunchanged residue asparagine in a construct
designated 12S.HN3. We also made a construct similar to
the arginine-to-glycine mutation, which we designate
12S.RG2. The ability to bind p300 is much reduced in the 12S.HN3mutantand is belowthe level ofdetectability in the 12S.RG2product(Fig.4, lanes 3 and4),suggesting that the presenceof bothpositively chargedresidues isimportant to stable p300 binding. The HN3 mutation has no discernible effecton pRB, p107, or p130 binding (Fig. 4, lane 4). The RG2 andLS20mutantproducts showreduced, but
consis-tently detectable, pRB- and p130-binding activity (see also
Fig. 8)andessentially wild-type abilitytobindp107.
pRB- and p300-binding sites in conserved region 1 do not
appear to overlap. Mapping of the p300- and pRB-binding
sites within conservedregion 1 has beenapproached sofar
with only relatively broad deletion mutants. These
con-structsindicated thatregion1isimportantforstablebinding of both of theseproducts,butdid littletodistinguishwhether
the same specific residues are involved in binding both
cellularproducts.Theonlydistinction sofarmade between
p300 and pRB binding requirements in region 1 is that
sequences in the C-terminal half of conserved region 1 are
less important for binding pRB than for binding p300; a
linkerinsertion mutationdeletingresidues 61to 85 disrupts
p300 bindingbuthas little effecton pRBbinding (79).
An important consideration when analyzing functional
sites that lie very close together is that deletion mutations may affectboth sites because of structural constraintseven
when the deletion lies entirely within a single site. An alternativeto usingdeletion mutations tomapbinding sites
directly is to use antibody competition experiments. This
approach has the advantage of permitting the assay of
protein interactions on a wild-type molecule after deletion
mutants have been used to map the antibody-binding
epitopes. For example, it has been shown previously that
antipeptide serum raised against an N-terminal peptide
blocksp300, butnotpRB, binding(82).
Toaddress thequestionof whetherp300andpRBbinding
sites in
region
1actually
involveoverlapping
sequences,we tested theabilityofasetofmonoclonal antibodiesbindingtodifferent epitopes within this region to block the stable interaction of eitherproduct.Thefeasibilityofthisapproach
is supported by previous observations noting that different
subsets ofElA-associatedproteinscould be
immunoprecip-itated with some of these antibodies (32). The associated
protein patterns seen in immunocomplexes with region
1-specific
monoclonal isolateswerecompared
with thepat-tern seen in
immunocomplexes
with monoclonal isolateM73,whichbinds neartheElA C terminus and is compat-VOL. 67,1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.118.240.73.256.2]480 WANG ET AL. rf-- f11- 4)
p300-- W
p300- l-4
p130- P3
p107- p13- __
p1O5RB-
plO5RB-E...~
EIA|
L SW
2 3 4
r)
p300-
9-F
p130-
p1O5RB-EIA. g
[image:5.612.74.288.70.249.2]5 6
FIG. 5. Exclusion of specific cellular protein associations by monoclonal antibody binding toElAconservedregion1. Coprecip-itation was assayed by immunoprecipitating Tran-35S-labeled in-fected HeLa cell lysates with ElA-specific monoclonal antibody series(31)M73, which binds near theElACterminus,orM37,M29, andMl, which bind within conserved region 1. The positions of p300, the ElA proteins, and the region 2-associated group of proteins are indicated at the left.
ible with the binding of all identified ElA-associated pro-teins.Testingof avarietyof monoclonal isolatespreviously generated by Harlow etal. (31) revealed three whose prop-erties were useful fordistinguishing p300 and pRB binding requirements (Fig. 5). Of these, two, M37 andM29,
inter-fered strongly with the binding of pRB and other region 2-associated proteins, but formed complexes containing readily detectable levels of p300 (Fig. 5, lanes 2 and 4). Conversely,monoclonal isolate Ml interferesstronglywith
p300binding but has little effectonthebindingof theregion 2-associatedset(Fig.5, lane6).
We mapped the epitope-binding sites of monoclonal
iso-lates Ml, M29, and M37 using a series of ElA deletion mutants. It has been shown previously that region 2 and exon 2sequences(i.e., residues C terminal toposition 120)
are not required for ElA product recognition by these
isolates (32; our additional unpublished data). The data obtainedfrom thepresentscreen that are mostsignificantfor
localizing the epitopes recognized by these antibodies are
presented in Fig. 6. Deletion of residues spanning
collec-tively theregions from positions2 to67 and 86to120 does
notdelete theepitope recognized byMl, suggestingthat the
Mlepitope is containedwithin residues 68 to 85. Consistent
with this conclusion, Ml fails to bind an ElA product
deleted for residues 81 to 120. By a similar analysis, we
conclude that M29 and M37 recognize epitopes contained
within residues 37 to 50, or 15 to 50, respectively. In addition, it is likely that the M37 epitope occurs near the
middle of the 15-to-50 range because deletions of either 15to
35 or 38 to 67 disrupt the epitope. Moreover, none of the
A
r M M29 M37 M37
0
Co ° N o co n E E 3 o a
:rj c 2 'n D r-t w )6r- -j
_ 4+ + -4- _ + -_ 4- - + _ + _ _ _ _ _- + +* + + +
B
Mappingot monoclonalantibody epitope recognition sites.
