0022-538X/85/050247-09$02.00/0
Copyright© 1985, American Society forMicrobiology
Molecular Cloning and Partial
Sequencing
of Hepatitis A
Viral
cDNA
DAVID L. LINEMEYER,l* JOHN G. MENKE,' ANTONIA MARTIN-GALLARDO,' JOSEPH V. HUGHES,2
ALEXANDER YOUNG,' AND SUDHA W. MITRA'
DepartmentofBiochemical Genetics' and Department of Virology and CellBiology,2 Merck Sharp & Dohme Research
Laboratories,
Merck
Institutefor
Therapeutic
ResearchRahway,
New
Jersey
07065Received 19October 1984/Accepted 8 January 1985
HepatitisA viruswaspurified from infected monkey kidney cellcultures, and the viral RNA wasused to
synthesizedouble-strandedcDNA.ThiscDNAwascloned either after insertion intoaplasmid-primed synthesis
system or after insertion into the PstI site of pBR322. The resulting clones were mapped by restriction
endonuclease analysis and by cross hybridization of the viral inserts to generate a composite map which representedatleast97% of the viralgenome,lackingca.220bases fromthe 5'endof thegenome.Theclones were verified tobe hepatitisA virus specific basedon their positive hybridization to viral RNAand tototal hepatitisAvirus-infected cellularRNAfromaheterologousmarmosethost system. The nucleotidesequenceof 3,054 base pairs of cDNA homologoustothe5'half of the viralgenomewasdetermined,andanopenreading frame of 854 consecutive coding tripletswasidentified. Inaddition,sequenceswhich encode the VP-1 and VP-3 viral structural proteins werelocated in the nucleotidesequence.
Hepatitis A virus (HAV) is a small RNA virus which
generallycausessubclinicalliver disease butcan cause acute
hepatitis (24). Type A hepatitis outbreaks have been
attrib-uted to contaminated food and water, and the disease is
transmitted predominately byafecal-oral route asthevirus
is shed infecalsamples ofinfected individuals (6). Thevirus is perpetuated either by poor environmental and personal
hygiene,asis prevalent inlower-socioeconomic-levelgroups,
orbya reservoirof nonepidemic cases which areclinically
unrecognized.Thevirus is oftenspread byyoungchildren in
daycare centers oramongmalehomosexualsandisreported
toberesponsible for20to40% of sporadic hepatitiscasesin
urban adults(7).
The incidence of hepatitis A is decreasing in developed
societies, creating larger numbers of unprotected
individu-als. Prophylaxis relies on passive immunization with
im-mune serumglobulin. All HAV virions regardless oforigin
or strain are
immunologically indistinguishable;
thus, thereis only one serotype (8). Development of active vaccines
with killed or live attenuated HAV to protect against the
reservoir of subclinical nonepidemic cases has been made
possible by in vitro cultivation of the virus (21, 22).
How-ever, an alternative antigen for active vaccination against HAVwould be desirable.
It is only recently that HAV has been characterized in
sufficient detail to allow its classification as a picornavirus
with the characteristics of the enterovirus group (9). The
virion has no envelope, is 27 nm in diameter, and has a
bouyant density of 1.34 g/ml in CsCl (27). The particles
contain a single-stranded RNA genome with a
polyadenyl-ated[poly(A)] tractlocatedatthe 3' end (5, 29);assuch, the
genomecanfunctionas amessenger to supportthesynthesis of viral proteins (16). By analogy to other picornaviruses, the genome should contain a large open reading frame for translation of a polyprotein which is proteolytically proc-essed into the virus proteins (25). As with other
picorna-viruses, four structural proteins have been described with
molecular weights of 32,000 to 33,000 (VP-1), 26,000 to
*Correspondingauthor.
29,000(VP-2),22,000to27,000 (VP-3), and 10,000to14,000 (VP-4). VP-1 appearsto be the major surfacecomponentof
thevirus and contains atleastoneneutralization epitopefor
HAV(4, 10).
In an effort to produce a subunit vaccine to HAV by recombinant DNAtechnology,wereporthere the molecular
cloningof the HAV genome. Twopreviousreports ofHAV
cloning have been described. Von der Helm et al. (31)
described cDNA clones of up to 1,000 base pairs (bp)
synthesized by using an oligodeoxythymidylic acid
[oligo(dT)], primer and RNA extracted from HAV purified
fromfecalmaterial. These clones were shown to hybridize
to
32P-labeled,
HAV-specific RNA derived fromHAV-in-fected PLC/PRF5 cells and to express a product which
reacted ina radioimmunoassay.
By using virus extracted from infected marmoset livers,
Ticehurst et al. (30) reported cloning HAV cDNAs which
represented atleast99% ofthe viralgenome. These clones
were shown to hybridize to RNA from HAV-infected Afri-can green monkey kidney cells but not to RNA from
uninfected cells. In addition, the nucleotide sequence of
some500 basesfromthe 3' endofthe genomewasreported. In this paper we report the molecular cloning of HAV cDNA
representing
at least 97% ofthe viral genome. Alsopresentedis the nucleotide sequenceof3,054bp fromthe 5'
half of the genome and the localization of the sequences which encode the VP-1 and VP-3viral structural proteins.
MATERIALSANDMETHODS
GrowthandpurificationofHAV. LLC-MK2 cells(a
mon-key kidney cellline)were infected withthe CR326 strain of
HAV(21)at amultiplicityof0.1 to0.5 andweremaintained
at35°C in Eagle minimal essential medium (Earle salts; Flow
Laboratories, Inc.) with 0.2% fetal calf serum and 2 mM
glutamine for 21 to 28 days. The infected cells were then
washed and scraped in phosphate-buffered saline and pel-leted. The cells were suspended in lysis buffer (10 mM
Tris-hydrochloride [pH7.5], 10 mM NaCl, 1.5 mM
MgC92,
1% NonidetP-40),subjectedto two cyclesofsonication for 20 s, and incubated on ice for 10 min. Cell debris was
247
on November 10, 2019 by guest
http://jvi.asm.org/
removed by centrifugation at 10,000 x g for 20 min. The supernatant was removed and sodium-Sarkosyl (SLS) was
addedto0.5%. Aftera30-minincubationat37°Candabrief
sonication, theviral supernatantwaspelleted througha20%
sucrose shelf in 0.1% SLS and TNE (10 mM
Tris-hydro-chlroide [pH 7.4], 150 mM NaCl, 1 mMEDTA) for20 h at
100,000 x g. After centrifugation, the viral pellet was
sus-pended in0.5% SLS inTNE,sonicated,andincubated for30
minat37°C.Viruswasbanded on 25 to45% preformedlinear
cesium chloride gradients by centrifugation for60 h.
Frac-tions were collected and assayed for their refractive index
and forhepatitis Aviral antigenby radioimmunoassay.The
majorviral peaks were pooled, diluted 20-fold in TNE with
0.1% SLS,and pelleted.
RNA extraction. The CsCl gradient-purified virus was
suspended in TNE containing 20 mM EDTA and 1% SLS.
The suspension was extracted with an equal volume of
phenol saturated with 0.1 M Tris-hydrochloride (pH 7.4) at
65°C. The aqueous layer of the extraction was further
extracted twice with equal volumes ofCHCl3-isoamyl alco-hol(24:1) and precipitated with 0.2 M sodium acetate (pH 5.5) and 2 volumes of ethanol at -20°C. The ethanol
precipitatewas collected by centrifugation and dissolved in
sterile distilled water.
Total cellular RNAs from uninfected and HAV-infected
LLC-MK2 cells or marmoset liver cells were extracted as
describedpreviously (3).
