0022-538X/84/060733-06$02.00/0
Copyright© 1984, American SocietyforMicrobiology
Effects
of
Deletions
on
Expression
of the
Herpes
Simplex
Virus
Thymidine
Kinase Gene from the Intact Viral Genome: the Amino
Terminus
of the
Enzyme
Is
Dispensable
for
Catalytic
Activity
M. E. HALPERNAND J. R. SMILEY*
Department ofPathology, McMaster University, Hamilton, Ontario, Canada L8N 3Z5 Received 17October1983/Accepted 21 February 1984
We havetransferredtwodeletionsaffectingthe 5' endoftheherpessimplexvirusthymidinekinase(TK)
geneinto the intact viralgenome.One,extending from -12 to+189, hadnoeffectonTKmRNAsynthesis andonlyasmalleffectonTKactivity, althoughthefirst27codons of theTKpolypeptideweredeleted. The other, extendingfrom -85 to +85, severely impaired TK mRNAsynthesis. We conclude that the amino terminus of the TK polypeptide is dispensable for catalytic activity, and that expression ofTK in viral infections requires someof the same promoterelements used in uninfected cells.
Theherpessimplex virus(HSV)geneforthymidinekinase (TK) is a delayed early, or a viral gene (2). As such, its expression during lytic viral infections requires the prior synthesisandcontinuousactivityoftheimmediateearly,or a,protein ICP4 (3, 6,7, 18,26, 27). ICP4appearsto act as a positive transcriptional activator for all ofthe non-a HSV
genes;however, its mechanism of action remains unknown. Thisrequirement for ICP4 function isremarkably tight,asno TK RNA canbe detected by RNA-driven
hybridization
inthe absence of a protein synthesis (7). In contrast to the situationduringlyticviralinfections,TKisexpressedinthe
absence of a proteins when microinjected into Xenopus oocytes (14) ortransfected intomousefibroblasts (29). The
reasonsfor this difference remain unclear andmayberelated
tothefactthatbothoftheseexpression systemsinvolve the delivery ofthe TK gene into cellsoutofcontext, andbyan artificial route. This a-independent expression has in fact
provided a valuable system for studying HSV promoter
function. Usingexpression inXenopusoocytes as anassay,
McKnight and co-workershave systematically assessed the
effects of mutations onthe expression of microinjectedTK genes (13-16). The results demonstrate that expression in uninfected cellsdependsonatleast threeseparate upstream
regions mappingbetween -105and -18 with respecttothe
major transcription initiation site. The question arises whetheror not these same sequences are alsorequired for expression during viral infections. Part of the answer has recently emerged from studies ofmutantTK genesresident
in biochemically transformed cells. It has been known for
some time that HSV TK genes present inuninfected cells remain responsive to the action of a proteins supplied by
superinfecting HSV (4, 5, 8). We have recently reported
results that,when takenin combinationwith those ofZipser
et al. (30), suggest that this transactivation phenomenon requires the integrity of at least two separate upstream
regions thatoverlap the constitutive promoter used in oo-cytes (23). In this paper, we report on the effects of two
deletions on theexpression ofthe TK gene from the intact
HSV chromosome. The data demonstrate that expression requires sequences mapping between -12 and -85. It is
thereforelikelythat thepromoterelements used inartificial
assay systems correspond to, or form a subset of, the
elementsusedduringnatural viralinfections. Inaddition,we
* Correspondingauthor.
find that theamino-terminal45residues of the TK polypep-tide are dispensable forcatalytic activity, and thatdeleting all of the nontranslated leader of the wild-type TKmRNA
haslittle effect on expression.
MATERIALS AND METHODS
Virus and cells. HSVtype 1(HSV-1)strain KOS wildtype and the TK-deficient deletion mutant d2 (21) were used in thisstudy. Virusstocks wereprepared and titrated onVero cells as previously described (22). Mouse LtA cells, an adenine phosphoribosyl transferase-deficient derivative of the thymidine kinase-deficient (TK-) line LM (TK-) (4), were obtained from R. Hughes, Roswell Park Memorial
Institute, Buffalo, N.Y. Vero and LtA cells were grownin Alpha minimal essential medium (GIBCO Laboratories) supplemented with 5 and 10% fetal bovine serum,
respec-tively.
