Copyright © 1986, American Society for Microbiology
Efficient and Accurate In Vitro Processing of Simian Virus
40-Associated Small RNA
NISSIMHAY,' ORNA AMSTER-CHODER,' AND YOSEFALONI12*
DepartmentofGenetics, Weizmann Instituteof Science, Rehovot 76100,
Israel,'*
andDepartmentof Molecular Biology,Princeton University, Princeton, NewJersey 085442 Received 6 March1985/Accepted3September1985
Nucleiwere isolated from simian virus 40(SV40)-infected cells witha hypotonic, detergent-free bufferand incubated in vitro in a high-ionic-strength buffer containing [a-32P]UTP. Thelabeled viral RNAs produced wereanalyzed by gel electrophoresis together with 3-h-labeled viralRNAsextractedfromSV40-infectedcells. The invitro-synthesizedRNAcontainedamajorRNAspecies of 62to64 nucleotides thatappearedonthe gel
at the same position as in vivo-synthesized SV40-associated small RNA (SAS-RNA). Analyses of the in vitro-synthesized 62- to 64-nucleotide RNA by hybridization to restriction fragments and by the use ofan
SAS-RNA deletion mutant clearly identified it as SAS-RNA. The intensity of the band of the in vitro-synthesized SAS-RNA increasedwithanincreasein thelabeling timeorwhenashortpulsewasfollowed bya chase. Moreover, the SAS-RNA band disappeared when ITP replaced GTP in the transcription reaction mixture. These results indicate that SAS-RNA is processed from a precursor molecule and that an RNA secondary structurecould bean elementrecognized bythe processingenzyme.
After
simian virus
40 (SV40)infection,
RNApolymerase
II transcribes both SV40 mRNAs and 62- to 64-nucleotide (nt) RNA, known as SV40-associated small RNA (SAS-RNA)
(6-8).
It was suggested that SAS-RNAis
specifically
processed from the noncoding sequence in long RNA
tran-scripts which
areinitiated atnormal late promoters (6). TheSAS-RNA-coding
region is located about 170 nt's down-streamfrom
thepolyadenylation site (0.17 map unit) of the late mRNAs (see Fig. 1A for its map location). Thus, the processing reactions which theSV40
primary late RNAtranscripts undergo yield
two RNAspecies: (i)
maturepoly(A)+
mRNAsand(ii) SAS-RNA.
SAS-RNA hasunique
uncapped 5' and poly(A)- 3' ends (6), so
its
processing
reaction
musttherefore
behighly specific.
To
directly address
thequestions of whether SAS-RNA is
a
processing
product and,if
so,how theprocessing
enzymesrecognize
theSV40
primary late RNA substrates andaccom-plish
suchprecise maturation,
anin vitroprocessing
system mustbedeveloped.
For this
purpose, weprepared
anisolated-nucleus
system from SV40-infected cells. In vitroincubation
of the nuclei in alow-ionic-strength
buffer leads to the prematuretermina-tion of
SV40 latetranscripts
andtothesynthesis of
a94-to 98-nt attenuator RNA (seeFig.
1Afor its
maplocation)
(1, 4,5, 13-15, 20, 22, 23); incubation
in ahigh-ionic-strength
buffer
leadstothesynthesis of long
viral RNAtranscripts (5,
13-15, 20).
In the present communication, we show that when the nucleiare
incubated
inahigh-ionic-strength
buffer foralong
timeorwhentheyare
pulse-labeled
with[a-32P]UTP
and the label is then chased with a highconcentration
of UTP, efficient andaccurateprocessing
of SAS-RNA can occur.BSC-1
cells were infected withSV40
(30 to 50 PFU percell)
aspreviously described
(18).At 42 to48 hpostinfection,
thecellswerewashed andcollected in cold
hypotonic
buffer(50
mMTrishydrochloride
[pH7.9],
1.5mM
MgC92,
1 mMdithiothreitol)
bycentrifugation for
1 min at 1,000 x g.Nuclei
wereisolated bysuspending 5 x107
cellsin 10 ml of*Correspondingauthor.