Synthesis and cloning of 3' cDNA. The initial HAV cDNA
cloneswere prepared byamodification ofthe procedure of
Okayama and Berg (20). Briefly, the viral RNA was
an-nealed to oligo(dT)-tailed plasmid pBR322-simian virus 40
(map units 0.71 to 0.86), and cDNA was synthesized by
avianmyeloblastosis virus(AMV) reversetranscriptasewith
theoligo(dT) tail as primer. This first strand of cDNA was
oligodeoxycytidylic acid [oligo(dC)]-tailed by usingterminal
transferase, restricted with HindIII, and annealed to an
oligodeoxyguanylic acid [oligo(dG)]-tailed linker pBR322-simianvirus40 (map units 0.19 to 0.32). Inacombined triple
reaction, the RNA was removed by RNase H, the second
cDNA strand wassynthesized byDNApolymeraseKlenow
fragment with the linker as primer, and the cDNA was
ligated with T4DNAligase. Thisreaction mixturewasthen
transformed into Escherichia coli HB101. The resulting
cloneswereselectedby resistancetoampicillinand screened
by hybridizationto a
32P-labeled
HAV cDNApreparedfromthe purified viral RNA by using reverse transcriptase and
calfthymusDNAfragments asprimer.
Primer extension cDNA synthesis and cloning. Arestriction
fragment of one of the initial HAV cDNA clones was
purified by electroelution (17) from an agarose gel and
denatured by boiling for 10 min and quick cooling in ice
water. The denatured fragment was usedas a primer after
annealingorhybridization tothe viral RNA. Forannealing,
160 ngofprimerwasaddedto ca. 1
Rg
ofpurifiedHAVRNA in 20 ,ulof water, heated to 70°C for 10min, and cooled to37°C. For thehybridization, 140 ngofprimerwasaddedto1
,ugofpurifiedHAV RNAin80%formamide-0.4MNaCl-0.01
M PIPES (piperazine-N,N'-bis(2-ethanesulfonic acid [pH
6.4])-2 mM EDTA and incubated at 47°C for 3.5 h. The
formamide was removed byfour sequential ethanol
precip-itations as previously described (15). The first strand of
cDNA was then synthesized with 1.5 U of AMV reverse
transcriptase per ,ul in the presence of50 mM
Tris-hydro-chloride (pH 8.0)-0.34 mM dCTP-1 mM dGTP-1 mM
dATP-1 mM dTTP-10 mM 2-mercaptoethanol-0.8 ,uCi of
[a-32P]dCTP-0.7
UofRNasinfor30 minat42°C.Thesecondstrand was synthesized by using the same triple enzyme
reactiondescribed aboveforthe initialclones. The
double-stranded cDNAwasoligo(dC)-tailed by usingterminal
trans-ferase, annealed to pBR322 which was oligo(dG)-tailed at
thePstI site, andtransformed into competent E. coli RR-1
(17). Theresultingclones were selected for
tetracycline-re-sistant, ampicillin-sensitive growthand screened forpositive
hybridizationtothe 32P-labeled cDNAprobe prepared from
HAV RNA.
Analysis of cDNA clones. Toensurethat thecDNA clones
were generated from HAV genetic information, selected
clones were labeled with
[cx-32P]dCTP
by nick translation(17) and hybridized to uninfected and HAV-infected
LLC-MK2 cellular RNAor marmosetliver RNAboundto nitro-cellulose membrane filters (29). In addition, hybridizations
were performed with RNA extracted from virus purified
from the livers of infected marmosets or from infected LLC-MK2 cells.
The inserts of the initial clones isolated after plasmid
priming were analyzed by restriction enzyme digestions
after releasefromthe vector byPstI-PvuII double-enzyme
cleavage.
The clones isolated afterprimer
extension wereanalyzed after release from the vectorwithPstI digestion.
The clones were organized on a genomic map by analysis
with multiple restriction enzyme digestions and Southern
blot
analysis
(28) with various cloned32P-labeled inserts as probes.Analysis of nucleotide sequence. Nucleotide sequencing
was performed with clones T28-18, T28-71, T28-77, and
T28-94 by the procedure of Maxam and Gilbert (18). In
addition,
Sau3A and TaqI fragments ofclone T28-77 weresubcloned into phage M13 (19) and sequenced
by
thedid-eoxynucleotide
chain termination procedure describedby
Sanger
et al. (26).Also,
in afew areasof this 5' half ofthe genome,oligonucleotide
primerswere synthesizedand usedfor direct
sequencing
ofthepurified
viral RNA(32).Enzyme and radioisotope sources. Restriction endonucle-ases were obtained from Bethesda Research
Laboratories,
NewEngland
Biolabs,
orInternationalBiotechnologies,
Inc.AMV reverse transcriptase was obtained from Life Sci-ences, Inc.
Polynucleotide kinase,
terminaldeoxynucleoti-dyl
transferase,
andribonucleaseHwerefromP-LBiochem-icals,
Inc.The sourceof RNasinwasfromBiotec,
Inc. TheT4DNAligaseand DNApolymeraseKlenow
fragment
wereobtained from P-L
Biochemicals,
Inc., CollaborativeRe-search, Inc.,and New England BioLabs. Bacterial alkaline
phosphatasewasfromBethesda ResearchLaboratories.The
[a-32P]dCTP
and[y-32P]ATP
radioisotopes
were obtained from Amersham Corp.RESULTS
Plasmid-primed cDNA synthesis and cloning. The initial
clonesof HAV cDNA were obtained
by
using
a systemofplasmid-primed cDNA
synthesis
which has been described(20). Viral RNA was extractedfrom
purified
viralparticles
grown in LLC-MK2 cell cultures as detailed above. The HAVRNA
migrated
inmethylmercury-agarose
gels
at asizeslightly largerthanRNA from the
closely
relatedpoliovirus
(Fig. 1). Also,
possibly
duetothe slowergrowth
and lowertiters of HAV, it was not
possible
to isolate HAV RNA withoutasignificantamountofdegradationasshownby
thesmearof RNA below the band of
full-length
RNAin lane 1. This viral RNA(500 ng)wasannealedtotheoligo(dT)-tailed
plasmid (1,400 ng) derived from
pBR322-simian
virus 40(map units 0.71 to
0.86),
and the first strand ofcDNA wason November 10, 2019 by guest
http://jvi.asm.org/
synthesized by reverse transcriptase in the presence of
[32P]dCTP
with theRNA astemplate and theoligo(dT) tailasprimer. The cDNA was then tailed with dCMP, and the
second strandofcDNA was synthesized by DNA polymer-ase I with an oligo(dG)-tailed linker derived from
pBR322-simian virus 40 (map units 0.19 to 0.32) as primer after
removal ofthe RNAby RNase H treatment.
The first strand of cDNA ranged in size from 0.5 to 2.5
kilobases(kb) byalkaline-agarosegelanalysis(Fig. 2). With
the same conditions ofsynthesis, it was possible to obtain cDNA aslarge as 4kb from poliovirusRNA(Fig. 2, lane 3).
Synthesis of cDNA with different concentrations of KCI
(lanes 1 and 2) or by previous denaturation of the RNA
template with heat or treatment with methylmercury (data notshown) did not significantly increase the size of cDNA
obtained. Since at least 50% ofthe viral RNA was
appar-ently full-length,the reasonlongercDNAproductswere not
generated is not known atthis time.
After ligation, the cDNA was transformed into E. coli
HB101,and the clones were selected byampicillin-resistant
growthyieldingca. 104 transformants perjxgof cDNA. The
cloneswere screened subsequently by colony hybridization
to a representative HAV-specific probe prepared by the
synthesis of
[ot-32P]dCTP-labeled
cDNA from HAV RNAwith calf thymus DNA fragments as primer. Restriction
1
2
3
FIG. 1. Methylmercuric hydroxide-agarose gel electrophoresis of RNA extracted from purified virus particles. Viral RNA was
extracted frompurified HAV particles,asdescribedin thetext,and electrophoresedat60Vthrougha1%agarosegel containing 10 mM methylmercurichydroxide (17). To visualize the RNA, the gelwas
treated with0.2M ammoniumacetatecontaining 1 ,ug of ethidium
bromide perml and irradiated with UVlight. The RNA analyzed
was viral RNA from HAV (lane 1), RNA from human embryo
fibroblasts(lane 2), and viral RNA from poliovirustypeI(lane 3;a
giftfrom V. Racaniello).