Marker rescue. Mutations weretransferred from plasmids
into the viral genome exactly as previously described (21,
22). Eachmutantplasmidwasseparately cotransfectedwith
wild-type and d2 viral DNA into Vero cells. The resulting virus stocks were then screened for rescue products.
TK-deficientproducts were selected from the crosses with wild-type viral DNA byplaque purification on Vero cells in the presence of100 ,ugofaraT per ml (17).
TK+ productswere selected from the crosses with d2 viral DNA by serial passage in LtA cells in the presence of
hypoxanthine, aminopterin, andthymidine. After the initial
selections, DNAfrom each of the recombinants was
exam-inedby Southern blot hybridization (24) to verify the pres-enceoftheappropriate deletion; then the recombinant viral strains were plaque purified an additional three times and
expanded into high-titer stocks. Blotting and hybridization were as previouslydescribed (22).
Induction of TKactivity.Subconfluentmonolayers of LtA
cells growing in 75-ml flasks were infected with the virus strains andharvested 12 h later.TKactivity incellextracts was measured by using C14 thymidine, as previously de-scribed(23).
S1 nuclease transcript mapping. To map the transcripts produced in cells infected with the mutant virus strains, we used the transcript mapping techniques of Berk and Sharp (1), as modified by Weaver and Weissman (28). Vero cells were infected at a multiplicity of 10, and total cytoplasmic RNA was prepared 4 h later, using the procedure of Berk andSharp(1). Uniquely end-labeled, single-stranded hybrid-733
on November 10, 2019 by guest
http://jvi.asm.org/
ization probes were prepared by using T4 polynucleotide
kinase and [.y32P]ATP as described by Maxam and Gilbert (11). Afterstrand separation, approximately 2 x 104 dpm of
the eluted probe was coprecipitated with 10
pxg
ofRNA inethanol. The dried pellet was suspended in 30 ,ulof0.6 M NaCl-0.05 M HEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonicacid)-0.001 MEDTA (pH 7.5) andhybridized
for 12 hat50°C. ForSi digestions,thesamplewasaddedto
0.20 ml of ice-cold 150 mM NaCl-50 mM sodium acetate-5 mM ZnSO4 (pH 4.6) containing 2 x 103to 4 x 103 Uof S1
nuclease (Boehringer Mannheim Biochemicals). After 1 h at 37°C,the reactions were stoppedbytheaddition ofEDTA to 20 mM, extracted twice with phenol-chloroform (1:1), and
ethanol precipitated. Digestions with exonucleaseVII were carried out in 200
[lI
of 0.3 M KCl-0.1 MTris-hydrochlo-ride-0.1 M EDTA (pH 7.4) for 1 h at 45°C, using 2 U of
exonuclease VII (BethesdaResearchLaboratories)per reac-tion.
