Copyrightq1997, American Society for Microbiology
Induction of AIDS by Simian Immunodeficiency Virus Lacking
NF-
k
B and SP1 Binding Elements
PETR O. ILYINSKII, MEREDITH A. SIMON, SUSAN C. CZAJAK, ANDREW A. LACKNER,
ANDRONALD C. DESROSIERS*
New England Regional Primate Research Center, Harvard Medical School, Southborough, Massachusetts
Received 16 September 1996/Accepted 9 December 1996
Rhesus monkeys (Macaca mulatta) were infected with five strains of simian immunodeficiency virus (SIV) derived from SIVmac239 containing deletions (D) or substitutions (subst) in NF-kB and Sp1 binding sites. We have shown previously that mutations in these regions still allow efficient SIVmac replication in primary lymphoid cell cultures (P. O. Ilyinskii and R. C. Desrosiers, J. Virol. 70:3118–3126, 1996). Two animals were inoculated intravenously with each mutant strain of SIVmac239: DNFkB, DSp1234, DNFkBDSp1234, sub-stSp12, and substSp1234. All but one of the infected animals showed an early spike in plasma antigenemia, maintained high virus burdens, and had significant changes in lymphoid tissues, and six died with AIDS within the first 60 weeks of infection. One of the animals infected with the SIV strainDNFkBDSp1234 showed lower levels of plasma antigenemia and lower virus burdens; the other animal infected with this same mutant strain died with AIDS 17 weeks after inoculation. No consistent novel mutations or reversions were detected in proviral sequences derived from the animals infected with the deletion mutants and the substSp12 mutant by 20 weeks postinfection. Point-mutated sequences were partially deleted in both animals infected with the substSp1234 strain. These results indicate that the NF-kB and Sp1 binding sites are not essential for the induction of AIDS by SIVmac239. They also provide indirect evidence for the importance of a novel enhancer element in the U3 region of the SIVmac long terminal repeat that is located immediately upstream of the NF-kB binding site within the C-terminal region of thenefcoding sequence.
Binding sites for the transcription factors NF-kB and Sp1 are critical for efficient initiation of transcription of human immunodeficiency virus type 1 (HIV-1) (2, 6, 10, 11, 18, 21, 24). These elements are located in the U3 region of the long ter-minal repeats (LTRs) just upstream of the TATA box. Partial deletion or substitution of these sequences significantly impairs transcription of HIV-1, and their elimination completely abol-ishes viral replication (14, 20, 25). HIV-2 and the closely re-lated simian immunodeficiency virus of macaque monkeys (SIVmac) differ from HIV-1 in this respect (4, 22). These viruses possess additional enhancer elements just upstream of the single NF-kB binding site (1, 5, 7, 9, 15, 16, 23, 27). The upstream enhancer element is able to compensate for a com-plete loss of NF-kB and Sp1 binding sites to maintain a signif-icant level of transcription (9). Mutant strains of SIVmac239 bearing changes in the four Sp1 elements and the single NF-kB site were capable of replicating in lymphoid cells in vitro to a level and with kinetics similar to those of the wild-type parental strain. Even the deletion of all NF-kB and Sp1 elements yielded a virus capable of replicating in lymphoid cells similar to wild-type or with only a slight delay (9).
Analysis of the replication of SIVmac mutants in primary macrophages yielded results somewhat different from those obtained in lymphocytes. Mutation of the Sp1 elements, either by deletion or by point substitutions, had profound effects on SIVmac’s ability to replicate in macrophages (9). Thus, the relative importance of the NF-kB and Sp1 elements appears to vary from one cell type to another.
Here we describe the properties of SIVmac239 variants with mutations in the NF-kB and Sp1 elements following
experi-mental infection of rhesus monkeys. The properties of these mutant viruses in lymphoid cells were more predictive of their in vivo behavior than their properties in macrophages. Our results indicate that high virus loads and disease can occur with SIVmac239 in the complete absence of NF-kB and Sp1 binding elements. Furthermore, our results provide indirect evidence for the importance of the novel enhancer element just up-stream of the NF-kB binding site in the SIVmac proviral ge-nome.