hypotonic
buffer (17) andpipetting
them up and down 10 times withaPasteurpipette, followed by centrifugation for
2 min at 1,000 x g. This step wasrepeated twice. The nuclei(approximately
3 x107)
weresuspended
in atranscription
reaction
mixturecontaining
5 mMKCl,
1.5 mMMnCl2,
1 mMdithiothreitol,
12.5% glycerol,
100 mM(NH4)2SO4,
30 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES)-NaOH (pH 7.9), a400 ,uMconcentration
eachof
ATP,
GTP,
andCTP,
S to 20 ,uM UTP (asindicated
in the figure legends), and 300 to 500,uCi of
[a-32P]UTP
(400mCi/mmol; Radiochemical Centre, Amersham, England) in
a finalvolume of 0.6 ml.Transcription
wascarried
out at30°C
for various
times,
asindicated in
thefigure legends,
andstopped by
theaddition
of
100 ,ugof RNase-free
DNase(Worthington
Diagnostics, Freehold, N.J.)
perml for
1min at30°C, followed by extraction with
phenol-chloroform (19).
The aqueous
fraction
waspassed through
aSephadex G-25
syringe
column to removefree
labelednucleotides. After
ethanol
precipitation,
theprecipitate
wassuspended
in TKM (25mMKCl,
2.5mMMgCl2,
50 mMTrishydrochloride [pH
6.7]), 200 ,ug of RNase-free DNase per mlwas
added,
andincubation
was carried outfor
1 h at4°C.
The DNase treatment wasstopped by
phenol-chloroform extraction,
and the RNAwasprecipitated with ethanol.
SV40-specific
RNA wasisolated by
hybridization
toandelution from
SV40
DNA on nitrocellulose filters.Hybridization
was carried out in70% formamide-0.3
MNaCl-10
mMHEPES-NaOH
(pH
7.5)-1
mM EDTA-0.1% sodium dodecyl sulfate at37°C
for 40to48 h. At the end of the incubation
time,
thefilters
werewashed once with 0.5x SSC (lx SSC is 0.15 MNaCl plus 0.015Msodium
citrate),
incubated inasolution of35%
formamide-0.01 M NaCl-1 mM EDTA-10 mM HEPES-NaOH(pH 7.5)at
37°C
for 30 min and then washed three times with 0.5x SSC. The bound RNAwaselutedby
incubating
thefilter inasolution of90% formamide-10
mM HEPES-NaOH(pH 7.5) for
1 hat37°C.
For thepreparation
of
in vivo-labeledRNA,
infected cellswere labeledfor
3 h with32P,
(5mCi/107 cells)
inphosphate-free
medium. Nu-clear andcytoplasmic
fractionswereprepared by
using
thehypotonic
bufferprocedure
described above.32P-labeled
402
on November 10, 2019 by guest
http://jvi.asm.org/
.; ;.
A
B4
e
72:
;(2844)
i
-122EARLY
s
(2905)
-90
059~~ ~ ~ ~ ~ ~ ~ C9
-76
\37 67
FIG. 1. Genomic map ofSV40 (A) and identification of SAS-RNAproduced inisolated nuclei by size analysis (B). (A) The map or indicates thepositionsof the primary late RNA transcript (we have 4 1-
cNJ
suggestedthat it terminates after transcription around the genome atE
-S 3 n the attenuation siteat0.74 mapunit[13])and thespliced early and O late mRNAs(inner arrows). The map position of attenuator RNA (Att. RNA)is indicated between 0.72 and 0.74 map unit. The map position of SAS-RNA is indicated between residues 2844 and 2905
(25)
in thepolarity
ofthe late mRNAs. athrough
e, Restriction fragments obtained by cleaving the viral DNA with HpaI, EcoRI, andBglI restriction enzymes (3). Ori, origin of replication. (B) For in vivoexperiments, viralRNAwasextracted and purified fromtheii
*cytoplasmic fraction of infected cells labeled for 3 hwith
32p;.