FIG. 2. Alkaline-agarose gel analysis ofviral cDNA. Plasmid-primed cDNA synthesis was performed as described in the text. Aftersynthesis of the first strand of cDNA, asamplewasethanol precipitated, suspended inwater,anddigested with PvuIItorelease thevector. The mixturewas electrophoresedon a1% agarosegel
containing 30 mMNaOH and 2mM EDTA. Thesynthesisof HAV
cDNAwasinthepresenceof 100 mMKCI (lane 1)or150 mMKCI (lane 2). Also shown is cDNAsynthesized from poliovirus RNA in the presence of 100 mM KCI (lane 3). The location of X-HindIII
fragments migratingin thesamegelarenotedontherightin kb.
endonuclease analysis of the hybridization-positive clones revealed a family of cloned cDNAs which all appeared to
havethesame terminus,presumably originatingfrom the 3' poly(A)tractof the RNAgenome,and extended for various lengths (datanotshown). Thelargest HAV insert obtained
was2.3 kb in length, from clone a18.
Hybridizationof cloned cDNAtoHAV RNA. Toverify the authenticityof theclones,afew cloneswereusedtoprepare
32P-labeledprobes bynicktranslation(17),and theseprobes
werethen hybridized by Northern analysistouninfectedand HAV-infected cellular RNAs as well as to viral RNA. An exampleof thehybridizationwithaprobe preparedfromone
of theclones is shown inFig.3. Theclonehybridizedwellto
RNAof viruspurifiedfrom either HAV-infected LLC-MK2 cells (lanes 1 and 2) or livercells of HAV-infected marmo-sets (lane 7). In all cases the cloned DNA hybridized to a
band of 7.5 to 8 kbas well as smallerdegraded RNA. The clone also hybridized to total cellular RNA extracted from eitherHAV-infectedLLC-MK2 cells(lane 4)orlivercells of
HAV-infected marmosets (lane 6). Importantly, there was nodetectable hybridizationofthe cloned cDNAto uninfec-tedLLC-MK2 total cellular RNA(lane 3)ortototalcellular RNA from livers of uninfected marmosets (lane 5). In the lastcases,althoughthetotalcellular RNApreparationswere largely degraded, the HAV specificity of the hybridization was obvious, indicatingtheauthenticity of the clones.
Additional cDNA synthesis and cloning. Restriction
en-zyme analysis of the largest clone, a18, indicated the
pres-1
2
3
-4.3
-2.2
-0.6
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.391.490.70.332.2] [image:3.612.121.240.339.630.2]1234
56 7
FIG. 3. Hybridization of cloned cDNAto various RNA prepa-rations. RNA preparations were electrophoresed through a 1% agarose gel containing 10 mM methylmercuric hydroxide as de-scribed in the Fig. 1 legend. The gel was then soaked in three
changesof 10 mM sodiumphosphate (pH6.8)for 10mineach,and the RNA was transferred to a nitrocellulose membrane as previ-ously described (29). The membrane filter was hybridized with a
12P-labeled probe prepared by nick translation (17) ofa 3' HAV cDNAclone. The clone used, a12, is homologoustoclone a18but lacksca. 300bpatthe 5'end. The RNApreparations hybridized by
theprobeincludetwodifferentpreparationsof RNA extracted from viruspurifiedfrom HA V-infected LLC-MK2 cells(lanes1and2)or
from viruspurifiedfrom the livers of HAV-infectedmarmosets(lane 7). Total cellular RNAs analyzedwerefrom uninfected LLC-MK2 cells (lane 3), HAV-infected LLC-MK2 cells (lane 4), liver cells fromuninfected marmosets (lane 5),and liver cells fromn HAV-in-fectedmarmosets(lane6). Viral RNAswere runat20 ng,and total cellular RNAswere runat1 ~Lg.
enceoftwoPvulI sitesneartheextreme5' endof this clone
(see Fig. 5), which when cleaved generated a 280-bp DNA fragment. This fragment was purified by electroelution (17)
from a 1.5% agarose gel ofPvuII-digested clone a18 DNA and used toclonefurther HAV cDNA sequencesbyprimer extension. Thepurified fragmentwaseither annealedtoviral RNAbyheatingto90'C and slowcoolingorwashybridized
tothe RNAat 420C in thepresence of80% formamide(15).
Thefirst strand of cDNAwassynthesized bytheaddition of
reverse transcriptase. Thesynthesis of the second strand of cDNA was accomplished by the addition of RNase H to
degrade the RNA, DNA polymerase I to catalyze the synthesis, and DNA ligase to join internal pieces of the second strand. The cDNAwassizefractionatedon asucrose
gradient, and two pools of cDNA were collected, a larger
poolwith cDNAranginginsize from less than 0.5toca.8kb and asmallerpoolof sizes less than 2.5 kb as shown inFig. 4. The cDNA in thepoolenriched for larger-sized products
wasclonedbytheaddition ofpoly(dCMP)tails,annealingto
poly(dG)-tailedPstI-cleaved pBR322,andtransformation of
E. coli RR-1. Positive clones wereselected by
tetracycline-resistant, ampicillin-sensitive growth and screened by
hy-bridization to the representative HAV cDNA probeused in the initial cloning. Of 220 tetracycline-resistant clones, 37 hybridized to the HAV cDNA probe. However, only fourof these hybridized to a probe preparedby nick translation of the 280-bpfragment primer, indicating that the majority of the clones were generated by nonspecific priming of the AMV reverse transcriptase. The clones contained inserts ranging in size from ca. 0.3 to 4.2 kb. As with the initial clones, the authenticity of a number of these clones was examined by preparing radiolabeled, nick-translatedprobes from the cloned inserts and hybridizing them to RNA. Amongtheclones analyzedwere T28-77 and T31-2 (seeFig. 5). All ofthe clones examined were found to hybridize to HAV-infected cellular RNA but not to uninfected cellular RNA (data not shown) verifying the HAV specificity of these clones.
Since themajority of the clones seemed to be generatedby nonspecific priming of thereversetranscriptase, the synthe-sis of cDNAfrom HAV RNA was examined in the absence ofprimer orafterannealing an unrelated primer. cDNA of almost genome length was synthesized in the absence of primer (Fig. 4, lane 3). The size and yield of this cDNAwas evenlarger than that generated by cDNA synthesis primed with a 340-bp fragment of clone T28-71 (lane 4) or with a
375-bp fragment of pBR322 DNA (lane 5). Indeed the
initiation of synthesis of these latter three cDNAs was
shown to berandomby hybridizing these [32P]cDNAs tothe cloned HAV cDNAs cleaved with restriction enzymes in
1
2
9.6-
6.5-4.3
2.2
1.4-3 4
5
-9.6
6.5
4.3
2.2
1.4
0.6
FIG. 4. Methylmercuric hydroxide-agarose gel electrophoresis of HAV cDNA prepared by primer extension. Double-stranded
cDNAwassynthesized from purified HAV RNAby usingaDNA
fragment primerasdescribed inthetextandwaselectrophoresedas
described in the legendto Fig. 1. The280-bp fragment generated fromthe 5'endof clonea18bydigestion ofthe insertwithPvuIlwas
usedasprimer, andtheresultingcDNAwassize fractionatedon a
5to25%sucrosegradient containing10 mMTris-hydrochloride (pH 8.0),50 mMNaCl,and 5 mM EDTA inanSW50.1rotor at190,000
xg for 6 hat 4°C. Pools were made of the faster (lane 1) and slower
(lane 2)migratingcDNA. In aseparateexperiment, double-stranded
cDNAwas preparedwith no DNA primer (lane 3), a340-bp
frag-mentgenerated fromthe 5'endof cloneT28-71 by BamHI-HincII digestion (lane 4),or a375-bp EcoRI-BamHI digestion fragment of pBR322 (lane 5).Twiceasmuchreactionsamplewasloaded inlanes 4and 5asin lane 3. Thelocations ofX-HindIIIand4X174-HaeIII
DNAfragments migratingin thegelsarenotedatthesidesin kb.
