JOURNAL OFVIROLOGY, JUIY1991, p. 3864-3872 0022-538X/91/073864-09$02.00/0
Copyright ©1991,AmericanSociety forMicrobiology
Overlapping
Retrovirus U5
Sequence
Elements
Are
Required for
Efficient
Integration
and
Initiation of
Reverse
Transcription
DAVID COBRINIK,t ASHOKAIYAR, ZHENG GE,MICHAEL
KATZMAN,t
HAN HUANG,§ANDJONATHAN LEIS*
Department ofBiochemistry, Case Western Reserve University SchoolofMedicine, Cleveland, Ohio44106 Received 4February 1991/Accepted 9 April 1991
Asecondary structureinthe 5' noncoding regionof avianretrovirusRNA,calledtheU5-leader stem, was
shownpreviouslytohavearole in initiation ofreversetranscription (D. Cobrinik,L. Soskey, andJ. Leis, J. Virol.62:3622-3630, 1988). Wenowshow thatanadditional RNAsecondarystructureneartheU5 terminus, called the U5-IRstem,isalsoimportantforreversetranscription. Mutations thatdisruptthe U5-IR stemcause
areplicationdefect associated with bothadecreaseinsynthesis ofviral DNA in infected cells andadecrease
ininitiation ofreversetranscriptioninmelittin-permeabilized virions. Structure-compensatingbase substitu-tionsintheU5-IRrestorereversetranscriptionefficiency. In viralDNA, U5-IRsequencesareincludedinthe U5term'inalregionthat functionsas aviralintegrationdonor site. When basesubstitutionsareintroducedinto thesesequences,areducedefficiencyofintegrationinvitro andinvivois observed. Theseobservationsindicate thatU5-IRsequenceshaveastructural role inreversetranscriptionofviral RNAandasequence-specificrole in theintegration ofviral DNA.
Theearlysteps ofretrovirus replicationconsistofreverse
transcription ofviral RNAandintegrationof viral DNAinto cellular DNA. Reversetranscriptionis initiatedbyextension ofaspecifictRNAprimerannealedtosequencesnearthe 5' endofthe viralgenome.Anumber ofcomplexsteps result in theproduction of double-strandedlinear viral DNA contain-inglongterminalrepeat(LTR)sequencesin thecytoplasmof infectedcellsaswellaslinear and circular viral DNA forms incellnuclei (8). The linearform hasrecentlybeenshownto be the primary substrate for integration. This reaction in-volves cleavage of the U5 and U3LTRterminibythe viral INproteinfollowed byligationtocellularsequencesthatare
also likelyto becleaved byIN (5, 12, 15, 21, 26).
Avianretrovirusparticlesareinfectiousasearlyas10min after budding (30), yet there is no detectable reverse tran-scription priorto cellinfection (4, 13, 31). The mechanism that restricts reverse transcription in virions is notknown. However, we have recently found thatan RNA secondary
structurecalled the U5-leaderstem affects theefficiency of initiation andhave proposed that thisstructure servessucha
regulatory role (7).
TheU5-leader stem isapotential RNA secondary
struc-ture consisting ofsequences in the middle of U5 and in the leader region adjacent to the primer-binding site (7). Muta-tions which disrupt this structure impairreverse
transcrip-tion in infected cells, while mutations which alter the
se-quence but retain the stem structure have no effect (7).
Studies withpermeabilized virions have localized the defect to the initiation ofreverse transcription. Disruption of the
U5-leader stemstructurecauses adecrease in incorporation
* Correspondingauthor.
t Present address: WhiteheadInstituteforBiomedical Research, Cambridge, MA 02142.
tPresentaddress: DepartmentofMedicine, TheMilton S. Her-shey Medical Center, Pennsylvania State University College of
Medicine, Hershey, PA 17033.
§ Present address: Department ofPathology and Immunology,
Case WesternReserve University School of Medicine, Cleveland, OH44106.
of the firstdeoxynucleotides fromthe3'OH terminus ofthe tRNATrP primer, while the amount of thetRNATFP primer annealedtothe RNA primer-binding site is equivalenttothat found inwild-type virions (7).U5-leaderstemstructuresalso
appeartobe conserved among most retrovirus families (1, 7).
The U5-leaderstemof avianretrovirusesis in aregion of RNA whichcanpotentially formnumeroussecondary struc-tures (14, 17). In this studyweinvestigated whetherone of these other structures, termed the U5-inverted repeat
(U5-IR)stem(see Fig. 1A), could also affect initiation ofreverse
transcription. In viralRNA, thisstructureis composed ofan
invertedrepeatsequenceadjacenttotheprimer-binding site. In viral DNA, the U5-IR stemfalls within the U5 terminal
sequences that arerecognized by the IN protein and func-tionsas aviralintegrationsite(21, 22). Thus, mutationsthat
disrupt the U5-IR stem structure and alter its nucleotide
sequencecouldalso result indefectstointegration.Ourdata indicate that U5-IRsequences are indeed requiredfor both replication functions.
MATERIALSANDMETHODS
Reagents. The Klenow fragment of DNA polymerase I (4
U/4l),T4DNAligase(2,000U/pul),T4polynucleotide kinase (10U/pl),andvariousrestrictionenzymes werefrom
Boehr-ingerMannheimBiochemicalsorNewEngland Biolabs, Inc.
[cx-32P]dATP (800 Ci/mmol), [a-32P]dCTP (3,000 Ci/mmol), and [y-32P]ATP (6,000 Ci/mmol) were from New England
NuclearCorp. pTZ18 DNA wasobtained from Pharmacia.
Thermostable DNA polymerase (Pyrostase; 4 U/,l) was
fromMolecularGeneticsResources.
Construction of U5-IR viral mutants. pRCAS-B and pRCAS-neo were generously provided by S. Hughes
(Na-tional Cancer Institute-Frederick Basic Research Facility). pRCAS-B is the pRCAS vector (20), except that it has a
central EcoRI fragment (spanning gag andpol genes)
de-rived from a cloned Bryan high-titer Rous sarcoma virus
(RSV) genome. pRCAS-neo contains the neomycin phos-photransferase gene inserted into the pRCAS ClaI cloning
3864
Vol.65,No. 7
on November 10, 2019 by guest
http://jvi.asm.org/
site. pDC1O1B was derived from pRCAS-B by deletion of the EcoRV fragment between nucleotide 3655 (28) and pBR322 nucleotide 189. pDC102 was constructed by diges-tion of RCAS-neo with Sacl, partial digesdiges-tion with RsrII,
formation of blunt ends with the Klenow enzyme, and ligation toHindII-digested pTZ18. The gag-pol EcoRI frag-ment was then replaced with that of the Bryan high-titer clone to form pDC102B. U5 mutations were introduced into pDC1O1B by insertion of synthetic deoxynucleotides be-tweentheRsrII and BstEII sites. All clones were confirmed by sequencing.