To detect TK-related transcripts, we used two different probes: one was aHinfl-EcoRI fragment end labeled at the Hinfl site located at +811; the other was an RsaI-PvuII
fragment end labeled at the RsaI site at +351. ICP4 tran-scripts were assayed by using a 290-base SalI-EcoRI
frag-ment labeled at the Sall site (9). The portions of the probe
protected from nuclease digestion were sized on
polyacryl-amidesequencing gels. DNAsequencingwasby the method
ofMaxam andGilbert (11). RESULTS
Rescue of deletion mutations. The genome of HSV is 150
kilobases in length, making direct site-specific mutagenesis difficult. Therefore,to study theeffectsofprecisely defined
deletions on TKexpression from theintact viralgenome,we decidedto use a two-stepapproach that has beenpreviously described (21). Briefly, mutations were introduced into an HSV-1TK gene cloned into abacterialplasmid, then trans-ferredinto the viral genomethroughhomologous
recombina-tion invivo. The resulting rarerecombinant virusesbearing
themutant allele were thenisolated byusingan appropriate
selection system. We had previously isolated a number of
deletions affecting the TK upstream region (23). Some of
thesemutations had no effect ontheexpressionor
transacti-vation of the TK gene when introduced into mouse fibro-blasts; others reduced expression and eliminated the re-sponsetoproteinsofsuperinfectingTK-virus. Asitwas not clear in advance what effects these same mutations would
have on expression from the intact viral genome, each
deletion was recombined with both wild-type HSV and a TK-deficientdeletion mutantvirus, HSVd2(21; seeFig. 1). The products of the former cross were screened for TK-deficientrecombinants, and the products ofthe latterwere screenedforwild-typerecombinants, asdescribed above.In all, we have attempted to rescue six deletions in this way, but have succeeded withonly two: Al andA35 (Fig. 1). Al
lacksresiduesextending from -11to +189onthe
wild-type
sequence (23; Fig. 1). The deletion leaves intact the three promoter elementsdefinedbyMcKnightandcoworkers
(13-16) butremoves allofthewild-type nontranslatedleader and the firstpotential initiation codon ofthe TK
polypeptide.
In contrast, A35 (-85 to +85)leaves the TKcoding
sequence intactbut deletes all threeofthepromoter elements thatare required for expression in Xenopus oocytes. As describedbelow, the Al deletion has little effect on TK
expression
from the HSV genome, whereas A35 reduces expression approximately 50-fold. This result parallels the effects of these same deletions on expression in uninfected
cells,
inA
HSVWT
Bam Pvu1 AUG PvuII Bam
x
,/&35
I
TK'VIRUS
B
HSV d2 *4
I
TK*VIRUS
c
-100 +1 +100 +200
AUG
WT AUG
AUG
A35
FIG. 1. Rescue ofdeletions. (A) TheA35 deletionwas rescued by recombination between wild-type HSV DNA and a plasmid
bearingthe deletion.(B)Alwasrescuedbyrecombination with TK-deficient HSV d2 DNA.(C)Thedeletionendpointsarediagrammed, using an expanded scale, with respect tothe threetranscriptional control elements identified byMcKnight and co-workers. In addi-tion, the major transcription initiation sites forwild-type and Al virusare indicated. For the sake ofconsistencywiththeliterature, the mapsarepresentedinthe ILorientationof the HSV genome. which Al had no effect on
expression
ortransactivation,
whereas A35 wasdefective for both (23).
TK+
recombinants were recovered from the Al x HSVd2 cross, andTK-recombinantswere recoveredfrom the A35 x
HSV+
cross,suggestingthat Al does not
dramatically
affect TKexpres-sion from the viral
chromosome,
but A35 does. To demon-stratethat themutantviruses recovered from thesecrossesactually
hadacquired
theinput mutations,
DNAfrom each wascleaved withacombination of PvuII andSmaI,
andtheTK-related
fragments
wereanalyzed
by
Southern blot hy-bridization. Deleted versions of the1,400-base
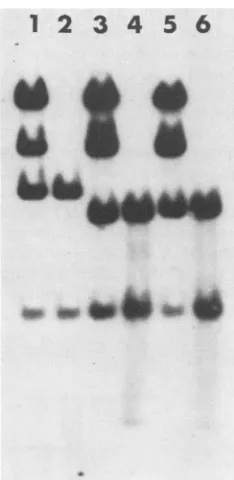
PvuII-SmaI TKfragment
werepresent in both isolates(Fig.
2),
and these noveldeletion-bearing fragments
comigrated
withthecorre-sponding fragments
oftheinput
plasmids.
Additional analy-ses showedthat Alvirusretained the EcoRI site at-80,
asexpected, and A35 virus lacked this site
(data
notshown).
We conclude,
therefore,
that both rescueproducts
haveincorporated
the mutationsoriginally
carriedby
theplasmid
DNA in the cotransfections.Although
we are not yet certain for the reasons for the failureto rescuetheotherfourdeletions,
it may be relatedto the fact that theplasmids
bearing
these four mutations all contain less than 130 bases ofhomology
with the viral chromosome upstream ofthe deletion(these
deletions wereinducedin thePvuIITK
fragment).