MAb Binds Fails to Epitope maps Coprecipitates Coprecipitates
bind within p105RB p300
p107 pl30
MI dl.2-36
dl.38-67
d086-120
M29 d012-36 d51-151-6
d1.81-120 AA: 68-85 +
dl.38-67 AA:37-50
M37 d1.1-14 dl.15-35 AA: 15-50
dl.51-116 dl38-67 22-46] 4
pm.LS20
pmDA21 pm YH47
pm.YG47 pm.LP49
FIG. 6. Mapping of monoclonal antibody epitope recognition sites. (A) Ability of a group of monoclonal antibodies to recognize a series ofElAdeletionandpoint mutant products was assayed by immunoprecipitating lysates ofTran-35S-labeledinfected HeLa cells. The residues deletedorsubstituted in theElAproducts assayed are indicated above each lane. Thepresence (+) or absence (-) of the epitope targeted byeachmonoclonal isolate, indicated by the ability of the monoclonal isolate to precipitate the mutant product, is indicated below each lane. Thepositionofthe 12S.2-36 product, which is expressed at rather low levels (77), is highlighted (arrowhead) in the M29 panel to distinguish it fromnonspecific background bands. (B) Deduced boundaries of the epitopes recognized by each monoclonal antibody are tabulated, along withtheassociatedprotein pattern observed in each specific immunocomplex.
rO Oj
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.150.473.397.649.2]CELLULAR PROTEIN ASSOCIATIONS AT THE ElA N TERMINUS 481
riNa a
e-I V L > N
Y u>- >- 41 Oa
vU)C) U) UL cnU)
E "O
-
NO\~~~p300
N\ \N92 plO5RB
68
-... " n
_ - - ~~~EIA
2 3 45 6 7
00
co
anN
O
: D IQ
>- L
>-O cnuiuruU)e _N N NN_
200-- 300
p130
-p107
92- plO5RB
68
-45- EIA
8 9 10I 112 13
U) U) U) U)
*9-p300-200
-pl3o
_p107
92- p105R8
68-45- EIA
14 5 16 17 18
FIG. 7. Association of cellular proteins with region 1 point mutantconstructs. Coprecipitation was assayed by immunoprecipitating
Tran-35S-labeled infected HeLa cell lysates with ElA-specific monoclonal antibody M73 (31). The positions ofp300, the ElA proteins, and
theregion 2-associatedgroupof proteinsareindicatedatthe right. The positions of molecular size markers (in kilodaltons)areindicatedat the left.
point mutations discussed above, and inthe section below, interferes with M37 recognition. Since these include
alter-ationsof residues 20, 21, 47, and 49, it isverylikely that the M37 epitope falls within themore narrow rangebordered by residues 22to46.
These results suggest strongly that p300 and pRB do not
actually bind to overlapping sequences in region 1; more likely, they bindtodistinct subregions. Binding of pRB, but notp300, appearstobedependentonsequences inthenear vicinity of residues 37to 50. Conversely, p300 binding,but notpRB binding, appears to bedependenton sequences in thenearvicinityof residues68to85.Overall,it isreasonable toproposethatpRButilizessequencesinthe N-terminalhalf ofregion 1,whilep300 bindingutilizessequenceslimitedto
the C-terminal half ofregion 1. Asimplemodel of how this
binding patternmaybe accommodated is suggested inFig. 11.
Binding of pRB, but not p300, is disrupted by specific mutation ofatyrosine residueatposition 47 withinconserved
region 1.The limitswepropose onthebindingsites ofp300
andpRBareconsistent with the observation cited above(79) suggestingthatsequencesfrom residues 61to85arefarmore importantforp300 binding than forpRB binding. Theyare also consistent with previous suggestions that the region
from residues 41to49 of ElAregion 1 might playarole in pRB bindingbased on homologyto sequencesin the HPV type 16 E7product,which also contains ahomologof ElA
region 2 (57) and associates with pRB (16). Within the
E7/E1Aconservedregion represented byElA residues 41to
49,thereoccurthree leucine residues withcloselyconserved spacing (57).There is alsoatyrosineresidue in both ElA and E7 within thisregion,which could besignificantbasedonthe
rarity ofoccurrenceof this residue ingeneral,and in ElA in particular; this is the only tyrosine residue among the 243 residues of the AdS 12SElAproduct.
Toassess the involvement of thisregionin pRB binding, weconstructedseveral additional mutations. Leucine 49was changed to proline in a construct designated 12S.LP49.
Tyrosine47waschangedtoboth the somewhatstructurally
similar residue histidine (12S.YH47) and the less similar residue glycine (12S.YG47). In addition, to probe the in-volvement of more distant sequences, we changed the strongly conserved glutamic acid at position 55 to valine
(12S.EV55). This last mutation has the effect of introducing ahydrophobic residue intoa veryhydrophilic region. Asso-ciation with cellularproteinswasassayed by coimmunopre-cipitation from infected HeLa cell lysates (Fig. 7). As
predicted from the monoclonal antibody competition exper-iments, noneof these mutations hadapronounced effecton
p300 binding (Fig. 7,lanes 3to7, 10,and11). In addition, the pRB-binding activity of the 12S.LP49 and 12S.EV55
muta-tions, although sometimes reduced, was always readily detectable(Fig. 7, lanes 5, 6, and 11). However, pRB binding wasnotreadily detectedinthese conditions in themutants
with an altered tyrosine residue (Fig. 7, lanes 3, 4, 10, and 17). 12S.YH47inparticular showedaconsistent lack ofpRB association (Fig. 7, lanes 3, 10, and 17). This mutant is clearly defective in p130 binding aswell, but p107 binding remains essentially unaffected. The generation of mutations
at position 47 which severely disrupt pRB, but not p300, bindingsupportsthehypothesisthatp300 and pRB associate with distinct, nonoverlapping sequences within conserved region1.
Despitethestrongconservation ofthe leucineresidues in
thisregion, the Leu-49mutation did notseverely affect the
appearanceof thecoprecipitating products. This doesnot,of
course, exclude the involvement of the leucines in pRB binding; it is possible, for example, that no single leucine residue is individually essential for this activity, although
each may play a role. Overall, these results indicate that
pRB binding in region 1 involves contact with specific residues,andthat,attheleast, Tyr-47isamongthese.These
resultsareillustratedschematicallyin the modelproposedin Fig. 11.