MATERIALS AND METHODS
Plasmids.The SIVmac239 proviral clone is carried on plasmids p239SpSp59
and p239SpE39which contain the left and right halves of the genome, respec-tively (22). The clones used contained an open nef reading frame. The structures of SIVmac239 variants with mutations in NF-kB and Sp1 binding sites have been described by Ilyinskii and Desrosiers (9). Mutations were introduced into both LTRs.
Cells and viruses. Rhesus monkey peripheral blood mononuclear cells (PBMC) and CEMx174 cells were maintained as described earlier (8). The culture medium was changed twice weekly. Pelleted CEMx174 cells were trans-fected withSphI-digested 59and 39SIVmac plasmids by a DEAE-dextran pro-cedure (19). For stock preparation, virus was harvested at or near the peak of virus production (15 to 23 days posttransfection), filtered, and stored in aliquots at2808C. Stocks were assayed for the concentration of SIV p27gagantigen with a commercially available antigen capture kit (Coulter Corp., Hialeah, Fla.). All mutated SIVmac239 strains that were used in this study have been described in detail by Ilyinskii and Desrosiers (9).
Experimental infection of rhesus monkeys. CEMx174-derived DNFkB,
DSp1234,DNFkBDSp1234, substSp12, substSp1234, and wild-type SIVmac239 were used for intravenous inoculation of juvenile rhesus monkeys (Macaca mulatta). The virus inoculum for each infection contained 50 ng of SIVmac p27. Two animals were infected with each of the SIVmac mutants and with SIV-mac239. In addition, one animal was inoculated intramuscularly with 50mg of cloned proviral DNA of strain SIVDNFkBDSp1234 and one with SIVmac239. Plasmid DNA was mixed with DME-DEAE-dextran solution similarly to the in vitro transfection procedure. All experimental animals were housed in accor-dance with the guidelines set by the Committee on Animals of the Harvard Medical School and by those of the Committee on Care and Use of Laboratory Animals, National Research Council. Animal husbandry, handling of the mon-keys, and collection of specimens were performed essentially as previously de-* Corresponding author. Mailing address: New England Regional
Primate Research Center, Harvard Medical School, One Pine Hill Dr., Box 9102, Southborough, MA 01772-9102. Phone: (508) 624-8042. Fax: (508) 624-8190.
1880
on November 9, 2019 by guest
http://jvi.asm.org/
scribed (3, 8). Animals that became moribund were sacrificed, and complete necropsies were performed.
Virus load, plasma antigenemia, and measurement of antibody responses. Cell-associated virus loads in infected macaques were evaluated by quantitative cocultivation of PBMC with CEMx174 cells. PBMC were purified, counted in a hemocytometer, and subsequently cocultured with CEMx174 cells in various numbers (8). On day 21, the presence of SIV p27gagwas evaluated and the numbers of PBMC needed to recover SIV were calculated. The amount of SIV p27 antigen in plasma was quantified with the Coulter antigen capture kit. Animal plasma was used at dilutions of 1:20 and 1:100 for enzyme-linked im-munosorbent assay (ELISA) reactivity to purified whole SIVmac as described previously.
Isolation of cellular DNA.Macaque lymphocytes were pelleted, washed, and lysed in a small volume (50 to 100ml). DNA was isolated in a single microcen-trifuge tube for each sample by using the HRI AmpPrep kit (HRI Research, Inc., Concord, Calif.) and subjected to PCR amplification as described below.
Primers.All nucleotide primers were custom-made by GIBCO BRL (Grand Island, N.Y.) except for forward and reverse primers manufactured by Perkin-Elmer Cetus (Norwalk, Conn.). The primers used for PCR amplification of SIV U3- and R-regions from the infected animals were 59-AGACTTATGGGAGA CTCT-39(9153-9170) and 59-CAGAAAGGGTCCTAACAGACCA-39
(10246-10225) for the first round of amplification and 59-GGGATTTATTACAGTGC AAGAAGA-39 (9467-9490) and 59-GGCTAGGAGAGATGGGAACACACA -39(10185-10162) for the second round. The primers used for the amplification of the sequence coding for the N terminus of thenefgene were 59-CGAGAA GTCCTCAGGACTGA-39(9040-9059) and 59-GGGACTAATTTCCATAGCC A-39 (9612-9593) (first round) and 59-CTGAACTGACCTACCTACAA-39
(9056-9075) and 59-TTCCTGGTCCTGAGGTGTAA (9569-9550) (second round).