Incontrast,theplasmids
bearing Al and A35 both retainover600bases of
homology
withviralDNAupstreamofthedeletion.
Consequently,
one would expect ahigher
frequency
of recombination between the viral genome and these twoplasmids.
We cannot,on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.318.558.78.341.2]1
2 3 4 5 6
[image:3.612.115.232.75.315.2]_
FIG. 2. Southernblotanalysis of the mutant genomes. Plasmid and viral DNAs werecleaved with a tnixture of PvuII and SmaI. After electrophoresis through a 1.4% agarose gel, the fragments were blotted onto nitrocellulose and detected by hybridization to nick-translated pTK173DNA. Lanes: 1,pXlbearing the wild-type TKgene; 2,wild-typeviral DNA; 3, Alplasmid;4,Alvirus; 5,A35 plasmid; 6, A35 virus.
however,rule out thepossibilitythat the other four deletions arelethal, althoughweconsiderthistobe unlikely.
Expression of TK from the mutant viral genomes. To
accurately quantitate the levels ofTK enzymatic activity inducedbyAlandA35, TK-deficientLtAcellswereinfected
with either virusandthelevels ofTKactivityweremeasured
12h later. Al induced approximately 40%of the wild-type levelsofenzyme, whereasA35 inducedonlyca.3%(Fig. 3). As Al isdeletedfor the first27codons ofthe TK polypep-tide,thisresult demonstrates thattheaminoterminus ofthe
proteinisnotrequired forcatalyticactivity. A35 retains the entire TKcodingsequence, yetisseverely impairedfor TK
expression. This suggests that the A35 deletion interferes
with TK mRNA synthesis.
The effects of these deletions on TK mRNA synthesis
were assessed by Sl mapping. In the first experiment, infected cellRNA washybridizedtoasingle-strandedprobe
end-labeled at aHinfl site(+811)andextendingtotheEcoRI
site at -80. After hybridization and treatment with S1 nuclease or exonuclease VII, theprotected portions ofthe
probe were sized on a 3% polyacrylamide sequencing gel (Fig. 4A). To provide an internal control for variations betweeninfections and betweenRNApreparations, portions ofthese same RNApreparations were also hybridizedto a probe forthe aICP4mRNA(Fig. 4B)(seeabove). All of the
infected cell RNA preparations used contained roughly comparable amounts ofICP4 mRNA. RNA from wild-type
infections gave a majorprotected species of approximately 810 residues corresponding to TK transcripts initiating at +1. In addition, a somewhat weakersignal was observed,
suggesting an additional transcript starting at ca. +70.
Al-though avariety of minorTK-related transcriptshave been
observed in biochemically transformed mouse cells (20),
comparable RNAs have not been reported during lytic
infections. Wewish topointout, however, that both ofthe
high-resolution Si mapping studies ofTKmRNApublished
to date (12, 25) have employed hybridization probes that
would not detect initiations at +70. Consequently, we con-sider it possible that the signal at +70 represents a real transcript, rather than an artifact of nucleasedigestion.
RNA from Al-infected cells lacked the +1 and +70
species, asexpected, and insteadgave a new major hybrid-ization signal at ca. +200. The intensity of this signal was
comparable to that of the wild-type, suggesting that the
novel Al transcript was produced in normal amounts. The initiation site forthe mutant RNAcouldnotbemappedwith confidence in this experiment as the probe was prepared from wild-type DNA and the resolution of the gel was
insufficient to allow us to decide whether the signal was
produced by initiationupstream ordownstream ofthe
dele-tion. The fact that the Si and exonuclease VII digestion products comigrated could be rationalized with upstream
initiations ifthe upstreamportion of thetranscript was too
short toform a stable hybrid with theprobe.. Therefore, to map the Alinitiationsiteprecisely,anotherwild-typeprobe,
end-labeledat anRsaI site(+351) andextendingto thePvuII siteat -200, wasused. Inthis
case,
the position of the Al hybridization signalwasdetermined with referencetomark-ers generated by the Maxam-Gilbert sequencing reactions
carried out on thesame probe. The results (Fig. 5)
demon-stratethat the Al transcript initiates atresidue +198ofthe wild-typesequence,downstreamfromthedeletion endpoint. Incontrast to theessentially wild-type levels ofRNAfound in Al-infected cells, we were not able to detect any
TK-related transcripts in cells infected with A35 virus. We
estimate from this that the mutant transcripts, if present,
accumulate to less than 5% of the wild-type levels. This is consistent with theverylow levelsof TKenzymatic activity inducedby this mutant. Since the sameA35 RNA
prepara-tions contained normal amounts of ICP4 mRNA (Fig. 4B), we conclude thatA35 deletionreduces TKtranscription, or
(less likely) the stabilityorprocessing ofthe RNA.