Transfections were performed by the calcium phosphate
precipitation technique (27) with ligation products derived
from 7 ,ug ofdigested pDC102B and 5 ,ug of
HpaI-digested pDC1O1B (or its derivatives) per semiconfluent 60-mm-diameter dish of QT6 cells. The cells were fed 1 day after transfection and split on day 2, and selection was
initiated on day 2. Virus production was monitored by an exogenous reversetranscriptase assay (7) with the following
modifications: the final Nonidet P-40 concentration was
0.067%, and 15
RIJ
of virus was assayed per 30-,ul reaction mixture.Analysisofreversetranscription products inpermeabilized virions. Virus preparations and melittin reactions were as previously described (7), except that the reaction volumes were 30
RI.
The amount of virus used in each reaction was that which incorporated 0.5 pmol of dCTP in a 30-min reverse transcriptase assay performed on a 1/10 dilution of the recovered virus and supplemented with excess dCTP. Thefinal concentrationand specificactivity of[a-32P]dATPwere 2 ,uMand 330 Ci/mmol, respectively, when dATP was present as the only deoxynucleotide, and 25 ,uM and 40
Ci/mmol, respectively,whenitwasused in combination with 1 mM (each) dCTP, dGTP, and dTTP. The dATP was not rate limiting at these concentrations. The final melittin concentration was 7.5 ,ug/ml.
Analysis of integrated viral DNA. High-molecular-weight cellular DNA waspreparedby the method of Hirt (19). The DNA was sheared by passing it through a 21-gauge needle severaltimes, heatedto 55°C, layered onto a gradient of 10 to40% sucrosein 0.1 MNaCl-10mMTris HCI (pH8.0)-1 mMEDTA, and spunfor16 h at 25,000 rpm in anSW28 rotor at4°C. Fractions containingDNAof>25kbwerecombined,
precipitatedwith 2volumesof ethanol, and used in polymer-asechain reactions (PCRs). Primers complementary to the gag gene (nucleotides 2251 to 2270) and to the pol gene
(nucleotides 2667 to2583) (28) were used in PCRs with the
following cycling conditions: 92°C for2 min, 55°Cfor 90 s, and then72°C for2 min.Amplification of pRCAS-BDNA,at concentrations between 0.01 to 1 pg, was linear up to 25
cycles.Thecell-derivedDNAbeinganalyzed forviral DNA wasthusamplified for24cyclesasdescribed elsewhere(23)
by using Pyrostase anda Hybaid thermal cycler. Amplified
DNAproductsweresubjectedtoagarosegel
electrophoresis
and transferred to nylon membranes (27), and viral DNA sequences were detected by hybridization to
[32P]dCTP-labeledprobes(2 x 109
cpm/Vig).
Theprobeswereprepared by DNA polymerase I (Klenowfragment)
extension of primers complementary to the gagandpol
genesand DNAcontainingportions ofthese genes
amplified
from the viral clonepATV8 (6).IN protein cleavage reactions. Reactions were
performed
asdescribed previously (22).
Integrative recombination invitro. Substrate
oligodeoxy-nucleotidesrepresenting wild-typeor mutantU5 sequences
were prepared as described previously (22) and are as
follows:
U5wild-type plus strand 5'TGAAGCAGAAGGCTTCA3' U5 wild-type minus strand 3'TCGTCTTCCGAAGTAA5'
U5 S2 plus strand U5 S2minus strand U5 S4plus strand U5 S4minus strand U5 S4C plus strand U5 S4C minus strand
5'AGCAGAAGGCAACA3'
3'TCGTCTTCCGITGTAA5'
5'AGCAGAAGGGAAGA3' 3'TCGTCTTCCCTTCTAA5'
5'TCCAGAAGGGAAGA3'
3'A>CTTCCCTTCTAA5'
The underlined sequences indicate base changes from the wild type for the S2, S4, and S4C mutations (see Fig. 1B and C). After the plus and minus strands are annealed, the resultant duplexes have a two-base 5' minus-strand over-hang.
Integrativerecombinationreactions were carried out iri 10-,u reaction volume containing 1 pmol of each of the duplexes shown above,5' 32plabeled only in the plusstrar.,
(105
cpm/pmol); 25 mM Tris HCl (pH 8.3); 10 mM 2-me,captoethanol; 3 mM MgCl2; and 1 pmolof IN. The reaction mixtures were incubated at 37°C for 90 min. The reactions were stopped by the addition of 10 ,ul of sequencingloading buffer (22), and the mixtures were heated to90°C for 5 min andanalyzedon a20%polyacrylamide-7 M ureasequencing
gel (22).
RESULTS
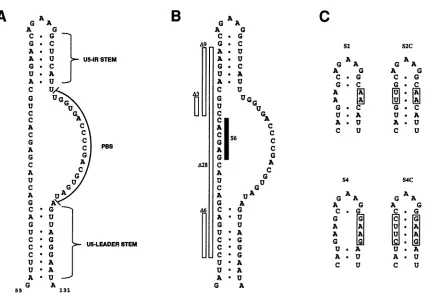
The RSV(Schmidt-RuppinA)RNAsequencefrom
nudle-otides 55to 131, which includes the primer-binding site and thepreviouslystudiedU5-leader stem structure (7), is shown inFig. 1A. This region also includes an additional secondary structure termed the U5-IR stem. This structure comprises
the inverted repeat sequencebetweennucleotides 81 and 101
(U5-IR) andcanpotentially form a7-bp stemwitha 5-base loop. Analyses ofthe RSV 5' noncoding region have indi-cated that the U5-IRstemisthermodynamicallystableand is conserved among all members of the avian sarcoma and leukosis retrovirusfamily forwhich sequenceinformationis available(1, 3,7). This structuralconservationsuggests that theU5-IR stemmay havean importantrole in viral
replica-tion. We sought to understand this role by analyzing the effectsofmutations in the U5-IR sequenceon viral
replica-tion.