For in vitroexperiments, nuclei isolated from infected cells were incubated in atranscription reaction mixture containing 300 ,uCi of[a-32P]UTP
(400 mCi/mmol)
and 6 ,uM UTP for 6 min. Unlabeled UTPwasthenaddedtoafinal concentration of 400,uM,andincubationat30°Cwas
190- continued foranadditional 30min. ThelabeledRNAproducedwas
180- purified. The in vivo- and in vitro-synthesized RNAs were analyzed
160- by gel electrophoresis. The arrowhead points to the position of
147- SAS-RNA. m, Size markers obtained bylabelingHpaII restriction
fragments of pBR322 DNA.
122- B
110-
B->D
*FIG. 2. Sizeanalysis of viral RNAs produced in isolated nuclei 9o- a - infected with either thewild-type (WT)ord12194strain ofSV40 (A) andhybridizationof the 62-to64-ntRNAwith restriction fragments 76 - ofSV40
(B).
(A)Nuclei isolated from cells infected witheitherthe76- C - e WT or
d12194
strain of SV40 were incubated in a transcriptiond-
reaction mixturecontaining
500,uCi
of[a-32P]UTP
(400
mCi/mmol)
67- d and 20 ,uM UTP for 20min at 30°C. Thelabeled RNAs produced
werepurified and analyzed by gel electrophoresis. The open arrow-head points to the position of attenuator RNA, and the filled e - arrowheadpointstotheposition of SAS-RNA. m, Size markers as inFig. 1B. (B)The RNA atthe position of SAS-RNAwas eluted from the gel and hybridized to a blot containing five restriction fragments (as indicatedin Fig. 1A).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.143.468.71.404.2]viral RNA was extracted from the cytoplasmic fraction (19), and viral RNA was purified by the hybridization-elution procedure described above for nuclear RNA.
Foranalysisof the RNA by gel electrophoresis, the RNA was denatured in 5 ,ul of 90%
formamide-10
mM HEPES-NaOH (pH 7.5) for 1 min at 90°C and subjected toelectrophoresis
on an acrylamide gel (bis/acrylamide ratio, 1:29) containing 7 M urea in TBE. Electrophoresis was carried out at a constant current of 20 mA for 4 to 5 h.Figure 1B shows that the RNA synthesized in vitro
contained
a major RNA species that appeared on the gel at the same position as the SAS-RNA synthesized in vivo (arrowhead) (6), suggesting accurate in vitro synthesis ofSAS-RNA.
Itis worth noting that in comparison with in vivoSAS-RNA
synthesis, SAS-RNA was synthesized in vitro at ahigh
level. We noted the appearance of a minor band above SAS-RNA. This may constitute SAS-RNA molecules longerby
afew nucleotides
or a different form of SAS-RNAspecies.
To
determine
whether the 62- to 64-nt RNA synthesizedin vitro was indeed SAS-RNA, we prepared nuclei from cellsinfected
with aviable
deletion mutant ofSV40(d12194),
inp c m
P19m
I-
180
-160
-147 -122
}>. -ll
* -76
* -67
FIG. 3. Sizeanalysis ofviral RNAs producedinisolatednuclei under either pulseorpulse-chase conditions.Nucleiwereincubated in atranscription reactionmixturecontaining500
p.Ci
of[a-32P]UTP
(400mCi/mmol)and 5,uM unlabeledUTPfor 5 minat30°C(pulse). After thepulse, 400,uMunlabeled UTPwasadded,andincubation wascontinued for 30 min (chase). The labeled RNAsproducedwere purified andanalyzed by gelelectrophoresis. p,Pulse;c,chase;m, size markers as in Fig. 1B. The open arrowhead points to the position ofattenuatorRNA, andthefilled arrowheadpoints tothe position ofSAS-RNA.