DISCUSSION
We have drawn two major conclusions from this work.
First, thetranscription ofTKfrom theintactHSV
chromo-x 0
LU
I--0
I
C') 0
I
C.) 5
4 3 2 1
TIME(minutes)
FIG. 3. TK enzymatic activity induced by mutant virus. LtA cells were infectedwith the indicated viruses and harvested 12 h later. TKactivity was measured inextracts prepared from equiva-lentnumbers of cells, asdescribed in the text.
on November 10, 2019 by guest
http://jvi.asm.org/
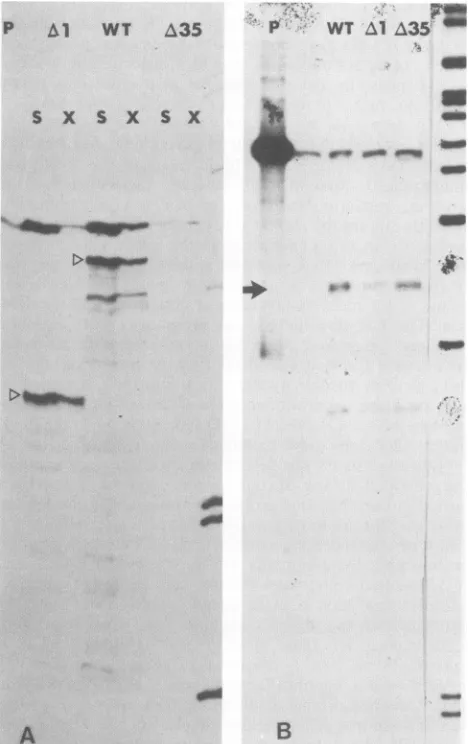
[image:3.612.313.550.515.693.2]P Al WT A35
S X S X S X
WT
*r 51The initiation site for the Al transcript is consistent with P WT Al A35 the proposed role of the Goldberg-Hogness sequence in
directing transcription initiation events to a site 25 to 30
bases downstream.The Al deletion leaves the TK Hogness * sequenceintact butdeletes the wild-type initiation site. The
>,_ mutant transcript, however, continues to initiate 28 bases
downstream, at asitethatordinarily lies within the body of the mRNA. Similar resultshave beenobtainedbyMcKnight - et al. withTK inuninfectedcells (15), and by others with a
variety of genes.
McKnight and co-workers (13-16) have identified three upstreamregionsnecessaryfor TK transcription in
uninfect-ed cellswithin the DNA sequences extendingfrom -105 to -18. A35 lacks two of these elements entirely and retains 4 g * ~ only a portion ofthe most distal element (-105 to -81),
qp
[image:4.612.70.304.76.448.2]A B
FIG. 4. Analysis of transcripts produced by wild-type and
mu-tant virus. (A) For TK transcripts, RNA prepared from cells infected withthe indicated virus washybridizedtoaHinfl-EcoRI TKprobe (P). After digestion with S1 nuclease(S)orexonuclease
VII(X),sampleswere run on a3% polyacrylamide sequencing gel. Themajor initiation sites areindicated by triangles. (B) For ICP4
transcripts, the RNA preparations usedin(A)werehybridizedtoa
SalI-EcoRI ICP4 probe (P). Afterdigestionwith Si nuclease, the
productswere run on an8%sequencing gel. Thearrowindicates the
approximately 180-base-long fragment protected byICP4 mRNA.