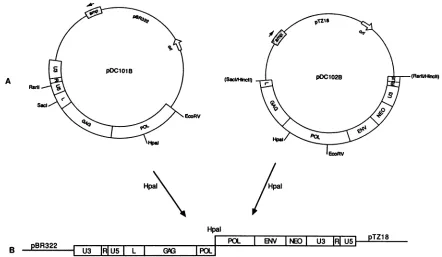
Production ofU5-IRmutantvirus.Thevirusvectorsystem used for these studies was derived from the pRCAS-neo
vector described by Hughes et al. (20). pRCAS-neo is a
Schmidt-Ruppin Astrain of RSV in which the viralsrcgene isreplacedby the neomycinphosphotransferasegene. Two
plasmids were derived from pRCAS-neo:
pDC1O1B,
whichcontains the 5' half ofpRCAS-neo, and
pDC102B,
whichcontains the
majority
of the viral genome but lacks se-quences5' oftheleaderregionSacl site and those 3' ofthe U5regionRsrIIsite in the downstreamLTR(Fig.
2). Both of these plasmids contain Bryanhigh-titer
viral sequences, which confer the high-titer growth phenotype, within por-tions of the gag andpol genes.To obtain virus-producer cells,
pDC102B
andpDC1O1B
were digested with
HpaI,
ligated to assemble afull-length
viraltranscriptionunit,andtransfected into
quail QT-6
cells. Cells thatexpressed
the virus-encoded neo gene were then selected in the presence of G418. Theadvantages
of thison November 10, 2019 by guest
http://jvi.asm.org/
3866 COBRINIK ET AL.
A A
G . G .-..
. C
. U
U U5-IRSTEM . c * A UW U G U G C\ C C C PBS G A C/ G/
GU/
uA A * G U* G US-LEADERSTEM
* G * A * A
* U
-A 131
B
A9 128 G A C G A A G U A C G u C C A C G A G C A U C A G C A G U C C C U U A G AA G * G . c .U * U . C * A . U U UGG UGA
AC
S6 C CC G A C G U GuA
A * G * U U * A * G * G * G * A * A A U AC
S2 S2C GA GAAA G A G
C .G C. G
G .C G C
A
G .C G. C
U .A U A
A.*U A.*U
C U C U
S4 S4C
A A
GA GA
A G A G
C .G C. G
G
M.ICi1
A A U .IA
A . U A.U
C U C U
FIG. 1. Predicted RNAsecondarystructuresfor the 5'noncoding regionofwild-typeandmutantRSV(Schmidt-RuppinA)RNAnearthe
primer-bindingsite(PBS). (A) Wild-typestructure.Numbersindicate nucleotides from the viral RNA 5' end. ThePBS, U5-leader stem,and
U5-IRstemareindicated. (B) U5-IRstemmutationsA3 and A9. TheA6,A28,and S6 mutationsaredescribedinthe text.Theyareanalyzed elsewhere(1, 6, 7).Sequence deletionsareindicatedbyopenbars;thesequencesubstitution isindicatedbythestippledbar.(C)U5-IRstem substitution and compensating substitution mutationsS2, S2C, S4, andS4C. Sequence substitutions are boxed, and the effects ofthese substitutionsontheU5-IR stem structureareshown.
system are that US mutations can easily be cloned into pDC1O1B (see MaterialandMethods), thattransfectionscan
be performed without additional subcloning steps, and that largeamountsofdefectiveviruscanbeproducedfromQT-6
cells. In addition, truncation of the downstream LTR of pDC102B ensures that mutations introduced into the
pDC1O1B U5 region cannot undergoreversion by recombi-nation with wild-type U5sequences.
Threetypesof mutations wereintroduced into the U5-IR stem: deletionmutations A3 andA9, substitution mutations S2 and S4, and structure-compensating substitution muta-tionsS2C and S4C (Fig. 1B and C). The A3 mutation does not disrupt the U5-IR stem but decreases the spacing be-tweenthe U5-IRandU5-leaderstemstructures.The A9,S2, andS4mutationsdisrupttheU5-IRstem,whiletheS2Cand S4C mutations retain the U5-IR stem structurebut alterits
sequence. We found that release of virus, as measured by
reverse transcriptase activity, from pooled producer cells
was roughly the same for the wild type and each of the mutant viruses described above and showed no correlation
with viral growth rates (data not shown). This strongly suggests that late replication events such as transcription,
splicing, translation, particle assembly, and budding were
notaffected by the U5-IR mutations.
Analysis of viral growth rates. Viruses with each of the mutations described above, obtained from QT-6 producer cells,wereusedtomeasuretherateof viral growthfollowing infection of fresh QT-6 cells. Reverse transcriptase assays
usingexogenous template primers were performed directly
on cell supernatants both to equalize the amount ofvirus used for infection andtomonitor theappearanceofprogeny
virus. Results represent the average of several infections which had nearly identicalrates of viralgrowth.
The substitutionmutationsS2andS4resultedinimpaired viralgrowth compared with the wild type (Fig. 3). TheA9 and A3 deletion mutations also resulted in impaired and delayed growth of virus (datanotshown). The viruses with structure-compensating mutations, S2C and S4C, displayed improved growthkinetics comparedtotheiruncompensated S2 andS4counterparts, butwerestillsignificantly defective in growth compared with thewild type(Fig. 3). The
differ-ences between S2 and S2C and between S4 and S4C were
reproduciblyobserved infour separate kinetic virusgrowth experiments. This partialrestorationof growthefficiency for S2C and S4C suggests that the U5-IR stem structurehas a
role inreplicationthat isatleastpartiallysequence
indepen-dent. However, the delayed growth observed with viruses containing structure-compensating mutations indicates that there is a major sequence-dependent replication function
encoded in the U5-IR.
Each ofthe virus mutants analyzed in this study is only partially defective. The eventual appearance of virus is
unlikely to be the result of genetic reversions, since the
progeny virus exhibited the same impaired-growth
pheno-type as that exhibited by virus derived from the original
producercells (datanot shown).
Analysis ofreverse transcription in permeabilized virions.
We have previously shown that reverse transcription is
A
G A C G A A G U A C G U C C A C G A G C A U C A G C A G U C C C U U A G 55 J. VIROL. rli 9 16on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.103.534.72.365.2]EcoRV
HpaI HpaI
Hpal
[image:4.612.88.530.79.336.2]r
POL I EWN INEDI U3 IRIU5I pTZ18FIG. 2. Strategy forproduction of virus containing U5 mutations. pDC1O1B (orU5 mutantderivatives) andpDC102B aredigested with
HpaI; theproductsareligated andtransfected into QT-6 cells.(A)Structure of pDC1O1BandpDC102B.Wide bars indicate viralsequences.