Q aS
a-
0-E 5D H
__w
22-
10- 90- 76- 67-
)PI-FIG. 4. Size analysis ofviral RNAsproduced in isolated nuclei incubated in thepresence ofeither GTP or ITP in the transcription reactionmixture.LaneGTPshows a standard transcription reaction mixture such as thatdescribedinthelegendto Fig.1B. In laneITP, ITP(400 ,uM) replaced GTP in thestandardtranscription reaction mixture.Transcriptionwasallowedtoproceedasdescribedin the legend to Fig. 2. The labeled RNAs produced were purified and analyzed by gel electrophoresis. The arrowhead points to the position oftheSAS-RNA.m,Sizemarkersasin Fig. 1B.
which the
coding
region ofSAS-RNA
hasbeen deleted (12), andcompared
thesyntheses
of SAS-RNA in mutant- and wild-type-infected cells. The absence of SAS-RNA in cells infected withdl2194 has beenpreviously demonstrated (6).Figure
2A, laneWT,
shows the appearance of a major band of 62-to64-nt RNA(filledarrowhead).
Traceamounts of 94- to 98-nt attenuator RNA(open
arrowhead)
and two additional bands of 125- and 155-nt RNAarealsorecogniz-able, together
with viral RNAofheterogeneous lengths.
The appearance of the 125- and 155-nt RNAbands
was not always reproducible. The 125-nt RNA was not SV40 spe-cific;it was presumably trappedon the filterduring hybrid-ization(see also reference6).Figure 2A,
laned12194,
shows that when nuclei from these cells were incubated in vitro under the sameconditionsasnuclei fromwild-type-infectedcells,
nomajor
bandof 62-to64-nt RNAappeared.
Instead,
viral RNA molecules which were
longer
than those inlane WT were observed. Attenuator RNAwas notrecognizable
in the RNA
preparation
fromdl2194-infected
cells. How-ever, because this observation was notreproducible
and because the present conditions were notoptimal
fortran-scription
termination at the attenuation site(13),
it isI..
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.396.478.69.396.2] [image:3.612.141.224.316.625.2]B.VA4554
B.
GGGACu
U G
U U
C G
C A
G A
A-U
U-A
CAA
Z790-G A
A-U G-C
U-AU2
GCU-2920
G-C
cU-A
AGC
A-U
2m -C-G dsbedin ALU
2770 4554 2930 2940
-; a-u
-UGAUCAUG UCUUAGCCAAUCU
AG-~-5Kcal
G-C
U-AA
AA-UG
AuG-U
AGs~s K col G-CUG AGU-c +7K0C
A-U G.C-GCGUUUCGA c
G-C -A u4RNA~
SAS-RNA A
(G-C
U C YGAU U-A U
GA.U9-AUC
U.
G-C UACu
U-A U
YCU
G-C -UUACGACU
U-A
CA-U GG-CA
A-U
G-C
U-U
G-C
U-A6-cU
A-U
C-G
A-U
5-UCAUG UCUUAGCCAAUCU
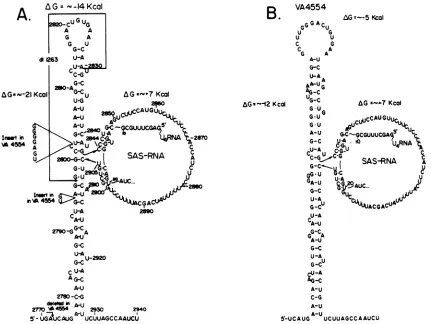
FIG. 5. Schematics ofputative RNA secondarystructures intheproximityof SAS-RNA of strains 776(A) and VA4554(B). Notethat SAS-RNA formsbulges and that residues 11 to 20 ofU4 RNA(10) base pair with the 5' and 3' ends of SAS-RNA. Note also that the involvement of the U4 ribonucleoprotein inprocessing-polyadenylation has been suggested (9). The sequences deleted in d1263 and the alterations in sequencesinVA4554areindicated(8). Changesin freeenergy(AG)werecalculatedbythe method of Tinocoetal. (24).
difficult to draw any conclusions concerning the role of SAS-RNA in the attenuationprocess.Ahypotheticalrole for SAS-RNA inattenuation haspreviouslybeen discussed(4). Figure 2B shows that when 62- to 64-nt RNA was eluted from thegel and hybridizedtoablot of restriction fragments (as shown inFig. 1A), it hybridized exclusively with frag-mentc, spanning0.17to0.37mapunit. Thisfragmentof the viral genome includes the SAS-RNA-coding region (6).