PCR amplification, molecular cloning, and DNA sequencing.PCR amplifica-tion was performed essentially as described earlier (8). A thermal cycler pro-duced by MJ Research (Watertown, Mass.) was used for PCR amplification. Two rounds of 30-cycle reactions were used for the amplification of desired proviral sequences. All routine cloning procedures were performed essentially as de-scribed (8). Epicurian Coli XL1-Blue MRF9Supercompetent Cells (Stratagene Cloning Systems, La Jolla, Calif.) were used for the transformation and plasmid growth. PCR products were gel purified, treated with polynucleotide kinase, directly cloned into blunt-end cloning vector pUC18/SmaI (Pharmacia, Piscat-away, N.J.), and sequenced. DNA sequencing was performed with forward and reverse primers by using an ABI Prizm automated sequencer. Sequencing reac-tions were performed with the help of AmpliTaq FS sequencing kit from
[image:2.612.59.556.90.247.2]Perkin-FIG. 1. Plasma antigenemia in rhesus monkeys infected with SIVmac mutants.
TABLE 1. Summary of experimental infections of rhesus monkeys with SIVmac239 derivatives bearing mutations in transcriptional control elements
Animal
no. Viral strain inoculationMode of Infectiousagent Status, 68 wk pia
131-94 SIVDNFkB Intravenous Virus Alive, high virus loads
104-94 SIVDNFkB Intravenous Virus Died, 15 wk pi
117-94 SIVDSp1234 Intravenous Virus Euthanized, 16 wk pi
118-94 SIVDSp1234 Intravenous Virus Alive, high virus loads
360-93 SIVDNFkBDSp1234 Intravenous Virus Alive, low virus loads
146-94 SIVDNFkBDSp1234 Intravenous Virus Euthanized, 17 wk pi
66-94 SIVDNFkBDSp1234 Intramuscular DNA Euthanized, 11 wk pi
179-94 SIVsubstSp12 Intravenous Virus Euthanized, 14 wk pi
183-94 SIVsubstSp12 Intravenous Virus Euthanized, 31 wk pi
185-94 SIVsubstSp1234 Intravenous Virus Euthanized, 60 wk pi
187-94 SIVsubstSp1234 Intravenous Virus Alive, high virus loads
346-93 SIVmac239 Intravenous Virus Euthanized, 13 wk pi
418-93 SIVmac239 Intravenous Virus Euthanized, 52 wk pi
101-94 SIVmac239 Intramuscular DNA Euthanized, 14 wk pi
api, postinoculation.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.60.555.478.714.2]Elmer Cetus which was used according to manufacturer’s recommendations on a Hybaid Omnigene thermal cycler (SunBioscience Inc., Branford, Conn.).
Nucleotide sequence analysis. Nucleotide sequences were analyzed and aligned by using MacVector version 4.1.4 software (International Biotechnolo-gies, Inc., New Haven, Conn.). The sequence of SIVmac239 has been published previously (22).
Histology.Tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin for routine histologic examination. Adjacent sections were used for in situ hybridization and/or immu-nohistochemistry.
In situ hybridization.In situ hybridization was performed with formalin-fixed paraffin-embedded sections by using DNA probes labeled with digoxigenin by random priming (Boehringer Mannheim, Indianapolis, Ind.). Hybridizations were performed under denaturing conditions to localize both DNA and RNA, followed by immunodetection as previously described (8). As a negative control, sections were hybridized with plasmid pUC19 labeled at the same time as the test probes. Positive controls were tissues previously demonstrated to contain viral nucleic acid of interest. The SIV probe is the combination of plasmids p239SpSp59and p239SpE39, which provide essentially the entire SIVmac239
genome (22). The cytomegalovirus probe is a 9.2-kb fragment containing the macaque CMV immediate-early gene and 39flanking region (provided by Peter A. Barry, University of California, Davis, Davis, Calif.). Probe labeling was determined by comparison to controls following the manufacturer’s protocol (Boehringer Mannheim).