on November 10, 2019 by guest
http://jvi.asm.org/
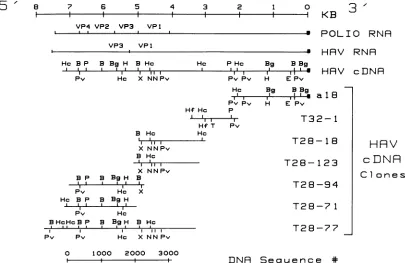
[image:4.612.87.273.68.334.2] [image:4.612.320.555.390.566.2]8 7 6 5 4 3 2 1 0 3'
I I I I I i K B
VP4 VP2 VP3 VP1
.. . .~~~~~~~~~~~~~~~
POLIO RNR
VP3 VP1
Hc B P B Bg H B Hc
Pv Hc X NNPv
a HRV RNR
Hc PHc Bg BBg
',' ,' ,i'§ HRV cDNR
Pv Pv H E Pv
Ho Bg BBg
PvPv H E Pv
Hf Hc P
', * ', T32-1
Hf T Pv
B Hc Hc
II ',i, T28-1 8
X NNPv B Hc
', 'i,,
T28-123
X NNPv B P B BgH B
. III --Jr T28-94
Pv Hc X
Hc B P B Bg H
III I I T28-7 1
Pv Hc
B HcHcB P B BgH B Hc
,
IS
l,lII
| | , l,X,,,T28-77
Pv Pv Hc XNNPv
0 1000 2000 3000
DNR
Sequence *FIG. 5. Schematic representation of restriction endonuclease cleavage sites onHAV cDNAclones. The viral RNAfrompoliovirus is shownforcomparisonwith the 3' poly(A)attheright (N)andthe locationofthe sequenceswhich encode the structuralproteins (12).The viral RNA from HAV is shown with the 3' poly(A) at the right and thelocation ofthe sequenceswhichencode theVP-1andVP-3proteins
(see text). Below the HAV RNA is shownacomposite restrictionmapofthe HAVcDNA, and thelocations of the cDNA inserts of the various clonesdescribed. The oligo(dG)-oligo(dC)tails at the ends of the cloned insertsare notshown. The lineat thebottom shows the location oftheregion ofcDNAwhich has beensequenced along with the basesequence numbers. Restrictionenzyme sitesidentified are
BamHI (B),BgIII(Bg), EcoRI (E), HindIII (H),HincIl (Hc),Hinfl(Hf), PstI (P), PvuII (Pv), NcoI (N), TaqI (T), and XbaI (X).
Southern analyses (28; data not shown). Similar results of
nonspecific primingwerereported by Kupperetal. with foot
and mouth disease virus (FMDV) when cDNA synthesis
originating from various regions covering about 75% of the
genome were found after
priming
with oligo(dT) (14). Mapping of the cloned cDNAs. Restriction maps wereprepared by using a number ofthe larger clones. By
com-paring these maps and by examining patterns of cross
hybridization with labeled inserts and
restriction-enzyme-cleaved DNAfrom otherclones inSouthern analyses (data
not shown), thepositions of the cloned inserts were aligned onthe map(Fig. 5). Asexpected, clonea18mappedto one end ofthe genome since it wasgenerated by the
plasmid-primed cDNA synthesis which required a poly(A) stretch
likethat present at the 3' end ofthe viral genome (30) for
priming.
Attheopposite
endofthe map, itwas foundthatHc BPv P
_1 11 1I
B
clone T28-77 had a 550-base inverted repeat at its 5' end
which was probably generated by a "snap-back"
synthe-sized
by
reverse transcriptaseduring
first-strandsynthesis.
Not including these repeated sequences, the clones
over-lapped to form a map of 7.3 kb; clones a18, T31-2, and
T28-77 were sufficientto producethe entiremap.
Partial nucleotidesequence of the HAV genome. The DNA sequencesof clones T28-71,T28-77, andT28-94 were
deter-mined by the Maxam and Gilbert technique of
chemical
sequencing
(18).Inaddition,M13cloneswereprepared fromfragments of clone T28-77 and were sequenced by the
dideoxynucleotide
termination method (26). All ofthere-gions were sequenced two or three times to ensure the
correctness of the sequence. In a few cases the sequence
was verified by direct sequencing of the viral RNA from
specific synthesized oligonucleotide primers (32). The
se-Bg Hc H
lI I l
B X HcN N
I1
IIPv
1000
I~~~~~~~~~. ... I...v
I~~ II
2000 3000
2 OOO
OOO.
3.I.
<4-4
-FIG. 6. Strategy of sequencingHAVcDNA.This schematicrepresentation shows the locations, directions, andextentsofDNAsequence
information obtainedby the Maxam and Gilbert technique (18) (solid arrows), the Sanger technique (26) with M13 clones of T28-77 (dotted arrows), anddirectRNAsequencing(dashed arrows). Also shownarethe locationof restrictionenzymecleavage sitesasdescribed inthe legendtoFig.5. For the locationof the sequenced regiononthe viralgenome, seeFig. 5.
HRV
cLDNR
Clo n e s
0
5'/
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.107.512.70.333.2] [image:5.612.136.498.596.686.2]252 LINEMEYER ET AL.
0 *0 27 00%4
CTC TCC CCTTGC CCT AGG CTC TGG CCG TTG CGC CCG GCG GGT CAA CTC CAT GAT
L-j
0000** ** 81 108
TAG CAT GGA GCT GTA GGA GTC TAA ATT GGG GAC GCA GAT GTGTOGGAC GTC ACC
L~~~~~~~~~J~~L.