TheTyr-47residue is the second individual residue iden-tified in the ElA products at which substitution severely impairs pRB bindingwithoutseverely impairingthebinding
of the pRB-homologous product, p107; mutation of the
Cys-124residueinregion 2,inaconstructdesignated pm928,
results inasimilarphenotype (49, 77) (Fig. 7,lanes 7 and18). Whileneither 12S.YH47nor12S.928isseverely impairedin the p107 binding property, the combination of these two single point mutations, inavirusdesignated 12S.YH47/928, abrogates p107 binding (Fig. 7, lane 13). p300 binding re-mainsunimpaired in the 12S.YH47/928 mutant. Curiously,
the combination ofpm928 and pmRG2 in the virus desig-4g
-30
-VOL.67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.149.475.77.236.2]TABLE 1. Virusimmortalization assay
Virus No. of colonies, averaged from multiple plates (% of wild type), in expt: Avg activity
1 2 3 4 5 6 7 (%)
Mock NDa 0 0 0 0 0 0
d1312 0 ND 0 0 0 0 0
12S.WT 73 110 81 36 31 66 66 100
12S.RG2 0 0 0 0 0 0 0 <1
12S.HN3 ND ND 6 (7.4) 1(2.8) 2(6.5) 4(6) 4.5 (6.8) 5.9
12S.LS20 ND ND 6 (7.4) 14(38.9) 6(19.4) 12(18) 7(10.6) 18.9
12S.DA21 ND 18(16.3) 44(54.3) 26(72.2) 29(93.5) 51(77) 46(69.7) 63.8
12S.YH47 89(122) 37(33.6) 31 (38.2) 6(16.7) 19(61.3) 57(86) 42(63.6) 60.2 12S.YG47 29(39.7) 22(20.0) 38 (46.9) 20(55.5) 31 (100) 64(97) 57(86.3) 63.6
12S.LP49 41(56.1) ND 54(66.7) 34(94.4) 60(196) 75(106) 76(115) 100
12S.EVSS 87(119) ND 58(71.6) 40(111) 63(200) 91(138) 83 (126) 100
12S.928 0 0 0 0 0 0 0 <1
aND, not done.
nated 12S.RG2/928 has a similar effect on p107 binding. While neither of these point mutations alone has a severe
effect onp107association(Fig.4, lane 3;Fig. 7, lanes 7, 16, and18), the combination impairsp107bindingseverely(Fig.
7, lane 12). The same phenotype was observed previously when the deletion of residues 2 to 36 was combined with the pm928 mutation (77).
Cosegregation of immortalization activitywithrequirement for specific amino acid residues in p300 binding. In the followingexperiments,weused thesingle-residue mutations described abovetoanalyze moreprecisely the links between
the bindingofspecific cellularproducts and ElA biological
activity. It has already been reported that substitution of
residue2has severedisruptive effects onbothp300binding and theElA transformation functionassayedincooperation
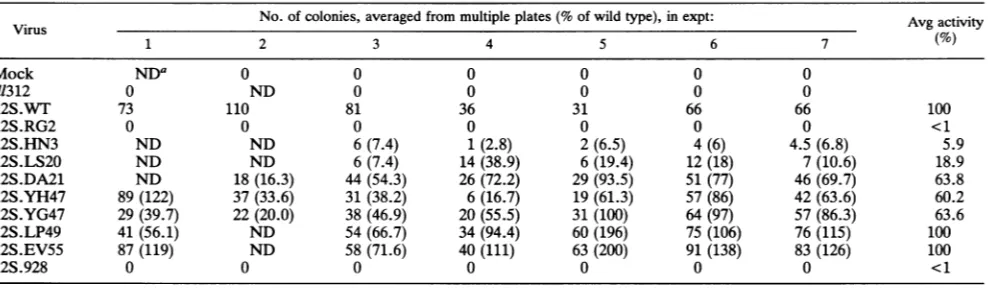
withthe ras oncogene (79). Our results extend and confirm thisobservation.Impairment of theElA-dependenthost-cell
immortalization function (Table 1) correlates closely with
impairment of p300-binding activity. 12S.HN3 and
12S.LS20, which show clearly reducedp300-binding
activ-ity, also show reduced immortalization activity, to levels
averaging, respectively,5.9 and18.9%ofwild-typeactivity.
12S.RG2,which shows nodetectable p300-binding activity,
also showsnodetectable immortalization activity. The bio-logical phenotype of thesemutants correlates more closely with theirp300 bindingdeficiencies than to partial reduction
inpRBbindingbecause 12SHN3,atleast, hasnodiscernible
defect in its region 2 associations (Fig. 4). The 12S.DA21
mutation, although relatively close to important
p300-bind-ing residues, shows no disruption of p300 binding and maintains consistently high levels of immortalization
activ-ity, on average, more than 60% of 12S.WT activity. 12S.LP49 and12S.EV55, which also show little disruption of protein associations, show essentially no loss of immortal-ization function.
Notably, the 12S.YH47 and 12S.YG47 mutations have
only a mild effect on the BRK immortalization function,
eventhough they clearly impair the pRB binding pattern in HeLa cells. Their protein association pattern appears as defective for pRBbinding as that of 12S.928 (Fig. 7), yet in clear contrast to the loss of immortalization function in 12S.928(includedin Table 1 for comparison), they retain as much as60% ofwild-type activity.
Protein association in BRK cells. We have previously assessed the cellular protein associations of a variety ofElA mutants in both HeLa and BRK cells (49, 66, 77, 80) and foundnosignificantdifferencesdependent on host cell type.