These results strongly suggestthat 62- to 64-nt RNA is the SV40-encodedSAS-RNA. Furtherevidence forthe identifi-cation of 62- to 64-nt RNA as SAS-RNA came from an
RNase T1 fingerprint analysis, which yielded the expected patternbasedonthe known basesequenceof the SAS-RNA-coding region (N. Hay, H. Skolnik-David, and Y. Aloni, unpublished results).
To determine whether SAS-RNA isproduced in vitro bya specific cleavage mechanism or is rather an independent transcription product, the following experiments were
per-formed. Isolated nuclei of SV40-infected cells were
incu-bated in vitro in a high-ionic-strength buffer [100 mM
(NH4)2SO4]
under either pulse or pulse-chase conditions.Thelabeled viral RNAswerethen purified and analyzed by gel electrophoresisasinFig. 1B. Figure 3,lanep,shows that duringa5-min pulsewith[a-32P]UTP, 94-to98-ntattenuator
RNA (open arrowhead) and long viral RNA molecules of heterogeneous lengths were synthesized. A faint band of SAS-RNAwasalso observed. Theproduction of SAS-RNA
mayindicatetranscription froman independentpromoteror
processing of SAS-RNA from precursor nascent chains. Figure 3, lane c, shows that after a 30-min chase with
unlabeled UTP, a different pattern of viral RNA species emerged. The 94- to 98-nt attenuator RNA almost disap-peared,theaverage sizeof the viral RNAdecreased, anda majorband of 62-to64-nt SAS-RNAappeared(filled
arrow-head). Since the UMP residues of SAS-RNA were labeled only during the pulse and since the labeled SAS-RNA species accumulated after the chase, we conclude that
SAS-RNA is produced in vitro from long precursor mole-culesbyanefficient andaccuratecleavage mechanism. The cleavage probably occurs when the precursor molecule is stillnascent. In thiscase,thevarioussmall RNAmolecules which appeared after the chase (see Fig. 3, lane c) may representeither(i)labeled viral RNAsequenceswith their5' ends adjacent to the 3' end of SAS-RNA which were
transcribed fromaregionof the viralgenomelocated down-streamfrom theSAS-RNA-coding regionor(ii) degradation products.Theobservation that SAS-RNAaccumulated after the chase is indicative of its remarkable stability in our
isolated-nucleus system.
We havebeguntouse theabove-described in vitro isolat-ed-nucleus systemforanalyzingthe elements which consti-tute the signal for the SAS-RNA-processing enzyme. Our firstapproachwasto determine theimportance ofan RNA secondary structure. For this purpose, we replaced GTP AG=- -14KIcal
A.
AGaA-211
Inmirtin
NA4554
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.100.531.76.400.2]with its analog ITP in the standard transcription reaction mixture. In previous studies, ITP was shown to abolish transcription termination in bacteria and bacteriophages (2, 11) and atthe SV40 attenuator (13) by destabilizing RNA stem-and-loop structures. Figure 4 shows theproduction of long viral RNA molecules in the twotranscription reaction mixtures. However, SAS-RNA was produced in the tran-scription reaction mixture which contained GTP (arrow-head) but not in that which contained ITP. Based onthese results, we suggest that an RNA secondary structure may
constitute at leastpart ofthe signal for theendonucleolytic
enzyme. Alternatively, the G residue could be an essential
nucleotide in the sequence required for processing, orGTP
mayprovide the energy source forthe processingreaction.