*
*
LA~~~~~~5
* &J2TTG CAG TGT AAA CTT GGC TCT GTCTTCC ACAAGGGT AGG
to t
. 89 ... . 6
CTA CGGGTG ALA CCT CTT AGG CTA ATA CTT CTA TGA AGA GAT GCT TTG GATGAL
* 243 ** 270
GCA ACAGCG GCG GAT ATT GGT GAG TTG TTA AGA CAAAA$CCA TTCAACGCC GGA
297 to. too 324 CGACTGOCTCTC ATCCAGTOG ATG CATTGA GTG OATTGATTG TCA 000 CTG TCT
L-J
000@00 ~~~~~~351**
CTA gGTTT; ATCTCA GAC CTC TCT GTG CTTAGOGCA AAC ACC ATTTOOCCTTfA
... 405 *** 432
ATGGGATCCTOT GAG AGOGG0 TCC CTC CATTGA CAG CTGGACTOTTCTTTG 000
* . 459 s.. 486
CCT TAT GTG GTG TTT 0CC TCTGAG GTA CTC AGOGGC ATTTAGOTTTTT CCT CAT
513 540
TCT TAA ACA ATA ATG AAT ATG TCC AAA CAA CGA ATT TTC CAG ACT GTC 000 AGT Thr Ile MET Asn MET Ser Lys Gln Gly Ile Phe Gln Thr Val Gly Ser
L-J Lwj
567 594
GGCCTTGACCAC ATC CTG TCT TTG GCAOATATTGAOGAAG GA CMATG ATTCAG
Gly Lou Asp H1S Ile Lou Ser Leu Ala Asp Ile Glu Glu GluGlnMETIleGln
621 648
TCCGTT OTT AGOACT GCA GTG ACTGOT OCTTCT TAT TTT ACT TCT GTGGAC CAA Ser ValVal Arg Thr Ala Val ThrGly Ala Ser Tyr PheThr Ser Val AspGln
675 702
TCT TCAGTT CAT ACTOCT GAO OTTGGC TTA CAT CAA ATT GAA CCC TTG AAA ACC Ser SerVal His Thr Ala GluValGly LeuHis GlnIle Glu Pro Leu LysThr
729 756
TCTOTTGATAAACCT AOTTCT AAG AAG ACTCAG000GAO AAG TTT TTC CTG ATT Ser ValAsp Lys ProSer Ser Lys LysThr Gln Gly Glu Lys Phe PheLeu Ile
783 810
CAT TCT OCT OAT TGOCTC ACT ACA CATOCTCTA TTT CATGAAOTTGCA AAA TTG
H1i Ser AlaAsp Trp Lou Thr Thr His Ala Leu Phe His GluVal Ala Lys Leu
837 864
GAC GTGGTG AAA TTA TTG TAT AATGAO CAGTTT GCC GTC CAAGOTTTG TTG AGA Asp Val Val Lys Leu Leu Tyr AsnGlu Gln Phe Ala ValGlnGly LeuLeu Arg
891 918
TACCAC ACA TATOCA AGATTTGGC ATTGAOATT CAA OTT CAG ATAAATCCC ACA
Tyr His Thr Tyr AlaArgPheGly Ile Glu IleGlnValGln IleAsn Pro Thr
945 972
CCCTTT CAG CAAGG0000 CTA ATTTOT OCTATGOTTCCT ALTGAC CAA AGT TAT Pro PheGln GlnGlyGly Leu Ile Cys AlaMET Val Pro Ser AspGln SerTyr
999 1026
GOT TCG ATAGCA TCC TTG ACTOTTTATCCT CATGOTTTG TTAAAT TGC AAC ATT
Gly Ser Ile Ala Ser Leu Thr Val Tyr Pro HisGly LeuLeu AsnCys Asn Ile
1053 1080
AAC MT GTGOTT AOAATAAAGOTTCCA TTTATT TAT ACT AOAGOT OCT TAT CAC
AanAsn Val ValArg Ile Lys Val Pro Phe Ile TyrThr Arg Gly AlaTyr His
1107 1134
TTT AAGOATCCACAGTATCCAOTTTOO GAATTA ACAATC AOAOTTTOOTCAGAO PheLys Asp ProGln Tyr Pro Val TrpGlu Leu Thr IleArg ValTrpSerGlu
1161 1188
TTOAAT ATTGOAACAGOA ACT TCAOCTTAC ACTTCA CTTAAT OTTTTAOCTAGO Leu Asn IleGlyThrGly Thr Ser Ala TyrThr Ser Leu AsnVal LeuAla Arg
1215 1242
TTT ACAOAT TTO GAOTTA CAT GOA TTAACTCCT CTT TCT ACA CAG ATG ATGAOA
Phe Thr Asp Lou Glu LouHisGly Leu Thr Pro Leu SerThr GlnMET METArg
---_eVP 3
1269 1296
AATGAATTTAGAOTTALTACT ACTGAA AATOTT GTAAATTTO TCGAATTATGAA
AsnGlu Phe ArgVal Ser Thr ThrGlu Asn Val ValLAn Leu Ser Asn TyrGlu
1323 1350
OATGCAAGOGCAAAA ATG TCT TTT OCTTTGOATCAGGAA OAT TOG AAM TCTOAT Asp Ala Arg AlaLys METOar Phe Ala LeuAspGln Glu AspTrp Lys Ser Asp
1377 1404
CCTTCCCAAGOTGOTGOAATTAAAATTACT CAT TTT ACT ACC TOG ACA TCCATT Pro SerGlnGly Gly Gly IleLys Ile Thr His PheThr Thr TrpThr Ser Ile
1431 1458
CCA ACC TTAOCTOCTCAG TTTCCA TTC AATOCTTCAOAT TCG OTTGOA CAA CAA
Pro Thr Leu Ala AlaGlnPhe Pro PheAsnAla Oar Asp Oar ValGlyGlnGln
1485 1512
ATTAAAOTTATT CCA GTGGACCCA TAT TTT TTC CAG ATO ACAAAC ACC AATCCT Ile LysVal Ile ProVal AspProTyr Phe PheGln MET Thr Asn Thr AsnPro
1539 1566
OATCAAAAGTOTATAACTGCC TTG OCTTCTATTTOTCAG ATG TTT TGC TTT TOG
Asp Gln Lys Cys Ile Thr AlaLeu Ala Ser IleCysGln METPhe Cys Phe Trp
--.-*- *@@@ *@@@*@--~~~~... ... ..
1593 1620
AGOGGAGATCTTGTTTTTOATTTTCAG GTTTTTCCAACC AAATAT CATTCAGOT ArgGlyAspLouValPhe AspPhe Gln Val Phe Pro ThrLys Tyr His SerGly
1647 1674
AGO TTGTTG TTTTGCTTTOTTCCT 000MTGAG TTGATAOAT OTTACTGGAATC Arg Leu LeuPhe Cys Phe Val ProGly Asn Glu Leu IleAsp Val ThrGly Ile
1701 1728
ACATTAAAA CAGGCAACC ACTOCTCCTTOTGCA GTGATGGACATT ACAGOAGTG
Thr Leu LysGlnAla Thr Thr Ala ProCys AlaValMETAsp Ile Thr Gly Val
1755 1782
CAGTCA ACCTTOAOA TTTCOT OTT CCT TOOATTTCTOAT ACA CCC TATCGAGTO
Gln Ser Thr LeuArg PheArg Val Pro Trp IleSOr Asp Thr Pro Tyr ArgVal
1809 1836
AATAGO TAC ACG AAO TCAGCACATCAAAAAOGT GAOTAT ACTGCCATT000AAG
Asn Arg Tyr Thr LysSer Ala His GlnLysGly GluTyr Thr Ala Ile GlyLys
1863 1890
CTTATT GTOTAT TOT TAT AATAGOCTO ACT TCT CCT TCT AATOTT OCT TCT CAT Leu Ile Val Tyr CysTyr AsnArg Leu.Thr Ser ProSer Asn Val Ala SOrHis
1917 1944
OTTAOA OTTAATOTTTAT CTTTCAGCAATTAATTTGGAA TOTTTTOCTCCT CTT Val Arg Val AsnVal Tyr LeuSer Ala Ile Asn Leu GluCys PheAla ProLeu
1971 1998
TAT CAT OCT ATG GAT OTTACCACACAGOTTGOAOAT OATTCAGGA GOTTTT TCA TyrHis Ala MET Asp Val Thr Thr Gln Val Gly Asp AspSer GlyGly PheSer
2025O'VP - 2052
ACA ACAOTT TCGACAGAGCAGAATOTTCCTOATCCC CAAOTTGOTATAACA ACT ThrThr Val OarThr Glu GlnAsn Val Pro Asp ProGln ValGly IleThr Thr
2079 2106
ATGAAG GACCTO AAA 000AAAGCCAATALG OGA AAG ATGOAT OTT TCAGOAGTO
METLys Asp Leu Lys Gly Lys Ala Asn Arg Gly Lys MET AspVal SerGlyVal
2133 2160
CAAGCA CCT GTGGOAOCTATC ACA ACA ATT GAGOATCCAGCATTAGCAAAGAAA
Gln Ala Pro Val Gly Ala Ile Thr Thr Ile Glu Asp Pro AlaLeu Ala Lys Lys
2187 2214
GTA CCTGAA ACG TTT CCT GAA TTGAAOCCTGCAGAO TCTAOACAT ACATCAOAT
Val ProGlu Thr Phe Pro Glu Leu Lys ProGly GluSOrArg His Thr SOrAsp
2241 2268