Thinlines indicateprokaryoticvectorsequences.TheU3, R, U5,and leader (L) noncodingregionsand thegag,pol,env,andneogenes are
indicated. The sizes of indicated regions of the viralgenome arenot toscale.ori,originof replication.(B)Structure of the ligation product
fromwhichboth theneogeneandfull-length viral RNAcanbe expressed.
substantiallyless efficient when theUS-leaderstem structure
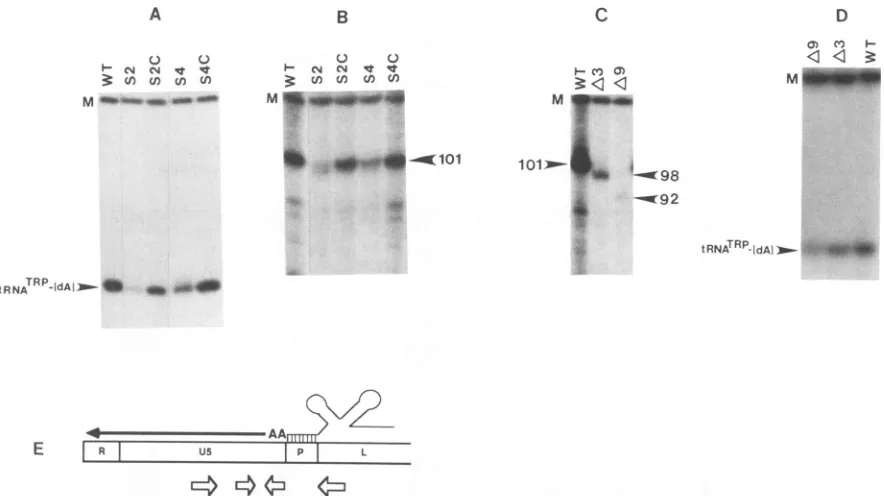
is disrupted (7). This was established by analyzing the
synthesis of the initial intermediate inreversetranscription, a 101-nucleotide cDNA (cDNA101) extending from the
tRNATrp primertothe 5'terminus of viral RNA, in melittin-activated virions (Fig. 4E). In order to evaluate whether mutations in the U5-IR stem also affect reverse
transcrip-tion, equal amounts of wild-type and mutant virus, deter-mined by an exogenous reverse transcriptase assay, were
incubatedwith melittin and [t-32P]dATP in the presence of
dCTP, dGTP,anddTTP. cDNAswereisolated from virions
and treated with alkali to remove the covalently linked
tRNATrp, and the amount ofcDNA101 was determined by
polyacrylamide gel electrophoresis (Fig. 4B). The substitu-tionmutations S2 and S4 resulted in decreased amounts of
cDNA101synthesized,withalargerdecreasebeing observed for the S2 virions. Structure-compensating mutations
re-stored thereverse transcription efficiency,with that ofS4C restored essentially to the wild-type level. Only a partial
restoration was obtained with S2C (Fig. 4B). These
differ-ences arenotduetoapackagingdefect inS2CandS4C (see below).
ThedeletionmutationsA3 and A9 also resulted inreverse
transcription defects. Because of the deletion of U5
se-quences, the cDNA productsinthesecases werethree and nine bases shorter than that of the wild type, respectively (Fig. 4C). The A9 mutation resulted in barely detectable amounts of cDNA92, while the A3 mutation resulted in reduced levels of cDNA98 compared with the wild type. These results correlated with the growthcharacteristics of A3 and A9.
The first two deoxynucleotides incorporated onto the 3'
OH terminus of the tRNATrP primerare dAMPs (Fig. 4E).
Thus, initiation ofreverse transcription can be determined by monitoring the appearance ofa dA-tRNATrP formed in
virions when[ao-32P]dATP is included in the melittin reaction mixtures as the only deoxynucleoside triphosphate. The
relative amounts of dA-tRNATrP produced with the wild type and each of the mutant viruses (Fig. 4A and D) were
similar to the amounts of cDNA intermediate products obtained when all deoxynucleotides were present. These results indicate that disruption of the U5-IR stem impairs initiation of reverse transcription in melittin-activated
viri-ons.
Analysis offull-length double-stranded viral DNA in the cytoplasmofcells 12 h after infection with each of the U5 mutant viruses described above yielded results parallel to
thoseobserved inFig. 4; theamount of viral DNAdetected bySouthern blottingcorrelated with the amounts of strong-stop cDNA anddA-tRNATrPproduced in melittin-activated virions(data notshown). Theseresults cannotbeexplained by a RNA packaging defect, since equal amounts of wild-type and mutant virus (asjudged by reverse transcriptase
activity measured with exogenous template primers)
con-tainedequivalentamountsofviral RNA(as judged byadot blot assay using a labeled viral cDNA probe [data not
shown]). Taken together, these results indicate that the
U5-IRstem structure, like the U5-leader stem structure, is necessary for efficient initiation ofreverse transcription in
infected cells.
Effect of U5 mutationson integration in vivo. The in vitro
and in vivoanalyses described above revealed thatreverse
transcription was notdefective for the S4C mutantandwas
only partially defective for the S2C mutant. Since these
A
Rsai.
B 2
on November 10, 2019 by guest
http://jvi.asm.org/
3868 COBRINIK ET AL.
E
0.
I-5;
c)
w
Cn
E-0.
cn
z
r
E
w
cn
w w UJ
C.
0
C.)
cn
:
cn
z :
w
cn
Cl)
w w
2000
1000
0
2000
1000
0
l
0
10 20
DAYS AFTERINFECTION
10 20
DAYS AFTERINFECTION
FIG. 3. Viral growthratesfollowing infection of QT-6 cellswith equal amounts of wild-type and U5-IR substitution mutant virus. Theviruswasdetectedbyassayofreversetranscriptase activityin
cell supernatants as described in Materials and Methods.O, wild-typevirus. (A) *,S2 virus;O,S2C virus. (B) A,S4 virus; A, S4C
virus.
viruses displayaconsiderable reductioningrowth efficiency
(Fig. 3), replication steps other than reverse transcription
seemed likelyto be affected. We felt that integration could be impairedin suchmutants, since theS2, R2C, S4,andS4C substitutions alterthe U5 terminal sequence recognized by
the viral INprotein (22). Toevaluate this possibility, the in
vivo integration efficiency formutantviruswascompared to
that ofthe wild type.