Nevertheless, because the biological activity of the 12S.YH47 and 12S.YG47 mutants is so striking in BRK
cells, we assessed the ability of these mutant products to associate with pRB specifically in these cells. While the
protein association pattern of most of the single-residue
mutantproductsdescribedhereis similar in BRK andHeLa cells (Fig. 8A), thepattern of the 12S.YH47 and12S.YG47
mutant products is notably different; in BRKcells, these mutantsappearreadilyable tobindpRB (lanes7 and8). The 12S.928product (Fig.8A, lane 11) remains defective for pRB binding as shown previously (77). Inasmuch aspRB phos-phorylation is cell cycleregulated, andonlythe
underphos-phorylated forms are generally thought to have
growth-suppressing activity (reviewed in reference78),theabilityof the 12S.YH47 and 12S.YG47productstobindpRB in these conditions may indicateaselective loss of theabilitytobind
morehighly phosphorylatedpRBforms,forms that would be
expected to predominate in the veryrapidly cycling HeLa
cells. At the relatively early time after infection (10 h) at
which the lysates in Fig. 8Awereharvested, the RB prod-ucts, especially in the mutant-infected cells, are still
rela-tively underphosphorylated, as evidenced by the relative
absence ofmultiplepRB bands inpRB-specific
immunopre-cipitations (Fig. 8B). At later times afterinfection, the RB
products are largely phosphorylated and show multiple bands inpRB-specific immunoprecipitations(Fig. 8C,lanes
19 to 21). Atthistime, the 12S.YH47 and 12S.YG47
prod-ucts still show pRB binding, although with an apparent preference for underphosphorylated forms(Fig.8C, lanes 14 and 15). However, this result must be considered with
caution, as even 12S.WT showsan apparentpreferencefor
underphosphorylatedforms (Fig. 8C,lane 13).
At early times postinfection in BRK cells, the 12S.928
product shows little abilitytobind p107 (Fig. 8A, lane 11).
However, atlater times thisability is readily apparent(Fig. 8C, lane16). 12S.YH47 and 12S.YG47 bind p107 readilyin
BRKcellsatbothearly and late times(Fig. 8A, lanes 7 and
8;Fig. 8C,lanes 14 and15). Despitetheabilityof12S.YH47
and 12S.YG47 to associate with pRB in BRK cells, the
phenotype of the 12S.YH47/928 construct is the same in
BRKasin HeLa cells: it is severely deficient in both pRB
andp107binding (Fig. 8C, lane17).Thus, even inBRKcells,
theTyr-47residue appearstoplayatleastastrongstabilizing
role in theregion 2 associations. It appears, moreover, that
p107 association is notcompletely independent ofregion 1
function;it mayonly beindependent of region 1ifregion2
remains unaffected.
on November 9, 2019 by guest
http://jvi.asm.org/
CELLULAR PROTEIN ASSOCIATIONS AT THE ElA N TERMINUS
A C
8-10HOURS 30-32HOURS
0 l- r-cmo 0 0( 00r L<0 0
E A xJCN OJj(NJOJ\U N N
_M
-.; n agoW m -p300-p107
= p1O5RB
68-45-i
_
_M u _4 EIAXE ; x:,
C\.;
Ct
2 0
92 N
20C --
!
92-68--'
45- u
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
B
IT It Go
~: 00
I\ C\ \I_ iC N N °9
RB
FIG. 8. ElA association with cellularproducts in BRK cells. Coprecipitation was assayed byimmunoprecipitating Tran-35S-labeled infected BRKcelllysateswith ElA-specific monoclonalantibody M73 (31) (AandC)orpRB-specific antibodyXZ133 (39) (B and C). Cells were labeled from either 8 to 10 h (A and B) or30to 32 h (C) after infection. The positions of p300, the ElA proteins, and the region
2-associatedproteinsareindicatedattheright of each panel. The positions of molecular size markers (in kilodaltons)areindicatedatthe left.
Induction of DNA synthesis. One of the most intriguing aspects of ElA biological functions is that ElA-mediated inductionof cellular DNAsynthesiscanbe dissociated from the immortalization function (38, 52, 77, 84). Both the p300-linked and thepRB-linked active sites of12S.WTare required to induce extended proliferation in primary cells,
but either site alone is sufficient for the induction of cellular DNAsynthesis. Theability toinduce expression of theG1 productsnecessarytosupportDNAsynthesis is therefore a redundant function in the ElA products. The new point mutations described here confirm andextend this analysis.
Each of the single-residue mutations is highly effective in
inducing cellular DNA synthesis. This includes 12S.RG2 (Fig. 9A), despite its severe immortalization and p300-bindingdefects. The kinetics of inductionareslightlyslower for 12S.RG2 than for 12S.WT (Fig. 9B); a similar delayed induction phenotype was noted previously in region 2 mu-tantssuchas 12S.928(77, 84).
An important question in considering the phenotype of
region 2mutations such as pm928is whetherundetectable
residual pRB bindingvia region 1 couldbe contributing to
theproficiency in induction of DNA synthesis. Strong evi-dence againstthis possibilityisprovided bythe 12S.YH47/
928 construct. Although 12S.YH47/928 encodes mutations
seriously affectingbothpRB-binding regions andbindingof all the knownregion2-associatedproteins, it isasproficient ininducing DNAsynthesis asis 12S.928 (Fig. 9A), strongly supportingthe conclusionthatbindingofpRB and theother
region 2-associated proteins is dispensable for ElA-medi-ated induction of DNA synthesis. In clear contrast to the abilities ofindividualN-terminal andregion2pointmutants toinduce DNAsynthesis, adoublepointmutant, 12S.RG2/
928, combiningpmRG2withpm928, virtually abrogates this activity (Fig. 9A). This construct demonstrates with great sensitivity the existence inthe ElAproducts of dual
mech-anisms for interveningin cellcycle controlpathways.