Heuverswyn et al. (16), Alwine and Khoury (8), and Sadofsky and Alwine (21) have suggested that a putative
secondarystructurecould exist in single-strandedDNAorin
late mRNAs in the proximity ofthe SAS-RNA-coding
re-gion. The existence of this structure could explain the observations that strain d11263, which lacks sequences
up-stream of the SAS-RNA-coding region (see Fig. 5A for the
sequences deleted), did not produce SAS-RNA, whereas strain VA4554, in which several sequences are inserted upstream ofthe SAS-RNA-coding region, leading to a less
stable secondary structure, produced three tofivetimes less SAS-RNA (8). We present here schematics of alternative secondary structures for the RNAs of the two wild-type strains 776 and VA4554 (Fig.5A and B, respectively). Inour
proposed secondary structures, SAS-RNA forms bulges in stem-and-loop structures. In these structures, the cleavage sites seem to be in a position easily accessible to the
processingenzymes. Furthermore, the conformational rear-rangements which accompany the excision of SAS-RNA
would result inadecrease infreeenergyfrom -14kcal (1cal
= 4.184 J) to -21 kcal and from -5 kcal to -12 kcal for strains776andVA4554, respectively. The -7-kcal decrease in free energy could provide all or a portion of the energy
required for the endonucleolytic reaction.
We have noticed that residues 11 to 20 in U4 RNA are
complementary to sequences at the 5' and 3' ends of SAS-RNA and can therefore base pair with them (Fig. 5A
and B). This base pairing can help stabilize the secondary
structures (Fig. 5A and B), in which the 5' and 3' ends of SAS-RNAareheldin closeproximity. Cleavagecouldoccur either by nuclease activity associated with the U4 ribonucleoprotein or through an RNase directed to the
properform ofthe secondary structure.
It seems thata common feature of mRNA splicing,
proc-essing-polyadenylation, and processing ofSAS-RNAis that the cleavage sites are primarily defined by one ofa set of nucleotide sequencesbutthat theiractualfunction is
modu-lated by the secondary and tertiary structures of the RNA andpossibly by proteins associated with them. Ourpresent
success inreproducing the processing ofSAS-RNA in vitro
isafirststepinprovidinginformationregarding thedetails of this mechanism and thereby of pre-mRNA processing.
Wethank 0. Resnekov for help in preparingcytoplasmic RNA, RuchamaLeiserowitz for technical assistance, and J. Feunteunfor providing straind12194. YosefAlonithanksA. J.Levineforhis kind hospitalityinallowing completion ofthiswork in hislaboratory.
This research was supported by U.S. Public Health Service
researchgrantCA 14995 fromtheNational InstitutesofHealth and
in part by the MINERVA Foundation, Munich. Part of the work reported in this paper was undertaken during the tenure of an American Cancer Society-Eleanor Roosevelt-International Cancer
Fellowship awarded to Yosef Aloni by the International Union AgainstCancer.
LITERATURE CITED
1. Abulafia, R., A.Ben-Ze'ev,N.Hay, and Y. Aloni.1984.Control oflate simian virus40transcriptionby the attenuation mecha-nismandtranscriptionally activeternarycomplexesare associ-atedwiththe nuclearmatrix.J. Mol.Biol. 172:467-487. 2. Adhya, S., and M. Gottesman. 1978. Control of transcription
termination. Annu.Rev.Biochem. 47:967-996.
3. Aloni, Y., R. Dhar,0.Laub, M.Horowitz,andG.Khoury. 1977. Novelmechanism forRNAmaturation: the leadersequencesof simian virus 40 mRNA are not transcribed adjacent to the codingsequences. Proc. Natl. Acad. Sci. USA74:3686-3690. 4. Aloni, Y., and N. Hay. 1983. Attenuation and modulation of
mRNAsecondarystructureinafeedback control
systemn
regu-lating SV40geneexpression. Mol. Biol. Rep. 9:91-100. 5. Aloni, Y., N. Hay, H.Skolnik-David,P.Pfeiffer,R.Abulafia,R.Pruzan, E. Ben-Asher, E. B.
Jakobovits,
0. Laub, and A. Ben-Ze'ev. 1983. Attenuationinthe controlofgeneexpression in animal viruses, p. 1-48. In A. Kohn and P. Fuchs(ed.),
Developments in molecularvirology, vol. 4. Martinus Nijhoff Publishers BV, Dordrecht, The Netherlands.
6. Alwine, J. C. 1982. Hybrid selection of small RNAs byusing simian virus 40 DNA: evidence that the simian virus 40-associated small RNAissynthesizedbyspecific cleavagefrom largeviraltranscripts. J. Virol. 43:987-996.