Equalamountsof eachmutant virus (measured byreverse
transcriptase activity) wereusedtoinfectQT-6 cells,andthe
high-molecular-weight Hirt pelletDNA (19), which contains
integrated provirus sequences, was extracted from cell
nu-clei after 12 h. In order tominimize the amount of
contam-inatingunintegrated viralDNA,high-molecular-weight DNA (length of>25 kb) was isolated by sucrose densitygradient
centrifugation as described in Materials and Methods. Viral
DNA was then detected by amplifying a 418-bp fragment fromthe pol region by the PCR technique and then
hybrid-izingthefragment by Southern blottingtoa32P-labeled viral
DNA probe. Various concentrations of pRCAS-B DNA (0.01 to 1 pg) wereamplified (Fig.5A) inparallel reactions.
This approach is similar to that previously described for
quantitating cellular mRNA levels by PCR (27).
The amount of viral DNA detected after 24 cycles of amplification for each of the U5mutantsis showninFig.5B. Comparison of the amount of amplified 418-bp fragment from the wildtype (Fig.5B, lane 4)with control DNA(Fig. SA,lane 2)indicatedthat approximately0.1pgof the former
was present before amplification. In the absence of virus infection, amplification of a 418-bp fragment was not de-tected (Fig. SB, lane 3). Theamounts ofS2, S2C, S4, S4C,
A9, andA3proviral DNAwereestimatedbydensitometryto
be 18, 25, 29, 44, 7, and 20% of the amount ofwild-type DNA, respectively (Fig. 5B).
A decrease in the amount of proviral DNA detected in cellsunder these conditions couldresult from eitheradirect effect upon integration or a decrease in the amount of the
linearviral DNAprecursortointegration synthesized during the early steps of viralreplication. Since the S4C virus does
not display a reverse transcription defect, we estimate that this mutationmustcausemorethanatwofold decreasein the efficiency ofintegration ofviral DNA. Since the S2, S4, and S2C mutants have defects in reverse transcription, the decreaseinintegrated proviruscannotbe attributedsolelyto
an integration defect. For example, the fivefold decrease in viral DNA detected for the S2C mutant might result from a
three- to fourfold decrease in integration efficiency and a
1.5-fold decreasein reversetranscription efficiency (Fig. 4).
Similar combined reverse transcription and integration
de-fects arelikelyto occurfor the S2and S4 viruses. Forthe A3
and A9 viruses, the reduction in proviral DNA is likely to result solely from defectsin reverse transcription, since the integration donor site sequences are intact in these mutants (21, 22).
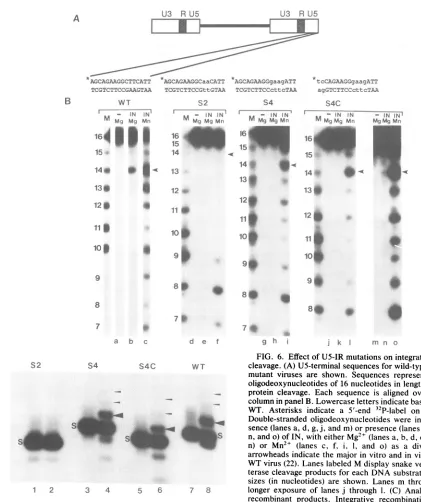
Effect of US mutations on integration in vitro. To further define integration defects produced by U5-IR mutations, duplex oligodeoxynucleotides corresponding to the 16 ter-minal US bases of these mutants were evaluated for their abilitytobenicked by theviral INproteinin vitro(Fig. 6A). Previous studies have shown that purified avian retrovirus IN protein nicks double-stranded DNA at the in vivo viral integrationsite, twobasesfrom the U3 and U5LTR termini (22). In this assay, cleavage ofa 5'-32P-labeled plus-strand
16-meratasitetwobasesfrom the US terminus is monitored by conversion toa 14-mer.
In the presence of
Mg2",
the S2, S4, and S4C mutations nearly abolished the ability of the US terminal sequences to be cleaved (Fig. 6B, lanes b, e, h, and k). However, some14-merproductcouldbedetected for theS4Csubstrate after
alongerexposure of theautoradiogram (Fig. 6B, lane n). In the presence of Mn2 , the endonuclease activity of IN was
20times higher than in the presence ofMg2+, but cleavage
occurred at manysites (lane c). When analyzed with Mn2 ,
cleavage of the S2 substrate was substantially reduced (lane
f). Cleavage of the S4 and S4C substrates was detected,
although thetotalamountofcleavage wasless than withthe wild-type substrate (lanes fand i). These data indicate that base changes in the US-IR stem decrease the efficiency of cleavage by IN and that the two-base substitution (S2) is
moredeleterious to cleavage thanthe four-base substitution (S4).
Oligodeoxynucleotides can also be used to demonstrate thejoiningreactioninvitro (12, 21). This is evidenced bythe
appearanceofoligodeoxynucleotides longer thanthestarting
substrate which arise by a self-integration reaction. The
substrates used in these experiments representLTR termini already nicked at the integration site. This allows one to examine primarily thejoining reaction, although some DNA
B
I
=13^
A A
A~~A
a &
A'-1
AN *..A1 -.,~A I .
I
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.65.298.79.426.2]A
N
3 U)enc en
M _ __m
B
0 0
NCM N T t
3: u) u cni
M _iuiat_
m
1*
0).owl go-loww o.w101iC
I-c) 0)
<
101F
i1-
989
D
0) CO
I-M-_
_1
:~-- 92
tRNATRP_LdAl 4I
tRNA P-IdA1 4 _
4 AA
t I RF US I P L
FIG. 4. Analysis ofreverse transcription products for wild-type and U5-IR mutant virus in melittin-activated virions. Shown aretRNATm primer extensionproducts formed after 30minofincubation of virions with melittin and[32P]dATPin the absence (Aand D) or the presence (Band C)of the other three unlabeled deoxynucleotides. The viruses (wild type [WT] orU5mutantsA3,A9, S2, S2C, S4, andS4C)are indicatedabove the respective lanes. The migration distances of thedA-tRNAT"',cDNA101, cDNA98,andcDNA92products are as indicated. Mdenotesa32P-labeled125-bp DNA fragment, added to each reaction upon termination, that serves as a control for the recovery and loading of reaction products.(E)Schematic representation of the initiation of reverse transcription. AA, initial dAMP residues incorporated onto the 3' OHterminus of thetRNATrpprimer; closed arrow,cDNA101reversetranscription intermediate; open arrows, inverted repeat sequences; P, primer-bindingsite; L, leader region.
cleavage also occurs and results in oligodeoxynucleotides migrating faster than the starting substrate.