A. B.
04
2
;0- 12S.WT
12S.RG2
10
-O
-MOCK
81012 16 20 24 32 40 hpi
p300 + - + _ +
p105-RB + _
FIG. 9. Induction of DNA synthesis in ElA mutant virus-in-fected primaryBRKcells. Primary BRK cells wereinfected at a
multiplicityof 10 PFUpercell. DNAsynthesiswasmeasuredasthe
incorporationof[3H]thymidine. (A)Infected cellswerelabeled with
[3H]thymidinefor 1 hat47to48 hpostinfection (hpi). Incorporation during this period in each infection is indicated as trichloroacetic
acid-precipitablecountsperminute.(B)Mock-12S.WT-,or
12S.RG2-infected cellswerepulsedwith [3H]thymidinefor 2 hatsuccessive intervals endingat thetimespostinfection indicatedon thex axis.
Incorporation duringeach interval ineach infection is indicatedon
theyaxisastrichloroaceticacid-precipitablecountsperminute. 200
-92
--p30C
-plO7 ] pIO5RB
E1A
VOL.67, 1993 483
6
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.152.474.72.344.2] [image:8.612.318.557.471.620.2]484 WANG ET AL.
p300 p!05RB
K
1'bir pO7-cyclinA-p33
r
lp130
N 1 2
I
_
pHIV.LTN-CTAT) l~~~~~
0 U) UO U)C U)
ZI N o No
> - A
*8 -Ac-
~~~CmC
--Cm
[image:9.612.94.259.72.260.2]2 3 4 5 6 7 8 9
FIG. 10. Enhancerrepressionfunction. The activity of the HIV
LTR enhancer is indicated by the ability ofareportergeneproduct toconvertchloramphenicol (Cm) toits acetylated forms (Ac-Cm).
Inthree independent experiments including theoneillustrated here,
the average CATactivity measured after cotransfection with 12S ElA mutant plasmids relative to activity in the absence of ElA
expression was0.25, 0.71, 0.72, 0.30, 0.28, and 0.33 for 12S.WT,
12S.RG2, 12S.HN3, 12S.LS20, 12S.DA21, and 12S.YH47/928,
re-spectively. Theaveragestandard deviationwas0.07.The
approxi-matelocationsof the single pointmutations areindicated by dots in
theupperbar diagram.
Enhancerrepression.Ithas been knownforsometimethat
ElArepresses theactivity ofavarietyofviral and cellular
enhancers (5, 18, 33, 34, 64, 67, 73, 74, 76). While some
studies have indicated a link between enhancer repression
andregion2function(47),moststudies have found that this
activitymapsprimarilytotheN-terminal active site(41,44, 60, 61,64, 66,75).Weused the pointmutations generated in
this study to examine the link between p300 binding and repressionof the enhanceractivity of the HIV LTR. Expres-sion was assayed with an HIV LTR reporter construct
drivingexpression of thebacterial CATgene (Fig. 10).
HIV LTR-driven CAT expression in HeLa cells is largely dependent on the presence in the LTR of two sequence
motifsclosely relatedtothe binding site forNF-KB(45). In theabsenceof thesesites(pNFA-CAT), the level of
expres-sion isverylow (Fig. 10, lane 1versuslane 2).
Cotransfec-tionof a 12S.WT expression plasmid, pl2S.WT, represses
expression of the HIV LTR CAT construct by about 75% (lane 4). Each of the mutant plasmids, pl2S.RG2 and
pl2S.HN3,encoding substitutionsofthe positivelycharged
N-terminal residues is severely impaired for the enhancer
repression function (Fig. 10, lanes 5 and 6). In contrast,
pl2S.LS20 and pl2S.DA21, affected at nearby residues,
repress almostaswell aswild type (Fig. 10, lanes 7 and 8).
Thedouble pointmutant 12S.YH47/928 retainsa
near-wild-type levelof repression activity (Fig. 10, lane 9), indicating
thatbindingof the region2-associatedsetofcellularproteins
is completely dispensable for repression of the HIV en-hancer. It is notable that pl2S.LS20 retains considerable
enhancer repression activity compared with pl2S.HN3,
whenin otherwaystheir phenotypes are moresimilar: they
both bind reduced levels of p300 and they both show
seriouslyreduced immortalization activity.
DISCUSSION
The work described here refines in several ways our
understanding
of therelationship
between ElAproteinse-quence
and the formation of its active sites. While theextreme N-terminal
ElAsequences
show relatively littleconservation
incomparison
withpreviously
recognizedcon-served
regions designated
regions
1,
2,
and 3,it is clear fromthe data described here that the function localizing to the
N-terminal
region
isdependent
onspecific
sequences.Sub-stitution with a
heterologous
N-terminal sequence of equallength
does not result in the maintenance of N-terminalfunction,
and certainsingle
amino acid substitutions greatly impair activity.The data described here
alsoimply
thatp300
and theregion
2-associated set of cellularproteins
do not bind toshared
oroverlapping
sequences
within conservedregion 1.The monoclonal
antibody
competition
experiments
inpar-ticular
support
thishypothesis.
Since immunoglobulinmol-ecules
bound to the ElAproteins
can be expected to causean
appreciable degree
of steric hindrance, the ability of themonoclonal
antibodies that bind within conservedregion 1 toblock
binding
ofonly
one class or the other ofE1A-associated
proteins argues
strongly
that the twoclasses bindto
separate
residues within
conservedregion
1. Indeed,conserved
region
1 isprobably
more accurately described astwo
nearly
contiguous
subregions
with independentfunc-tions. Determination of the
precise
boundary between
thesesubregions
willrequire
furtheranalysis,
butgiventhestrik-ing specificity
of the monoclonalantibody
exclusion, it ispossible
that residues toward the
middle of conservedregion1 are not essential
for eitherbinding
function. Theseresultssuggest
asimple
outline of ElA structure such as thatproposed
in
Fig.
11. Thebasic feature of this
model is thesuggestion
that conserved
region
1comprises
two distinctsubregions
thatparticipate
in the formation of two activesites.