some during natural viral infections requires sequences
located between -12 and -85. This conclusion is based on the factthatAl, lackingresidues -11 to +189, accumulates
wild-type levels ofa novel TK transcript and A35, lacking residues -85to+85,accumulates less than 5% of the normal levels. We believe that the defect in A35 is atthe level of transcription initiation, rather than at the level of mRNA processing,transport,orstability,fortworeasons. First,the
difference between Al and A35 strongly suggests that the
defect in A35 can be accounted for by the deletion of
sequenceslocated between -12 and-85. Thepositionof the
relevant sequences is therefore consistentwith a transcrip-tional defect and is more difficult to reconcile with other possibilities. Second, Al synthesizes normal amounts ofa truncated mRNA that lacks the first 197 residues of the wild-typetranscript. Therefore,thestructureofthe 5' endofTK
mRNA does notappeartobe critical for normalprocessing, transport, or stability.
WT A1 GG
XCT
C_s"
Us.
FIG.5.Fn_apn~ fteA~ ntainst.TeRApeaa
tions~~~~
usdi~~
i. eehbrdzdt a sIPu Kpoe After~Sinulaedgsin_hrtce rget eeszdo an~~8%glwt_epc~ oteprdcso h aa-ibrseunigraton are u n h aepoe
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.392.503.241.686.2]whereas Al retains all three (Fig. 1). The fact that A35 is
impaired stronglysuggests thattranscriptionin natural HSV infectionsrequiresatleast some of the samesequences used inuninfectedcells. Thisconclusion isnotespecially surpris-ing in view of our recent demonstration that sequences
located between -70 and -12 are required for the ICP4-mediated activation of TK genes resident in mouse fibro-blasts (23). We emphasize that the results of the present
study wereobtained by assayingthe effectsof deletions on
expression ofTKfrom its natural location within the intact HSV genome, eliminating thepotential artifacts associated
with other assay systems. One question that remains is
whether expression from the intact viral genome requires
sequencesin additiontothose needed inXenopusoocytes. The results obtained with Al rule out a requirement for
sequences between -11 and +189. However, it remains possiblethat sequences upstream of themostdistal
constitu-tive promoterelement play a role during infection. To test
this possibility, it will be necessary to modify sequences upstream of -105 while leaving the -105 to -18 region intact. This may prove to be difficult in practice as the
modifications might affecta neighboring essentialgene. The second major setof conclusions that we have drawn from our data is that the amino-terminal 45 residuesofthe
wild-typeTKpolypeptidearedispensable for catalytic activ-ity,and that the nontranslated leader of theTKmRNAdoes
not play a major sequence-specific role in translational initiation. These conclusions are basedonthe properties of virus bearing the Al deletion. This deletion removes all of
thewild-type nontranslated leader ofTKmRNAand the first 27 codons forthe TK polypeptide, but leaves the promoter
unaltered. Consequently, transcription in the mutant virus
initiatesatresidue +198ofthewild-typesequence, 44 bases upstream from what is ordinarily the second methionyl codon ofthe polypeptide. Presumably, translation initiates at this second site, which is the first AUG codon in the mutantRNA. As faras we are able todetermine, Al virus
accumulates wild-type levels of the truncated transcript. Because it also inducesapproximately40% ofthewild-type levelsofTKenzymaticactivity,weconcludethatthe amino-terminal45 residues ofthe wild-type enzyme are not
abso-lutely required for catalytic activity. Since we have not
directly measured the levels of the translated mutant TK
polypeptide,we are unable to decide from the present data
whether the approximately twofold reduction of activity
compared with wild-type is due to reduced
translation,
reduced activity ofthe truncatedprotein, or acombination of both factors.Nevertheless, because thedeletionhas
only
a relatively minor effect on the overall level ofactivity, it
seems safe to conclude that the truncated protein product retainsappreciableactivity,andthat the truncated mRNA is
translated at closeto the wild-typerate. This latter
conclu-sion implies either that the wild-type leader plays no
indis-pensable, specific role in translation, or that the novel 44-base leader ofthe Al transcriptis able to substitute for this function. At present we are unable to distinguish between these twoalternatives.