CACATG TCT ATT TAT AAA TTCATGOGAAGO TCT CAT TTT TTGTOTACTTTTACC His MET Ser Ile Tyr LysPhe METGly ArgSer HisPhe LouCys Thr Phe Thr
2295 2322
TTC AAT TCAAAT AAT AAA GAGTAC ACA TTTCCAATA ACCTTGTCT TCG ACTTCT
Phe Asn SOrAsn Asn Lys Glu Tyr Thr Phe Pro Ile Thr LouSer OarThrSer
2349 2376
AAT CCT CCT CATGOTTTA CCA TCA ACA TTAAGO TGOTTC TTCAATCTOTTTCAG Asn Pro ProHis Gly Leu Pro Ser Thr Leu Arg TrpPhe Phe Asn Leu PheGln
2403 2430
TTG TAT AOAGOACCATTGOATTTGACA ATT ATC ATC ACAOGAOCTACTqATGTG Leu Tyr OrgGly Pro LeuAsp Leu Thr Ile Ile IleThr GlyAla Thr Asp Val
2457 2484
OATGOAATG0CC TOG TTT ACTCCA GTAGOCCTTOCTOTTGAC ACC CCATOGGTG Asp Gly METAla Trp Phe Thr ProValGly Leu Ala Val Asp Thr Pro Trp Val
2511 2538
GAAAAOGAATCAOCTTTG TCT ATTOATTATAAA ACT0CCCTTOGA OCTOTTAOA
Glu Lys GluOar Ala LeuSer Ile AspTyr Lys Thr Ala LouGly Ala Val Arg
2565 2592
TTTAATACAAOL AOL ACA000 AAC ATT CAG ATTAOATTG CCATGOTATTCT TAT Phe AsnThr Arg ArgThr Gly AsnIleGln Ile Arg LeuPro TrpTyr Ser Tyr
2619 2646
TTA TATOCT GTOTCTOGAGCACTO OATGGC TTGCGA OAT AAGACAOATTCTACA Leu TyrAla Val SerGlyAla Loeu AspGlyLouGly Asp Lys Thr AspSOr Thr
2673 2700
TTTGOATTGOTTTCC ATA CAG ATT GCA AAT TACAAC CAC TCTOATGAA TAT TTG PheGly Leu Val Ser IleGln Ile Ala Asn Tyr Asn HisSOrAsp GluTyr Leu
2727 2754
TCCTTT ALT TOTTATTTG TCT GTC ACA CAA CAA TCA GAO TTC TAT TTT CCTAOA
SOr Phe SerCys Tyr LouSer Val ThrGlnGlnSOrGluPheTyr Phe ProArg
2781 2808
OCTCCA TTAMATTCAAATOCTATG TTGTCC ACTGAGTCTATOATGALT AOAATT Ala ProLouAsnSOr AsnAla MET Leu Ser Thr Glu SerMETMETSerArg Ile
2835 2862
GCAOCTGOAGACTTGGAGTCA TCAGTOOAT OATCCTAOATCAGAOGAAGACAGA Ala AlaGlyAsp LouGluSerSOr ValAspAspProArgSer GluGluAsp Arg
2889 2916
AOATTTGAO ALTCATATAGAATOT AGOAAACCA TAT AAAGAA TTG AOATTG GAG
ArgPheGluSOrHis IleGluCys Arg Lys Pro TyrLys Glu LeuArgLou Glu
2943 2970
OTT000 AAA CA AOACTT AAA TATOCTCAGGAAGAGTTG TCA AATGAAGTGCTT Val Gly LysGlnArgLeuLysTyr Ala GlnGluGlu LeuSer AsnGlu Val Leu
2997 3024
CCA CCT CCTAGO AAAATGAAGG00 TTA TTTTCACAA GCCAAA ATT TCTCTT TTT
Pro Pro ProArg LysMET LysGlyLeu PheSer GlnAlaLys IleSerLeu Phe
3051 TAT ACTGAOGAACATGAAATA ATGAAA TTT TyrThr Glu Glu HisGlu IleMETLysPhe
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
quencingstrategyisshowninFig.6,and the DNAsequence
corresponding to the region ofthe HAV genome believed,
by analogytopoliovirus, toencode the structural proteins is
presented in Fig. 7. As found with poliovirus (12, 23) and with FMDV (1, 11), there wasalarge openreading frame for
translation beginningnearthe 5' end. In the DNA sequence
ofHAV, the open reading frame started atDNA sequence base 493 with Thr-Ile-Met-Asn-Met and continued until the end ofthe currently identified sequence, an open reading frame of 2,562 bases. A repeat of 30 bases wasfound in clone T28-77 beginning at position 1934,- but this was determined
tobecaused by areading mistake of thereverse
transcript-ase since this repeat was not present in the overlapping clones T28-71andT28-94 nor was it seenbydirect
sequenc-ing ofthe viral RNA. In addition to the sequence ofthe
structural gene region, the 3' end of clone al8 was se-quenced and found to contain a poly(A) tract (data not shown).
A
32P-labeled
oligonucleotide primer was prepared which was 18 bases in length and homologous to aunique region near the extreme 5' end of the sequenced genome. The sequence homologous to the primer was GGACTGGCTC TCATCCAG and was located from bases 271 to 288 in the HAVnucleotide sequence(Fig. 7). Theprimerwasannealed to HAV RNA, and cDNA was synthesized by reversetranscriptase in the presenceofactinomycinD(42p.g/ml)to
prevent snap-back. The size ofthis product analyzed by
electrophoresisthrough apolyacrylamide gel was520 bases
(Fig. 8)indicatingthat the 5'end of the genome was ca. 220
bases from the first base in the DNA sequence presented
here.
Correlationof amino acid sequence data. Inpoliovirusmost ofthe protease cleavages occur atGln-Gly pairs toprocess
thepolyprotein intothe individual mature peptides(12, 23).
Other cleavages occur at Asn-Ser or Tyr-Gly pairs. An
analysis of the proposed open reading frame of the HAV
sequence demonstrated none of the above pairs in the regions predicted for protein processing. Thus, it was be-lieved the protease cleavage sites in HAV were different from those in poliovirus. To locate the position of the sequenceswhichencodedvarious structuralproteins, amino acid sequence data was obtained from acrylamide-gel-puri-fied VP-1 and VP-3 peptides. The N-terminal amino acid sequenceanalysisofVP-1 determinedthat the 5' end ofthe VP-1 gene waslocated atDNA sequence base 1972. There was a directcorrelation between the next 12 aminoacids of
thereading frame(Fig. 7) andthosefoundby theamino acid
sequencing (data not shown). To further confirm the
pro-posedopenreading frame,amino acid sequenceinformation
wasobtainedfrom the mixture of CNBr cleavage peptides of
purifiedVP-1 andofpurified VP-3 (C. D. Bennett and J. V.
Hughes, unpublisheddata). The amino acid sequences of the
mixtures offragmentswerecompared to the DNA sequence.
The correlations found with the projected amino acid
se-quenceof the open reading frame are shown in Fig. 7.
1
2
p-1.35. 1.08- 0.87--0.6
-0.31
- 0.27-
0.28-.0
j.
..."W
NW '.. . j.