The
single-residue
substitution mutations described
hereconstitute
important
new
reagents
forprobing
the roles ofElA-targeted
cellular
proteins.
It is
already
clear
thatthe useof
N-terminal-specific
and
region
2-specific
mutations todisrupt
ElA
active sites has distinct advantages
overthe useof conserved
region
1 versus conserved
region
2mutations.The
12S.RG2/928
double substitution mutant is a particularly
elegant example
of the
precision
with which both
ElA activesites
can be inactivated. In
addition,
the 12S.YH47/928
mutation
is more
precisely
disruptive
than any
region 2mutation
studied
previously.
Together,
12S.RG2,
12S.928,
12S.RG2/928,
and
12S.YH47/928 represent
the most
preciseset of
inactivating
mutations so far available in
ElA.Collec-tively,
these mutants
represent
each of the distinguishable
phenotypes
so far
generated.
12S.RG2 abrogates
p300
bind-ing selectively.
12S.928
abrogates
pRB
binding
selectively
(p107 binding
is
relatively
unaffected,
while
p130
bindingisintermediate;
see also reference
25).
12S.RG2/928
abrogates
all known associations
dependent
on exon 1 of
12S,
while12S.YH47/928
abrogates
binding
of the entire
region
2-asso-ciated set without
disrupting
p300
binding.The
12S.HN3 and
12S.LS20
mutations show
an
interest-ing
distinction in ElA function. Both these
mutants
showreduced
p300
binding
and marked defects in immortalization
activity.
Nevertheless,
12S.LS20
retains
much of the
E1A-mediated
enhancer
repression
function,
while
12S.HN3
is asdefective
in
this
activity
as 12S.RG2.
Interestingly,
Subra-manian et al.
(69)
reported
a linker insertion
mutation
substituting
the
sequence
Arg-Ile-Arg
for Leu-19
thathas aon November 9, 2019 by guest
http://jvi.asm.org/
25 37 80 121 139
|
NM
la l 1b1 2 VI60 /55
'100
FIG. 11. Schematic model of cellular protein-bindingsites inElA
exon1. The 139 residues of the exon 1 sequences common to the12S and13Sproducts, sufficient forElA-induced proliferation of primary BRKcells,arerepresentedasindividual circles in the above diagram. Residues 81 to 120 are shaded gray to indicate that they are not essentialforElA biological activityorprotein binding functions (66) andarerepresentedas aloopbecause the ability to remove them all in a single deletion indicates that they do not contribute essential spacingbetweenconservedregions 1and 2. The region from residue 26to35canalsoberemoved as a single deletion without impairing ElA function(41),eventhough residuesoneither side of it combine
toformabindingsite forp300.Thisregion, therefore, is also depicted
as a loop and shaded gray (residue 35 is partially hidden behind residues 24 and 25 in this diagram). The unshaded regions in this diagram correspond to the unshaded regions in the bar diagrams shown here and inFig. 1. Conserved region2extends from residues 121 to 139. The (D)-L-X-C-X-E motif represented in Ad5ElA by residues121to126appearstobe adirecttarget forbinding by pRB,
p107,andprobablyp130(15, 17,25, 79).The conserved residues in
thismotif arehighlighted by internal dots in the model above. The remainder of conserved region 2 constitutes a casein kinase II substraterecognitionsitemotifseparatedfrom the pRB-binding site bytwoadjacentprolineresidues. Thesignificanceof the casein kinase II motif to ElA function is not known, but this motif apparently contributestothe function of the HPV E7 transforming gene product (3),whichcontains a direct homolog ofElAregion 2. Conserved region1is drawnasareversedlooptorepresent the genetic evidence implyingthattheupstreamhalfof conserved region 1 contributes to formationofthepRB-bindingsitewhile the downstream half
contrib-utes toformation of the p300-binding site. (Residue 79 is partially concealedbyresidue37 in thisdiagram.) Residues21, 49, and 55 are shaded graytoindicate that nonconservative substitutions at these positions have little or no effect on ElA biological function or protein-bindingactivity. Residues 2, 3, and 20 are highlighted with internaldotstoindicate their apparent involvement inp300binding. Interestingly,computeranalysissupports some features of this sche-matic outline. The Hopp and Woods (35) method predicts three strong hydrophilic peaks in the ElA 12S product, occurring at residues133to138, 55to60,and 100to105;thelattertwosites are strikinglysimilartothepeaksof thesimpleloops postulated from the genetic data. Inaddition, an interestingpatternemerges when the EIA proline distribution is superimposed onthis model. TheElA
productsareproline-rich;within thefirst139 residues, prolines occur
atpositions 33, 34,35, 40, 41, 54, 57, 67, 84,85, 87, 90, 92, 93, 99, 102, 109, 113, 117, 130, and 131. Fully half of these fall within the hypothetical loop postulated to extend from about residue 80 to residue 120 (on average, every fourth residue in this stretch is a proline).Sinceprolineresiduesareoften associated with the presence
of 3-turnsinproteinmolecules(10), this distributionmightindicate
thatthisregionformsatightlytwistedstructure.
biological phenotype very similar to that of the 12S.LS20
mutation described here. Their mutation was severely de-fective in a virus immortalization assay, but close to wild
type in itsabilityto repressenhanceractivity in CATassays
of the SV40 and immunoglobulin H enhancers;
p300
binding was not assessed. It is possible that 12S.LS20 retains the enhancer repression function to a greater extent than12S.HN3because ofaslightlygreater ability totitrate
p300,
butthe widedifferenceintheir enhancer repression activities compared with their more similar
p300
binding and virus immortalization activities raises the possibility that the dif-ference in their biological activities may be qualitative,rather than only quantitative. The difference between 12S.LS20 and 12S.HN3 may lie, not in the amount, but in thewaybywhichp300 is boundtothe altered ElAproducts;
for example, one mutant may have a more significanteffect than another on further p300 protein-protein associations. Resolving the role of p300 in the enhancer repression and immortalization functions will require much further charac-terization of p300, and substitution mutants such as 12S.HN3 and12S.LS20maybe particularly useful in distin-guishing the individual steps in these pathways.