Preston and co-workers (10, 19) have reported that in
addition tothe42,000-molecular-weight (42K)TK polypep-tide, infected cells alsosynthesizesmalleramountsofa39K anda38K TK-relatedprotein.Theseauthorshaveproposed thatthesmallerproteinsarisethroughtranslational initiation events at the second and third initiator codons of TK
mRNA, respectively. In studies to be reported elsewhere, Haar, Smiley, Marsden, and Preston have shown that Al virusfails to induce the 42K protein and overproduces the
39K species. Taken in combination with the present data, this demonstrates that the 39K protein retains close to full
catalytic activity. This conclusion agrees with that of
Rob-erts and Axel (20), who proposed on the basis of indirect evidence that anaberrantTKtranscript postulatedtoinitiate at +200encodesafunctionalenzyme. The functional signifi-cance of the 39K and 38K polypeptides produced during
wild-type infections remains unclear. ACKNOWLEDGMENTS
We thank HelenRudzroga for expert technical assistance, and RoyPerssonforagiftofanICP4plasmid.
This workwas supported by grantsfrom the Medical Research Council and the National Cancer Institute of Canada. J.R.S. is a
research scholarof the National CancerInstitute.
LITERATURE CITED
1. Berk,A.J.,and P. A.Sharp. 1977.Sizing and mapping of early adenovirus mRNAsby gelelectrophoresis ofSi endonuclease-digested hybrids. Cell 12:721-732.
2. Garfinkle, B., and B.R.McAuslan. 1974. Regulation of herpes simplex virus-induced thymidine kinase. Biochem. Biophys.
Res.Commun. 58:822-829.
3. Honess, R. W., andB.Roizman. 1974.Regulation of herpesvirus macromolecularsynthesis. I. Cascade regulation of the synthe-sisof three goups of viral proteins. J. Virol. 14:8-19.
4. Kit, S., and D. L. Dubbs. 1977. Regulation of herpesvirus thymidine kinase activity in LM (TK-) cells transformed by ultraviolet light-irradiated herpes simplex virus. Virology 76:331-340.
5. Leiden, J. M., R. Buttyan, and P. G. Spear. 1976. Herpes simplex virus gene expression in transformed cells. I. Regula-tionof the viral thymidine kinase gene in transformedLcells by products of superinfecting virus. J. Virol. 20:413-424.
6. Leung,W.C.1978. Evidence foraherpessimplex virus-specific factorcontrolling the transcription of deoxypyrimidine kinase. J. Virol. 27:269-274.
7. Leung, W.-C., K. Dimock, J. R. Smiley, and S. Bacchetti. 1980. Herpes simplex virus thymidine kinase transcripts are absent
from both nucleus and cytoplasm during infection in the
pres-enceof cycloheximide. J. Virol. 36:361-365.
8. Lin, S.-S., and W. Munyon. 1974. Expression of the viral thymidine kinase gene in herpes simplex virus-transformed L cells. J. Virol. 14:1199-1208.
9. Mackem, S., and B.Roizman. 1980. Regulation of herpesvirus macromolecularsynthesis: transcription-initiation sites and do-mains ofalpha genes. Proc. Natl. Acad. Sci. U.S.A. 77:7122-7126.
10. Marsden,H.S.,L.Haarr,andC. M. Preston.1983.Processing ofherpes simplex virus proteins and evidence that translation of thymidine kinase mRNA is initiated at three separate AUG codons. J. Virol.46:434-445.
11. Maxam,A.M., and W.Gilbert. 1980. Sequencing end-labelled
DNAwithbase-specific chemical cleavages. Methods Enzymol. 65:499-560.
12. McKnight, S. L. 1980.The nucleotide sequence andtranscript map of theherpessimplex virus thymidine kinase gene. Nucleic AcidsRes. 8:5949-5964.