7. Alwine, J. C., R. Dhar, and G. Khoury. 1980. A small RNA induced late in simian virus40lyticinfectioncanassociate with early viralmRNAs. Proc. Natl. Acad. Sci. USA 77:1379-1383. 8. Alwine, J.C.,and G.Khoury. 1980. Simian virus40-associated small RNA: mapping on the simian virus 40 genome and characterization of itssynthesis. J. Virol. 36:701-708. 9. Berget, S. M. 1984. Are U4 small nuclearribonucleoproteins
involved inpolyadenylation? Nature(London)309:179-182. 10. Busch, H., R. Reddy, L. Rothblum, and J. C. Choi. 1982.
SnRNAs, SnRNPs andRNAprocessing. Annu. Rev.Biochem. 51:617-654.
11. Farnham, P. J., and T. Platt. 1980. Amodel for
transcription
termination suggested by studieson thetrp attenuatorin vitro
using baseanalogs. Cell20:739-748.
12. Feunteun,J.,G.Carmichael, J. C.Nicolas, andM.Kress. 1981. Mutants carrying deletions in the two simian virus 40
early
genes. J. Virol. 40:625-634.
13. Hay, N.,and Y. Aloni. 1984. Attenuation inSV40as a mecha-nism oftranscription termination by RNA polymerase B. Nu-cleic AcidsRes. 12:1401-1414.
14. Hay,N., and Y. Aloni. 1985. Attenuation of latesimian virus40 mRNA synthesis is enhanced bytheagnoprotein and is tempo-rally regulated in isolated nuclear systems. Mol. Cell. Biol. 5:1327-1334.
15. Hay, N., H. Skolnik-David, and Y. Aloni. 1982. Attenuation in the control ofSV40 geneexpression. Cell 29:183-193. 16. Heuverswyn, H. V., C. Cole, P. Berg, and W. Fiers. 1979.
Nucleotide sequence analysis oftwo simian virus 40 mutants with deletions in
the
regioncodingfor thecarboxyl
terminus of the T antigen. J. Virol. 30:936-941.17. Jakobovits, E. B., andY. Aloni. 1980. Isolation and character-ization of various forms of simian virus 40
DNA-protein
com-plexes. Virology 102:107-118.18. Laub, O., and Y. Aloni. 1976. Transcription of simianvirus 40. VI. SV40 DNA-RNA polymerase complex isolated from pro-ductively infected cells transcribed in vitro.
Virology
75: 346-356.19. Penman, S. 1966. RNAmetabolism inthe HeLacell nucleus. J. Mol. Biol. 17:117-130.
20. Pfeiffer, P., N.Hay, R.Pruzan,E.B. Jakobovits,and Y.Aloni. 1983. In vitro prematuretermination inSV40late
transcription.
EMBOJ. 2:185-191.
21. Sadofsky, M.,andJ. C.Alwine. 1984.
Sequences
onthe 3' side of hexanucleotideAAUAAAaffectefficiency
ofcleavage
atthe polyadenylation site. Mol. Cell. Biol. 4:1460-1468.on November 10, 2019 by guest
http://jvi.asm.org/
22. Skolnik-David, H., and Y. Aloni. 1983. Pausing of RNA poly-merasemolecules duringin vivotranscriptionof the SV40 leader region. EMBO J. 2:179-184.
23. Skolnik-David,H.,N.Hay,andY.Aloni.1982. Site ofpremature termination of late transcription of simian virus 40 DNA. Enhancement by5,6-dichloro-1-i-ribofuranosylbenzimidazole. Proc.Natl. Acad. Sci. USA79:2743-2747.
24. Tinoco,I., P. N.Borer, B. Dengler, M. D. Levine, 0.Uhlenbeck, D. Crothers, and J. Gralla. 1973. Improved estimation of secondarystructureinribonucleic acids.Nature(London) New Biol. 246:40-41.
25. Tooze, J. 1981. AppendixA. TheSV40 nucleotidesequence,p. 799-841. In J. Tooze (ed.), DNA tumor viruses. Cold Spring HarborLaboratory,ColdSpring Harbor,N.Y.