The involvement ofa tyrosine residue in pRB binding to
ElA has interesting correlations in HPV E7. The HPV E7
proteins, which contain a direct homolog ofElA region 2,
also encode a region strikingly similar to the pRB-binding portion of conserved region 1 (57), including a tyrosine in HPV type 16 (HPV16) E7. Moreover, of the other three
tyrosine residues occurring among the 98 residues of the
HPV16 E7 product, two occur within the pRB-binding region homologous to ElA region 2. They are presumably
notessential for pRBbinding since they donot occurinAd5 ElA region 2, yet it isnotable that position 125 in theAd5
ElA (D)-121-L-122-X-123-C-124-X-125-E-126 motif,while
not completely conserved, is a tyrosine in almost every serotypicallydistinct ElAgene sequenced(sequences com-piled in references 1 and 43). Together, these data suggest
that tyrosine residuesmayplaya significantstabilizingrole
in pRB-binding proteins. This may have clinical relevance
since it appears that the oncogenicity ofHPVs correlates
with the stability of pRB binding to the E7 products (3, 23,
54).
The 12S.YH47 and 12S.YH47/928 mutants also reveal novel aspects ofp107 interaction with theElAproducts. It has previously been thought that conserved region 1 may not be a binding site for p107 since it is not required forp107
association. However, it appearsfrom the data reported here
that p107 binding is stabilizedbyspecificinteractionsinboth
regions2 and 1;p107association may only be independentof
region 1 if region 2 remains unaffected. Conversely, p107
binding can also bemaintainedin the absence of the region
2 cysteine residue, as evidenced by the 12S.928 mutant, even though this residue is critical for pRB association. p107
association with the 12S.928 mutant products shows an interesting kinetic aspect in BRK cells (in which ElA
expression induces synchronous progression through the cellcycle). Whilep107 is bound by the 12S.WT products as soon as they are expressed, the12S.928products bindp107 only after the cell cycle has advanced throughG1.Asthere
is a strongly cell-cycle-regulated component of E1A-p1O7 complexes, i.e., cyclin A (19, 21, 37), these kinetics may reflect an involvement of cyclin A in stabilizingp107 asso-ciation with the ElA products.
The 12S.YH47 and 12S.YG47 mutants also reveal new details of pRB interaction with the ElA products. For the
first time, a marked difference in the protein association
485
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.59.301.62.280.2]486 WANG ET AL.
pattern ofanElAmutant has appearedbetween HeLa and
BRK cells. In particular, pRB binding to the 12S.YH47 products is readily detectable in BRK cells, but sharply
impairedin HeLacells. This mayreflect a selective loss of the ability to bind phosphorylated pRB forms. However, evenwhen pRB is in ahighlyphosphorylated state inBRK
cells,themutantresidue47productsstill showpRBbinding, incontrast to their severe defect in HeLa cells. Thus, the
differencemaybemorerelatedtocelltypethantothe degree of pRB phosphorylation.Wearecontinuing studies aimedat
further defining the role of the Tyr-47 residue in binding
pRB.
ACKNOWLEDGMENTS
WethankFrancoiseChadrin, ReginaWhittaker, and Lisa Bianco forexcellent technical assistance; Jim Duffy, Phil Renna, and Mike Okler forpreparingthe photographsandillustrations; Arnold
Rab-son for supplyingthe HIV LTR-CAT constructs; and Ed Harlow
andQianjinHufor gifts ofmonoclonal antibodies.
This workwas supported by PublicHealth Service grants CA-55330and CA-53592 fromtheNational Institutes of Health.
REFERENCES
1. Allard,A., and G. Wadell. 1988. Physical organization of the
enteric adenovirus type 41 earlyregion1A. Virology
164:220-229.
2. Bandara, L. R., J. P. Adamczewski, T. Hunt, and N. B.La
Thangue. 1991. CyclinA andtheretinoblastomagene product
complexwitha commontranscription factor. Nature(London)
352:249-251.
3. Barbosa, M. S., C. Edmonds, C. Fisher, J. T. Schiller, D. R. Lowy, and K. H. Vousden. 1990. The region of the HPV E7
oncoproteinhomologous toadenovirusElAandSV40large T
antigen contains separate domains for Rb binding and casein
kinase IIphosphorylation.EMBO J. 9:153-160.
4. Berk, A. J.,F.Lee,T. Harrison, J.Williams, and P. A. Sharp.
1979. Pre-earlyadenovirus5 geneproductregulatessynthesis of early viralmessengerRNAs. Cell17:935-944.
5. Borrelli, E.,R.Hen, and P. Chambon. 1984.Adenovirus-2 ElA
productsrepressenhancer-induced stimulation of transcription. Nature(London) 312:608-612.
6. Boulanger, P. A., and G. E. Blair. 1991. Expression and
interactions of human adenovirus oncoproteins. Biochem. J.
275:281-299.
7. Cao,L., B. Faha, M. Dembski, L.-H. Tsai, E. Harlow, and N. Dyson. 1992. Independent bindingof theretinoblastoma protein
and p107 to the transcription factor E2F. Nature (London)
355:176-179.
8. Carlock,L. R., and N. C.Jones.1981.Transformation-defective mutant of adenovirus type5 containing a single altered ElA
mRNA species.J. Virol. 40:657-664.
9. Chellappan,S.P.,S.Herbert,M. Mudryj,J. R. Horowitz, and J. R. Nevins. 1991. The E2F transcription factor is a cellular targetforthe RB protein. Cell65:1053-1061.