As shown in Fig. 6C, large integrative recombination
products were detected in the presence of
Mg2"
with thewildtype (lane 8) but not with substrates containing the S2
mutation (lane 2). Such products, however, were detected
A B
(o 0
Cn CN4CM *
< cn aS 3~U) c) U U) <
418 _S _ _
~~~~~~~~418
12 3 1 2 3 4 5 6 7 8 9
FIG. 5. Quantitation of integrated wild-type and U5 mutant
provirus. (A) Amplification ofa418-bp fragment derived from the polgene,asdescribedinMaterials andMethods, by using pRCAS-B
DNA. Amplified DNA was analyzed on a 0.8% agarose gel and
transferredtoanylon membrane, and viralsequences weredetected
byhybridizationtorandomly primed32P-labeled cDNAasdescribed
inMaterialsand Methods. Theamountsof DNAamplifiedwere as
follows: lane 1, 1.0 pg; lane 2, 0.1 pg; and lane 3, 0.01 pg. (B) Chromosomal DNAs were isolated from cells 12 h after infection
with wild-type or U5 mutant viruses and used as templates for
amplification of the same 418-bp fragment. Equal amounts of
amplified DNAwereanalyzedon a0.8%agarosegelasdescribed for
panelA. The mutantviruses used for the initial infections of cells from whichthe DNA was isolatedare indicated above the lanes.
QT6indicatesDNAisolatedfrom uninfected cells.
with the S4 and S4C substrates (lanes 4 and 6), but in reduced amounts compared with the wild type. Densitomet-rictracing oftheautoradiogram indicates that the amount of
integrative recombinant product shown by the large arrow-head wasreduced42and63%for S4 and S4C, respectively.
Similar resultswereobtained in thepresence ofMn2+ (data notshown). Combiningthe invitro results displayed in Fig. 6B and C, the S2 mutation is predicted to cause a greater
integration defect than the S4 mutation because ofits se-verelycompromised nickingandjoining reactions.
DISCUSSION
Theexperiments described above support thenotionthat U5-IR sequences have roles in both reverse transcription
and integration. In viral RNA, U5-IR sequences form a
secondary structure necessary for efficient initiation of re-verse transcription, while in DNA these sequences are
specifically recognized by IN and are required for integra-tion. This represents an instance of a cis-acting element
possessing unrelated replication functions in its RNA and DNAforms.
The U5-IR stem RNA secondary structure, like the U5-leader stem, is a primary determinant for efficiency of initiation ofreverse transcription. There is a decrease in
initiationin melittin-activatedvirionswhen the U5-IR struc-tureisdisrupted andanimprovedreverse
transcription
andgrowth efficiency when the U5-IR stem is restored by compensatingmutations. Wecannotruleoutthe possibility
that the
compensating
mutations activated initiationinde-pendentlyof the
inhibitory
effect of the S2orS4substitution
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.92.534.73.321.2] [image:6.612.62.305.500.591.2]3870 COBRINIK ET AL.
U3 RU5 U3 RU5
AGCAGAAGGCTTCATT AGCAGAAGGCaaCATT AGCAGAAGGgaagATT t,
TCGTCTTCCGAAGTAA TCGTCTTCCGttGTAA TCGTCTTCCcttcTAA
WT S2
F
~~
IN IN IN INM Mg Mg Mn M MgMgMn
164
w416
15 14
-K
13 S1
12*
1051
9
8
7
a b c
13 12
11i
10
a
S4
- IN IN M Mg MgMn
16
15 '
14
f
13
1290 .1
119w
109..7
a
;cCAGAAGGgaagATT
igGTCTTCCcttcTAA
S4C
M IN IN - IN IN MgaMgMn MgMgMn
16j
15
14
13*
0
121
119
10*
810~8
81 ^
84
w
*
89
7
*-d e f g h
j
ik I mno
C
S2 S4 S4C WT
S
1 2 3 4 5 6 7 8
mutation. This appears unlikely, however, since the A9 deletiondisrupts the U5-IR stemstructurewithoutchanging the bases involved in the S2 and S4 mutations and also results in an initiation defect. Additional evidence that
supportsareplication role for the U5-IR stemisthe
conser-vation of this structure inseveral retrovirus families (1). Independent evidence supports a reverse transcription
rolefor U5-IRsequencesin murine leukemia virus (MuLV).
Murphy and Goff (24) have shown thatatleastonemutation
[image:7.612.69.490.69.571.2](dlAva2) constructed in the comparable region of MuLV RNAcauses areplication defect without affecting encapsi-dation of viral RNA. This mutation would disrupt the equivalent ofthe U5-IR stem. Another mutation, dlAva3, which wouldalso disrupttheU5-IRin MuLV, doesnothave
FIG. 6. EffectofUS-IRmutationsonintegrationandIN protein
cleavage. (A)U5-terminalsequencesforwild-type (WT)andU5-IR
mutant viruses are shown. Sequences represent double-stranded oligodeoxynucleotides of 16nucleotidesinlengthusedto assayIN protein cleavage. Each sequence is aligned over the appropriate column inpanelB.Lowercase letters indicatebasechangesfromthe WT. Asterisks indicate a 5'-end 32P-label on plus strands. (B)
Double-stranded oligodeoxynucleotides were incubated in the
ab-sence(lanesa,d,g,j,andm)orpresence(lanes b,c, e,f,h, i, k,1,
n,ando)ofIN,with eitherMg2+ (lanesa,b, d,e,g,h, i, k,m,and n) or Mn2+ (lanes c, f, i, 1, and o) as a divalent cation. The
arrowheads indicatethemajorinvitro and in vivocleavage site of WT virus(22). LaneslabeledMdisplaysnakevenom
phosphodies-terasecleavage productsfor each DNAsubstrate, and theproduct
sizes (in nucleotides) are shown. Lanes m through o represent a
longerexposure of lanesj through 1. (C) Analysis of integrative
recombinant products. Integrative recombination products were
analyzed as described in Materials and Methods by using 5' 32p_ labeledduplex U5oligodeoxynucleotide LTR substrates with
two-base 5' PoverhangsrepresentingtheWT(lanes7 and8)and the S2 (lanes1and2),S4(lanes3and4),and S4C(lanes5and6) mutations. Double-stranded oligodeoxynucleotides were incubated in the
ab-sence(lanes 1, 3, 5,and7)orpresence(lanes 2, 4, 6,and8)of IN. S denotesthe respective startingsubstrates. DNAproducts larger
than thestartingsubstratesareindicated bythearrowheads.
anyeffecton replication. However, closer inspection of the sequence in dlAva3 reveals that the potential to form an
alternative but smaller U5-IRstemexists.Thus, MuLVstem structures mayhave similar roles tothose demonstrated for avian sarcoma andleukosis virus.