13. McKnight,S. L.1982.Functionrelationships between transcrip-tional control signals ofthe thymidine kinase gene of herpes simplex virus. Cell 31:355-365.
14. McKnight, S. L., and E. R. Gavis. 1980. Expression of the herpes thymidine kinase gene in Xenopus laevis oocytes: an assay for the study of deletion mutants constructed in vitro. Nucleic Acids. Res. 8:5931-5948.
15. McKnight,S.L.,E. R.Gavis,R. Kingsbury,and R. Axel.1981.
Analysis of transcriptional regulatory signalsofthe HSV thymi-dine kinase gene: identification of anupstream controlregion. Cell25:385-398.
16. McKnight, S. L., and R. Kingsbury. 1982. Transcriptional control signals of a eukaryotic protein-coding gene. Science
on November 10, 2019 by guest
http://jvi.asm.org/
217:316-324.
17. Post, L. E., S. Mackem, and B. Roizman. 1981.Regulation of
alpha genes of herpes simplex virus: expression of chimeric genesproduced by fusion ofthymidine kinase with alphagene
promoters. Cell 24:555-565.
18. Preston, C. M. 1979. Control ofherpes simplex virus type 1
mRNAsynthesis in cells infected withwild-type virus or the
temperature-sensitivemutanttsK.J. Virol.29:275-284.
19. Preston, C. M., and D. J. McGeoch. 1981. Identification and
mapping oftwopolypeptides encoded withinthe herpessimplex
virustype1thymidine kinasegenesequences. J.Virol. 38:593-605.
20. Roberts,J.M., and R. Axel. 1982. Geneamplification andgene
correctioninsomaticcells. Cell 29:109-119.
21. Smiley, J. R. 1980. Construction in vitro and rescue of a
thymidine kinase-deficient deletion mutation of herpes simplex
virus. Nature(London) 285:333-335.
22. Smiley, J. R., B. S. Fong, and W.-C. Leung. 1981.Construction ofadouble-jointedherpessimplexviral DNAmolecule:
invert-edrepeatsarerequiredforsegmentinversion, and directrepeats
promotedeletions.Virology 113:345-362.
23. Smiley, J. R., H. Swan, M. M. Pater, A. Pater, and M. E. Halpern. 1983. Positive control of the herpes simplex virus
thymidine kinase gene requires upstream DNA sequences. J.
Virol.47:301-310.
24. Southern, E. M. 1975. Detection ofspecificsequences among
DNAfragmentsseparated by gel electrophoresis. J. Mol. Biol.
98:503-517.
25. Wagner, M. J., J. A. Sharp, and W. C. Summers. 1981.
Nucleotide sequence ofthe thymidine kinase gene ofherpes
simplex virus type 1. Proc. Natl. Acad. Sci. U.S.A. 78:1441-1445.
26. Watson, R. J., and J. B. Clements. 1978. Characterization of
transcription-deficient temperature-sensitive mutantsofherpes
simplexvirustype 1. Virology91:364.
27. Watson, R. J., andJ. B. Clements. 1980. A herpessimplexvirus
type 1 function continuously required forearly and late virus
RNAsynthesis. Nature(London)285:329-330.
28. Weaver, R.F.,andC.Weissmann. 1979. MappingofRNA bya
modificationof theBerk-Sharpprocedure:the5'termini of 15S beta-globin mRNA and mature 10S beta-globin mRNA have
identicalmapcoordinates. Nucleic Acids Res.7:1175-1193.
29. Wigler, M., S. Silverstein, L.-S. Lee, A. Pellicer, Y.-C. Cheng, and R.Axel. 1977. Transfer ofpurified herpesvirusthymidine
kinasegenetocultured mousecells. Cell 11:223-232.
30. Zipser, D., L. Lipsich, and J. Kwoh. 1981.Mappingfunctional
domains in thepromotorregionoftheherpesthymidinekinase
gene.Proc. Natl. Acad. Sci. U.S.A.78:6276-6280.
on November 10, 2019 by guest
http://jvi.asm.org/