10. Chou, P. Y., and G. D. Fasman. 1979. Prediction of a-turns.
Biophys.J.26:367-384.
11. Chow, L. T., T. R. Broker, and J. B. Lewis. 1979. Complex
splicingpatternsof RNAs from the early regions of adenovirus 2. J. Mol. Biol. 134:265-303.
12. DeCaprio, J. A., J. W. Ludlow, J. Figge, J.-Y. Shew, C.-M.
Huang,W.-H. Lee,E. Marsilio, E. Paucha, and D. M. Living-ston.1988.SV40largeT antigen forms aspecific complex with the product of the retinoblastoma susceptibility gene. Cell
54:275-283.
13. DeFeo-Jones,D., P.S.Huang, R. E. Jones, K. M. Haskell, G.A.
Vuocolo, M. G. Hanobik, H. E. Huber, and A. Oliff. 1991. CloningofcDNAs for cellularproteins thatbindto the retino-blastomageneproduct. Nature (London)352:251-254.
14. Devoto, S.H., M.Mudryj, J.Pines, T. Hunter, and J. R. Nevins.
1992. A cyclinA-protein kinase complex possesses
sequence-specific DNA binding activity: p33 2 is a component ofthe
E2F-cyclin A complex. Cell 68:167-176.
15. Dyson, N., P. Guida, C. McCall, and E. Harlow. 1992. Adeno-virusElAmakes two distinct contacts with the retinoblastoma protein. J. Virol. 66:4606-4611.
16. Dyson, N., P. Howley, K. Munger, and E. Harlow. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934-937.
17. Egan, C., T. N.Jelsma,J. A. Howe, S. T. Bayley, B. Ferguson, and P. E. Branton. 1988. Mapping of cellular protein-binding sites on the products of early-region1Aof human adenovirus type 5. Mol. Cell. Biol. 8:3955-3959.
18. Enkemann, S. A., S. F. Konieczny, and E. J.Taparowsky. 1990. Adenovirus 5 ElA represses muscle-specific enhancers and inhibits expression of the myogenic regulatory factor genes,
MyoDI and Myogenin. Cell GrowthDiff. 1:375-382.
19. Ewen, M. E., B. Faha, E. Harlow, and D. M. Livingston. 1992. Interaction of p107 with cyclin A independent of complex formation with viral oncoproteins. Science 255:85-87. 20. Ewen, M. E., Y. Xing, J. B.Lawrence, and D. M. Livingston.
1991. Molecular cloning, chromosomal mapping, and expres-sion of the cDNA for p107, a retinoblastoma gene product-related protein. Cell 66:1155-1164.
21. Faha, B., M. E. Ewen, L.-H. Tsai, D. M. Livingston, and E. Harlow. 1992. Interaction between human cyclin A and adeno-virus ElA-associatedp107protein. Science 255:87-90. 22. Figge, J., T. Webster, T. F. Smith, and E. Paucha. 1988.
Prediction of similar transforming regions in simian virus 40 large T, adenovirus ElA, and myc oncoproteins. J. Virol. 62:1814-1818.
23. Gage, J. R., C. Meyers, and F. 0. Wettstein. 1990. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16differin retinoblastoma protein binding and other properties. J. Virol. 64:723-730. 24. Gendelman, H. E., W. Phelps, L. Feigenbaum, J. M. Ostrove, A.
Adachi, P. M. Howley, G.Khoury,H. S. Ginsberg, and M. A. Martin. 1986. Trans-activation of the human immunodeficiency virus long terminal repeat sequence by DNA viruses. Proc. Natl. Acad. Sci. USA 83:9759-9763.
25. Giordano, A., C. McCall, P. Whyte, and B. R. Franza. 1991. Human cyclin A and the retinoblasoma protein interact with similar but distinguishable sequences in the adenovirus ElA gene product. Oncogene 6:481-486.
26. Glenn, G. M., and R. P. Ricciardi. 1985. Adenovirus 5 early region 1A host range mutants hr3, hr4, and hr5 contain point mutations which generate single amino acid substitutions. J. Virol. 56:66-74.
27. Gluzman, Y. 1981.SV40-transformed simian cells support the replication of earlySV40 mutants. Cell 23:175-182.
28. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant genomes which express chloramphenicol acetyl-transferase in mammalian cells. Mol. Cell. Biol. 2:1044-1051. 29. Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977.
Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59-72.
30. Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of human adenovirus5 DNA. Virology 52:456-467.
31. Harlow, E., B. R. Franza, Jr., and C. Schley. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region1A products. J. Virol. 55:533-546.
32. Harlow, E., P. Whyte, B. R. Franza, Jr., and C. Schley. 1986. Association of adenovirus early region 1 proteins with cellular polypeptides. Mol. Cell. Biol. 6:1579-1589.
32a.Hearing, P. Unpublished data.
33. Hen, R., E. Borrelli,and P. Chambon. 1985. Repression of the immunoglobulin heavy chain enhancer by the adenovirus-2ElA
products. Science 230:1391-1394.
34. Hen, R., E. Borrelli, C. Fromental, P. Sassone-Corsi, and P. Chambon. 1986. A mutated polyoma virus enhancer which is active in undifferentiated embryonal carcinoma cells is not repressed by adenovirus-2 ElA products. Nature (London) 321:249-251.




![FIG. 9.fectedmultiplicityincorporationtheduringinfectedintervalsIncorporationacid-precipitable[3H]thymidine Induction of DNA synthesis in ElA mutant virus-in- primary BRK cells](https://thumb-us.123doks.com/thumbv2/123dok_us/1304016.83580/8.612.152.474.72.344/fectedmultiplicityincorporationtheduringinfectedintervalsincorporationacid-precipitable-thymidine-induction-synthesis-mutant-virus-primary.webp)