"io
_
0.23- A
0.19- _
FIG. 8. Polyacrylamide gel analysis of oligonucleotide-primed
cDNA. An 18-baseoligonucleotide homologoustoHAVRNAnear
the5'end of thegenome waslabeled with32Pbyusing polynucleo-tide kinase and then was annealed to HAV RNA. First-strand cDNA synthesis was performed with reverse transcriptase, as
describedin thetextwith theaddition of42 ,ugofactinomycinD per ml. The resulting cDNA was heated 2 min at 90°C in 90% form-amide-1 mM EDTA and electrophoresed through an 8% poly-acrylamide-8 M ureagel with Tris-borate buffer (lane 2). Thegel
wasfixedin30% aceticacid-30%methanol, dried,andexposedto X-rayfilm. As molecular weight markers, HaeIII-digested iX174 DNA waslabeled with32Pbyusingkinase andwaselectrophoresed (lane1). Thesizesof the marker bandsareindicated in kb.
DISCUSSION
In this paperwe reportthe molecularcloning and partial
sequencing of cDNA prepared from purified Hepatitis A
virusRNA.The cloneswereobtained fromaseries ofcDNA
cloning experiments which yielded overlapping clones
rep-resenting atleast 97%oftheviralgenome.Thecloneswere
verified to be HAV-specific by labeling the inserts and
hybridizing them to various RNA preparations. These
la-beled inserts were shown to hybridize to RNA extracted from HAV grown in LLC-MK2 cells and to cellular RNA fromHAV-infected LLC-MK2cells,butnot tocellular RNA fromuninfected LLC-MK2cells. Inaddition, toensurethat
FIG. 7. Nucleotide sequenceofcloned HAV cDNAfrom the 5' structural generegion of the viral genome. Nucleotide sequence was
determined byusing clones T28-18, T28-71, T28-77, and T28-94 as shown in the schematic diagram in Fig. 6. In addition,thissequence was
verifiedby direct sequencing of the viral RNA in a few areas (see Fig. 6). An open reading frame of 854 consecutive coding triplets was
identified inthe sequence bycomputer analysis with the Intelligenetics software.Amino acids identified by stepwise Edman degradationof purified VP-1 and VP-3 products and matching with the above sequence are shown by underlining as follows (Bennett and Hughes,
unpublished data):VP-1, ;VP-3,---.The N terminus ofVP-1is indicated; however, the precise N terminusof VP-3 has not been
determined.Thelocationofpotentialinitiation codons for translation before this open reading frame areindicatedbybrackets, and those for
terminationcodons areindicatedby overlying dots. Asdescribed in the text, there are ca. 220 bases preceeding this sequence at the 5' end
ofthe viralgenome.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.403.474.70.346.2]the cloned sequences were HAV in origin, not from other sequences possibly found in infected LLC-MK2 cells, the labeled inserts were shown to hybridize to RNA extracted from HAV purified from the livers ofinfected marmosets. The labeled inserts also hybridized positively to cellular RNAextracted from thelivers ofinfectedmarmosets but not to cellular liver RNAfrom uninfected animals.
Arestriction enzyme map was prepared fromthe cloned inserts and was compared with amappublished recently by Ticehurst et al. (30) generated from clones of a different isolate ofHAV. There are many similarities in restriction enzyme sites between the two maps as well as many differences. Clearly, the degree of relatedness between the
two restriction maps is expected between two different
strains of the virus.
The clones we obtained do not extend to the extreme 5' endof theviral RNA andthereforedonot represent approx-imately the first 220 bases of the viral genome. The current
determination ofthe base sequence begins near the 5' end
andreaches a large open reading frame for translation after 493 bases. The first methionine in this large open reading frame is atthe third codon, and there is a second methionine at the fifth codon position. There is no data at present to indicate which methionine acts as the initiator methionine. Clearly, neither ATG codon isfollowed by a G; a G at this +4position has been suggested to be preferred for ribosome initiation sites in eucaryotic mRNAs by the findings of M. Kozak (13). However, both ATG codons have thepreferred A residue at the -3 position (13). The open reading frame continues for 2,562 bases to the end of theregionsequenced. Thelargest open reading frame in the first 493 bases contains only 20 amino acids after a methionine codon. If one examines the other two reading frames, there are only two open frames of significant size which follow methionine residues in the entire sequence presented, one of 56 amino acidsandasecond of55amino acids.It isunknownwhether anyof these or other open frames are used to encode protein products.
We have identified the locations ofthe sequences which encode the HAV structural proteins VP-1 and VP-3. This
was accomplished by comparing the predicted amino acid
sequence with amino acid sequence data generated from
sodium dodecyl sulfate-polyacrylamide gel-purified VP-1
and VP-3. Thiscomparison identified the amino terminus of VP-1 as the valine at DNA sequence position 1972. From CNBrcleavage products of VP-1, fragments beginningwith methionines at DNA sequence positions 2053, 2089, 2236, and 2437 were also identified as originating from VP-1. Amino acid sequencing of purified VP-3 showed theamino terminus to be blocked and unavailable for sequencing as purified; however, fragments beginning with methionines at DNA sequence positions 1237, 1312, 1495, 1552, and 1711
were identified from CNBr cleavage products of VP-3. It is
not possible at this time to determine the precise carboxy terminus of the VP-1 or the precise amino terminus of the VP-3. However, since VP-1 has been shown to have a molecular weight of 33,000, which would require a coding capacity of ca. 900 bases of DNA, the carboxy terminus mustbe near the histidine at DNA sequence position 2875. Since the molecular weight of VP-3 is 27,000, its amino terminus must be very close to the methionine at DNA sequence position 1237. These two structural proteins are in the same relative order on the genome as that found for poliovirus, but each one begins about 250 bases closer to the first methionine in the large open reading frame. In other words, the VP-3 and VP-1are shifted about 250 bases in the
5' direction of the HAV genome as compared with the
poliovirus genome.
Regardless of this apparent shift in the position of the structural gene sequences, the sequence ofthe clones indi-cates an overallgenomeorganization ofHAVwhichis very
similar to that of other picornaviruses. Also, as has been
describedfor poliovirus and FMDV (1), HAV has a stretch
ofpyrimidine residues
preceding
thefirst ATGcodonsin thelarge open reading frame. This structure may be an
impor-tant site for the recognition ofribosome binding in
picorn-aviruses. However, there is no significant homology
be-tweenthe DNA sequence ofHAVand that ofpoliovirusor
foot and mouth disease virus. Also, there is no obvious
homology between these viruses at the level ofthe amino
acid sequence based on the translation of the nucleotide sequences. In addition, the amino acid pairs which are
cleaved during processing of the polyproteins are different
for these viruses. In poliovirus, proteolytic cleavage occurs
8outof10timesatGln-Gly pairs,theC-terminalcleavageof
VP-1 being at a Tyr-Gly pair and that between VP-4 and VP-2being at an Asn-Serpair (12). The sequence specificity
for the proteolytic processing is much broader in FMDV,
where different amino acid sequences are found at three
cleavage sites in the structural proteins (1, 2). Even in
poliovirusthere must beadditional determinants, other than
the Gln-Gly sequence, to specify the cleavage sites since proteolysis does not occur at all Gln-Gly pairs in the poliovirus proteins or in every protein in the infected cell. The only cleavage site we have located in HAV occurs at a Gln-Val pair atthejunctionbetween the VP-3 and VP-1; this assumes of course that there is no additional processing of the N terminus ofthe VP-1 to generate the matureprotein. Although this is not exactly the same as the Gln-Gly pair used in poliovirus or the Gln-Thrused in FMDV, there is a similarity in the VP-3-to-VP-1 cleavage sites for these picornaviruses.
It hasbeen shown recently that atleast one neutralization epitope for HAV is present on the VP-1 (10). This was demonstrated bycross-linking virus-neutralizing antibodyto virus particles and analyzing which viral proteins were cross-linked. This suggests the ratherobvioususe ofatleast
the VP-1 protein or a peptide of it for development of an
HAV vaccine. Now, with the cDNA clones and identifica-tion ofthestructural genesequences, itshould bepossibleto producevaccines forHAV by recombinant DNA or in vitro synthesis techniques. We are currently working toproduce HAV structural gene products by in vitro expression of the cDNA clones and by in vitrooligopeptidesynthesisbased on the translation of the HAV cDNA sequence presented here.
