Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction
22
0
0
Full text
Figure
+7
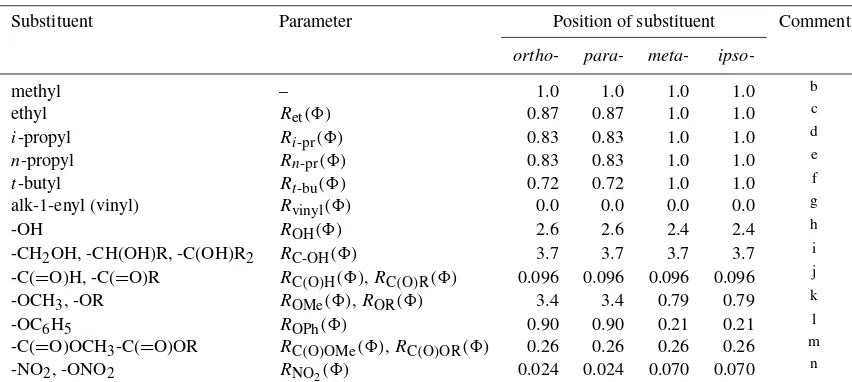
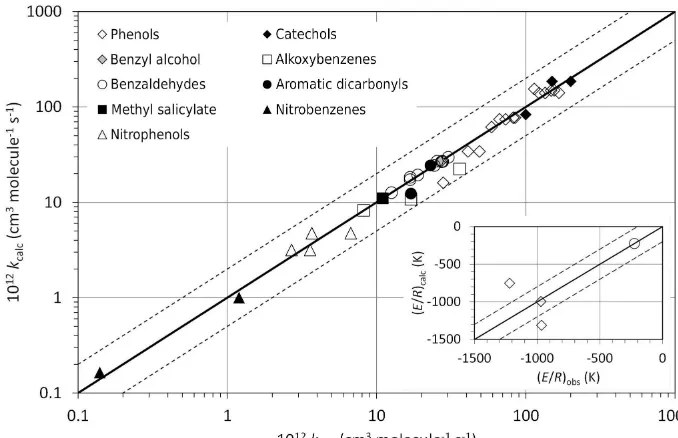
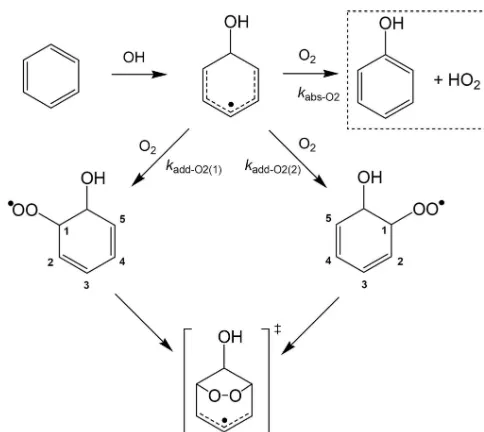
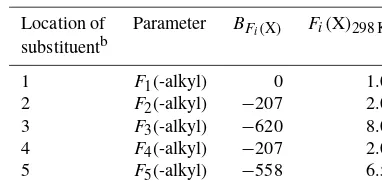
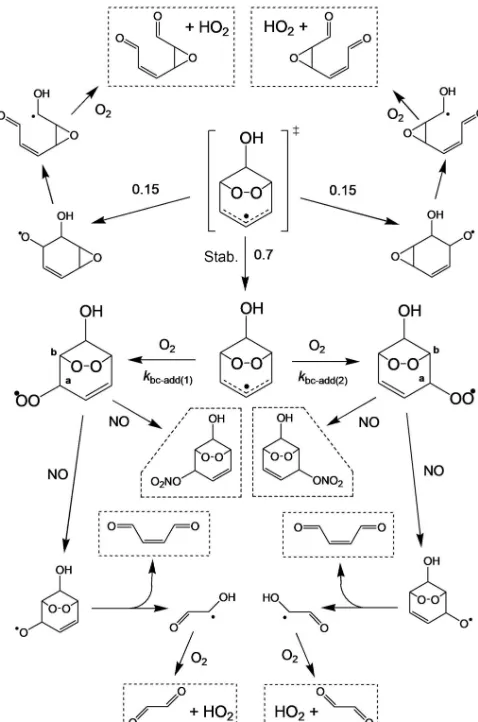
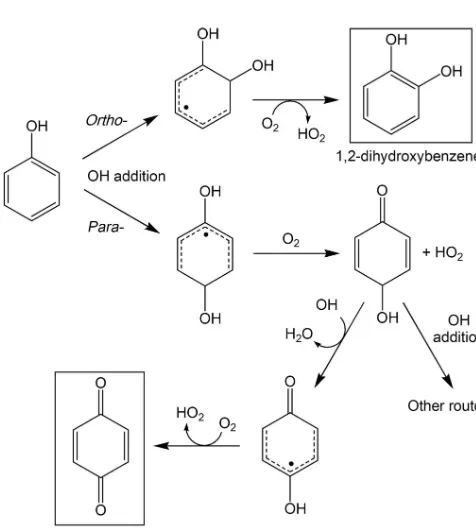
Related documents