In viral DNA, U5-IRsequences were found to be
neces-A
B
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
sary for efficient
integration
as well as for specific in vitronicking
at the viralintegration
site by purified IN protein(Fig.
5 and 6). These results were not unexpected, bothbecause mutations near the U5 terminus of other retrovi-ruses have been shown to cause integration defects (9, 10,
25, 26)
andbecausemutations inthisregionwerepreviouslyfound to inhibit in vitro
nicking
by theavian retrovirus INprotein (22). Furthermore,
Roth etal. (26) have shown thatU5 mutations which block
integration
also blockcleavage at theU5 terminusinvivo.Thecurrentstudy indicatesthat theU5-IR mutationswhich affect
integration
interfere withbothIN-catalyzed
cleavageandjoining reactions in vitro, but todifferent extents. The S2 substitution appears to induce a stronger defect than the S4 substitution. In the case ofthe
S2C
virus,
in which thereis no defect in reversetranscrip-tion,
wespeculate
that the ability of this virus to replicateand
integrate
in vivo(Fig.
3A and 5) might be due tocooperative
interactions between the IN protein bound tothe
wild-type
U3 LTR terminus and that bound to themutated U5 LTR terminus. Alternatively, other viral or
cellular
proteins
may contribute to the S2 integrationeffi-ciency
invivo,
orintegration
mediatedbyoligomeric
formsof viral DNA asdescribed
by Hagino-Yamagishi
etal. (16)mayoccur.
We have found that the S2 mutation caused greater
initiation and
integration
defects than the S4 mutation.Furthermore,
S4Cfully
restored the reverse transcriptionefficiency,
whereas S2C led toonly
apartial
restoration.These
findings
wereunexpected, since S2C has fewernucle-otide substitutions than S4C and forms a U5-IR stem of
similar
stability. Thus,
thepartial
initiation defect of S2C suggests that both the U5-IR stem structure and sequence affect initiation.Apparently,
the CTTC/GAAG U5-IR se-quence is favored overGTTG/CAAC,
which moreclosely
resembles the
wild-type
sequence, GAAG/CTTC (Fig. 1C).We
speculate
that this couldreflecttherole ofaproteinthatrecognizes
both the U5-IR stem structure and the GAAGsequence in either orientation in the stem. Although the same sequence
preferences
aredisplayed by
the INprotein
in in vitro
cleavage
andintegration reactions,
IN isunlikely
to be involved in initiation since it canbe mutated withoutaffecting
reversetranscription (29).
The
biological
consequencesoftheS2, S4, S2C, S4C,
and A9mutationscanbeexplained
onthe basis of eitherdisrupt-ing
the U5-IR stem structure,thereby affecting
initiation of reversetranscription,
oraltering
DNA sequencesrecognized
by
the INprotein
forintegration.
The A3 mutation ismoreenigmatic.
The sequences that are deletedby
this mutation are notrequired by
INforspecific
cleavage
of the U5 LTR terminus(22),
nordothey
appeartodisturb the basepairing
orsequenceoftheU5-IRstem.This suggests that features in addition tothe U5-IR stem and U5-leader stem structures
are
required
for maximalinitiationefficiency.
Oneintriguing
possibility
is that U5 sequences between the U5-leaderandU5-IR
stems,namely,
those substituted in the S6 mutation(Fig. 1B),
form basepairs
withcomplementary
sequencesin theTqlC
loop
of thetRNATrP
primer,
asinitially proposed by
Haseltineetal.
(17,
18)
andsupported
by
theexperiments
of Cordelletal.(11).
This appearstobe the case,since in vitro andinvivo studies indicate that thisU5-primer
interactionisimportant
forinitiationofreversetranscription
(1). Thus,
the A3 mutationmight impair
initiation ofreversetranscription
by altering
thespacing
between the U5-leader and U5-IRstructures,
thereby
affecting
the interactions between theTtlC
loop
of thetRNATrp
primer andU5.Thisis discussed inmoredetail elsewhere
(1).
Our previous analyses of a mutation that disrupts the US-leader stem (A6 [Fig. 1B]) revealed a defect in the initiation of reverse transcription in infected cells and in reactions ofmelittin-permeabilized virions but not in reac-tions of purified viral RNA and reverse transcriptase (7).
Thisraises the possibility that the US-leader and possibly the U5-IR structures are important for signalling the initiation of reversetranscription following infection in the context of the highly structured virion core. The role of viral proteins in such signalling is unclear, although it may be relevant that initiation ofpoliovirus replication also is likely to require a RNA secondary structure near the initiation site. In this case, poliovirus genes whose products could interact with the secondary structure in trans have been identified by analysis of second-site revertants (2). We have isolated second-site revertants for severalof the US mutations (Sa), and a similar analysis might identify a retrovirus gene product that interacts with the US-leader and U5-IR stem structures to regulate initiation of reverse transcription.
ACKNOWLEDGMENTS
Wethank H.-J. Kungforcritical comments on the manuscript. This workwassupported inpartby Public Health Service grant CA 38046 from the National Cancer Institute and by Cancer ResearchCentergrantP30CA43703.D.C.and H.H. aresupported inpartbyPublic HealthServicetraininggrantT32GM07250from the NationalInstitute of GeneralMedical Sciences.
REFERENCES
1. Aiyar, A., D. Cobrinik, Z. Ge, H.-J. Kung, and J. Leis. Submit-tedfor publication.
2. Andino, R., G. E. Rieckhof, and D. Baltimore. 1990. A func-tional ribonucleoprotein complex forms around the 5' end of poliovirusRNA.Cell63:369-380.