ACKNOWLEDGMENTS
We thank Carl D. Bennett for providing amino acid sequence analysis of thepurified VP-1andVP-3 proteins andJoanne Tomas-sini, Abner Schlabach, DarleneWilliams, Linda Stanton, and Paula
Giesa for the assistancein growth andpurificationofthevirus in cell
cultures. We also thank Joel Shapiro for synthesis ofthe
oligonu-cleotide primer.
A.M.G. is the recipient ofafellowship from the Ramon Areces
Foundation, Madrid, Spain.
LITERATURECITED
1. Beck, E., S. Forss, K. Strebel, R. Cattaneo, and G. Feil. 1983. Structure of the FMDV translation initiation site and of the structural proteins. Nucleic Acids Res. 11:7873-7885.
2. Boothroyd, J. C., P. E. Highfield, G. A. M. Cross, D. J.
Rowlands, P. A. Lowe, F. Brown, and T. J. R. Harris. 1981. Molecular cloning of foot and mouthdisease virus genome and
on November 10, 2019 by guest
http://jvi.asm.org/
nucleotide sequences in the structural protein genes. Nature (London) 290:800-802.
3. Chirgwin, J. M., A. E. Przybyla, R. J. MacDonald, and W. J. Rutter. 1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry
24:5294-5299.
4. Coulepis, A. G., S. A. Locarnini, and I. D. Gust. 1980. lodina-tion ofhepatitisAvirus reveals afourth structural polypeptide.
J. Virol. 35:572-574.
5. Coulepis, A. G., G. A. Tannock, S. A. Locarnini, and I. D. Gust. 1981. Evidencethat the genomeof hepatitisAvirusconsistsof
single-strandedRNA.J. Virol. 37:473-477.
6. Dienstag, J. L. 1981. HepatitisA virus: virologic, clinical, and
epidemiologic studies. Hum. Pathol. 12:1097-1106.
7. Dienstag, J. L., A.Alaama, J. W. Mosley, A. G. Redeker, and R. H. Purcell. 1977. Etiology of sporadic hepatitis B surface
antigen-negative hepatitis. Ann. Intern.Med. 87:1-6.
8. Dienstag, J. L., A. N. Schulman, R. J. Gerety, J. H. Hoofnagle, D. E.Lorenz, R. H. Purcell, and L. F. Barker. 1976.HepatitisA
antigen isolated from liverand stool: immunologic comparison of antisera prepared in guinea pigs.J. Immunol. 117:876-881.
9. Gust,I.D., A. G. Coulepis, S. M. Feinstone, S. A. Locarnini, Y. Moritsugu, R. Najera, and G.Siegl.1983. Taxonomic classifica-tionof hepatitisA virus.Intervirology 20:1-7.
10. Hughes, J. V., L. W.Stanton, J. E. Tomassini, W. J. Long, and E. M. Scolnick. 1984. Neutralizing monoclonal antibodies to
hepatitisAvirus:partial localization ofaneutralizing antigenic site.J. Virol.52:465-473.
11. Jacobson, M. F., J. Asso, and D. Baltimore. 1970. Further
evidenceontheformation ofpoliovirus proteins. J. Mol. Biol. 49:657-669.
12. Kitamura, N., B. L.Semler, P. G. Rothberg, G. R. Larsen, C. J. Adler, A. J. Dorner, E. A. Emini, R. Hanecak, J. J. Lee, S. van der Werf, C. W. Anderson, and E. Wimmer. 1981. Primary
structure, gene organization and polypeptide expression of
poliovirusRNA. Nature(London) 291:547-553.
13. Kozak, M. 1984. Point mutations close to the AUG initiator
codon affecttheefficiency of translation ofratpreproinsulinin vivo. Nature(London) 308:241-246.
14. Kupper, H., W. Keller, C. Kurz, S. Forss, H. Schaller, R. Franze, K. Strohmaier, 0. Marguardt, V. G. Zaslavsky, and P. H.Hofschneider. 1981. Cloning ofcDNA ofmajorantigen of foot and mouth disease virus andexpression inE.coli. Nature
(London)289:555-559.
15. Lamb, R. A., and C. J. Lai. 1980. Sequence of interruptedand
uninterrupted mRNAs and cloned DNA coding for the two
overlapping nonstructural proteins of influenza virus. Cell
21:475-485.
16. Locarnini, S. A., A. G. Coulepis, E. G. Westaway, and I. D. Gust. 1981.Restrictedreplication of human hepatitisAvirusin
cell culture: intracellular biochemical studies. J. Virol. 37:216-225.
17. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular
cloning:alaboratory manual. Cold Spring Harbor Laboratory,
ColdSpring Harbor, N.Y.
18. Maxam, A. M., and W. Gilbert. 1977. A new method for
sequencingDNA. Proc. Natl. Acad.Sci. U.S.A. 74:560-564. 19. Messing, J., R. Crea, and P. H. Seeburg. 1981. A system for
shotgun DNA sequencing. Nucleic Acids Res. 9:309-321. 20. Okayama, H., and P. Berg. 1982. High-efficiency cloning of
full-length cDNA. Mol. Cell. Biol.2:161-170.
21. Provost, P. J. 1984. In vitropropagation of hepatitisA virus,p. 245-261. In R. J. Gerety (ed.), Hepatitis A. Academic Press, Inc.,New York.
22. Provost, P. J., and M. R. Hilleman. 1979.Propagation of human
hepatitis Avirus in cell culture in vitro. Proc. Soc. Exp. Biol. Med. 160:213-222.
23. Racaniello, V. R., and D. Baltimore. 1981.Molecularcloningof
polioviruscDNAand determination of thecompletenucleotide sequenceofthe viral genome. Proc. Natl. Acad. Sci. U.S.A. 78:4887-4891.
24. Rakela, J., A. G. Redeker, V. M. Edwards, R. Decker, L. R.
Overby, andJ.W.Mosley. 1978. HepatitisA virus infection in
fulminant hepatitis and chronic activehepatitis. Gastroenterol-ogy74:879-882.
25. Rueckert, R. R., T. J. Matthews, 0. M. Kew, M. Pallansch, C. McLean, and D.Omilianowski. 1979. Synthesis and processing of picornaviral polyprotein, p. 113-125. In R. Perez-Bercoff (ed.),The molecularbiology ofpicornaviruses. Plenum Publish-ingCorp., NewYork.
26. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA
sequenc-ing with chain terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A.74:5463-5467.
27. Siegl,G., and G. G.Frosner. 1978. Characterizationand
classi-fication of virus particles associated with hepatitisA. I. Size, density,and sedimentation. J. Virol.26:40-47.
28. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separated by gel electrophoresis. J. Mol.Biol.
38:503-517.
29. Thomas, P. S. 1980.Hybridization of denaturedRNAand small
DNAfragments transferredtonitrocellulose.Proc.Natl. Acad. Sci. U.S.A. 77:5201-5205.
30. Ticehurst,J. R.,V. R.Racaniello,B. M.Baroudy, D.Baltimore,
R. H.Purcell, and S. M. Feinstone.1983.Molecularcloning and characterizationofhepatitisAviruscDNA. Proc.Natl. Acad. Sci. U.S.A. 80:5885-5889.
31. Von derHelm,K., E. L.Winnacker, F. Deinhardt, G. Frosner, V. Gauss-Muller, B. Bayer, R. Scheid, and G. Siegl. 1981.
Cloning of hepatitisAvirus genome. J.Virol. Methods3:37-43. 32. Zimmern, D., and P. Kaesberg. 1978. 3' Terminal nucleotide
sequence of encephalomyocarditis virus RNA determined by
reverse transcriptase and chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 75:4257-4261.