3. Bilofosky, H. S., C. Burks, J. W. Fickett, W. B. Goad, F. I. Lewitter, W. P. Rindone, C. D. Swindell, and C. S. Tung. 1986. The GenBankgeneticsequencedatabank. Nucleic Acids Res. 14:1-4.
4. Boone,L.R., and A. M. Skalka. 1981. Viral DNA synthesized in vitroby avian retrovirus particles permeabilized with melittin. I. Kinetics ofsynthesis andsize of minus- andplus-strand tran-scripts.J. Virol. 37:109-116.
5. Brown, B., B. Bowerman, H. Varmus, and J. M. Bishop. 1987. Correctintegrationof retroviral DNA in vitro. Cell 49:347-356. 5a.Cobrinik,D. Unpublisheddata.
6. Cobrinik, D.,R.Katz,R.Terry, A. M.Skalka,andJ. Leis. 1987. Aviansarcomaandleukosis viruspol-endonucleaserecognition of the tandem long terminal repeat junction: minimum site required for cleavage isalsorequired forviralgrowth.J.Virol. 61:1999-2008.
7. Cobrinik, D., L. Soskey, andJ. Leis. 1988. A retroviral RNA secondary structure required for efficient initiation of reverse transcription. J. Virol. 62:3622-3630.
8. Coffin, J. M. 1990. Retroviridae and theirreplication, p. 1437-1500. In B. N. Fields, D. M. Knipe, R. M. Chanock, M. S. Hirsch, J. L. Melnick, T. P. Monath, and B. Roizman (ed.), Virology, 2nd ed. RavenPress,Ltd., NewYork.
9. Colicelli, J.,andS. Goff. 1985. Mutants andpseudorevertantsof Moloney murine leukemiaviruswith alterationsatthe integra-tion site. Cell 42:573-580.
10. Colicelli, J.,and S. Goff. 1988. Sequence andspacing
require-mentsofaretrovirusintegrationsite. J. Mol. Biol. 199:47-59. 11. Cordell, B., R. Swanstrom, H. Goodman, and J. M. Bishop.
1979. tRNATmPas primerfor RNA-directed DNApolymerase: structural determinants offunction. J. Biol. Chem. 254:1866-1874.
12. Craigie, R., A.-T. Fujiwara, and F. Bushman. 1990. The IN protein of Moloneymurine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell 64:829-37.
on November 10, 2019 by guest
http://jvi.asm.org/
3872 COBRINIK ET AL.
13. Dahlberg,J. E., R. C.Sawyer,J. M. Taylor, A. J. Faras, W. E.
Levinson,H. M.Goodman, and J. M.Bishop. 1974. Transcrip-tion ofDNA from the 70S RNA of Rous sarcoma virus. I.
Identification ofaspecific 4SRNAwhich serves asprimer. J.
Virol. 13:1126-1133.
14. Darlix, J.-L., M. Zuker, and P.-F. Spahr. 1982. Structure-functionrelationship ofRous sarcomavirusleaderRNA. Nu-cleic AcidsRes. 10:5183-5196.
15. Fujiwara, T., and R. Craigie.1989.Integration ofmini-retroviral DNA: acell-freereaction for biochemical analysis of retroviral integration. Proc. Natl. Acad. Sci. USA 86:3056-3069. 16. Hagino-Yamagishi, K., L. A. Donehower, and H. E. Varmus.
1987.RetroviralDNAintegratedduringinfection byan integra-tion-deficientmutantof murine leukemiavirusisoligomeric.J. Virol.61:1964-1971.
17. Haseltine, W. A.,D.G. Kleid,A. Panet, E. Rothenberg, and D. Baltimore. 1976. Ordered transcription of RNA tumor virus
genomes. J.Mol. Biol. 106:109-131.
18. Haseltine, W. A., A. M. Maxam, and W. Gilbert. 1977. Rous
sarcomavirusgenomeisterminallyredundant:the 5'sequence.
Proc.Natl. Acad. Sci. USA 74:989-993.
19. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmousecellcultures. J.Mol. Biol. 26:365-371. 20. Hughes, S. H., J. J. Greenhouse, C. J. Petropoulos, and P.
Sutrave.1987. Adaptorplasmids simplify the insertion of foreign DNA intohelper-independent retroviral vectors. J. Virol. 61: 3004-3012.
21. Katz, R., G. Merkel, J. Kulkosky, J. Leis, and A. M. Skalka. 1990. The avian retroviral IN protein is both necessary and
sufficientforintegrative recombinationin vitro.Cell63:87-95. 22. Katzman, M., R. A. Katz, A. M.Skalka, and J. Leis. 1989.The
avian retroviral integration protein cleaves the terminal se-quences of linear viralDNA attheinvivo sitesofintegration.J. Virol.63:5319-5327.
23. Mullis, K. B., and F. A. Faloona. 1987. Specific synthesis of DNA in vitro viaapolymerase-catalyzed chain reaction. Meth-ods Enzymol. 155:335-350.
24. Murphy, J. E.,and S.Goff. 1989. Construction and analysis of deletion mutations in the U5regionof Moloney murine leuke-mia virus:effectson RNApackaging and reversetranscription. J. Virol. 63:319-327.
25. Panganiban, A.,and H. Temin. 1983.The terminalnucleotides of retrovirusDNA arerequired for integration butnotfor virus production. Nature(London) 306:155-160.
26. Roth, M., P.Schwartzberg, andS. Goff. 1989. Structureof the termini ofDNA intermediates in the integration of retroviral DNA:dependence ofINfunction andterminalDNAsequence. Cell58:47-54.
27. Sambrook, J.,E. F. Fritsch,and T. Maniatis. 1989.Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, ColdSpring Harbor, N.Y.
28. Schwartz, D.E., R. Tizard,and W. Gilbert. 1983. Nucleotide sequence ofRous sarcomavirus. Cell 22:853-869.
29. Schwartzberg, P., J.Colicelli, and S. Goff. 1984. Construction andanalysisofdeletion mutations in the pol gene of Moloney murine leukemia virus: a new viral function required for pro-ductive infection. Cell 37:1043-1052.
30. Smith, R.E. 1974. High specific infectivity avianRNA tumor viruses. Virology60:534-547.
31. Temin, H. M., and D. Baltimore. 1972. RNA-directed DNA synthesis andRNAtumorviruses. Adv. VirusRes.17:129-186. J. VIROL.