Immunohistochemistry.To examine the phenotype of the lymphoma in ani-mal 183-94, forani-malin-fixed sections were immunostained with antibodies directed against B cells (L26), T cells (CD3), and macrophages (HAM56) as previously described (28), using an ABC technique with diaminobenzidine as the chromo-gen (all primary antibodies from Dako Corp., Carpenteria, Calif.). Selected sections were labeled sequentially for T cells and B cells, using an alkaline phosphatase development technique for the second labeling (Vector Labs, Bur-lingame, Calif.) with Vector Blue as the chromogen.
RESULTS
Infection of rhesus monkeys with wild-type and mutant SIV.
Two juvenile rhesus monkeys in each group were infected with mutant SIV strains DNFkB,DSp1234, DNFkBDSp1234, sub-stSp12, and substSp1234 and with wild-type SIVmac239. One additional animal was inoculated intramuscularly with plasmid proviral DNA of DNFkBDSp1234 and one with SIVmac239 DNA (Table 1). Each of the mutated viruses bore the changes in both viral LTRs in order to avoid reversion to wild type during replication or through recombination. A number of parameters were monitored following virus inoculation to eval-uate the levels of virus replication and the virulence of the infection.
Plasma antigenemia.All infected animals showed a spike in plasma antigenemia 1 to 2 weeks after inoculation (Fig. 1 and Table 2) as is almost always seen in monkeys infected with the parental SIVmac239. Persistent plasma antigenemia was ob-served in several animals, all of whom proceeded to develop AIDS within the first year. All animals which maintained high levels of p27 in plasma (104-94, 101-94, 117-94, 179-94, 183-94, and 346-93) died or had to be euthanized within the first year because of poor health (see below). One animal, 360-93, in-fected with DNFkBDSp1234, was weakly positive only at the week 2 time point and has been negative ever since. Another
[image:3.612.58.299.90.243.2]FIG. 2. Frequency of infectious cells in PBMC of rhesus monkeys infected with mutant viruses. Viral load from every animal was graded on a scale from 0 to 14, depending on the number of PBMC needed to recover SIV (0, no recovery with 106PBMC; 1, recovery successful from 13106PBMC; 2 to 14, recovery successful from 333,333, 222,222, 111,111, 74,074, 37,037, 24,691, 12,345, 8,230, 4,115, 2,743, 1,371, 914, and 457 cells, correspondingly).
TABLE 2. Plasma antigenemia in rhesus monkeys infected with SIVmac239 TCEamutants
Animal
no. Virus strain
SIVmac p27 concn (ng/ml) at week postinoculation
0 1 2 3
131-94 SIVDNFkB 0.0 6.8 2.2 0.0
104-94 SIVDNFkB 0.0 1.8 2.5 2.0
117-94 SIVDSp1234 0.0 0.7 0.5 0.6
118-94 SIVDSp1234 0.0 1.6 0.5 0.2
360-93 SIVDNFkBDSp1234 0.0 0.0 0.3 0.0 146-94 SIVDNFkBDSp1234 0.0 2.4 0.7 0.0
179-94 SIVsubstSp12 0.0 5.4 4.0 1.5
183-94 SIVsubstSp12 0.0 12.5 2.3 4.0
185-94 SIVsubstSp1234 0.0 1.4 0.8 0.0
187-94 SIVsubstSp1234 0.0 0.6 0.1 0.0
346-93 SIVmac239 0.0 3.6 1.5 0.8
418-93 SIVmac239 0.0 1.9 0.7 0.7
aTCE, transcriptional control element.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.58.555.457.700.2]animal infected with theDNFkBDSp1234 mutant 146-94 had a high initial spike of plasma antigenemia and became positive again in the later stages of infection (Fig. 1 and Table 2). Aside from animal 360-93, there was no substantial difference in the levels or kinetics of appearance of plasma antigenemia be-tween the animals infected with the SIV mutants and those infected with wild-type SIVmac239. These also did not differ from the levels or kinetics of appearance observed in previous studies with wild-type SIVmac239 (3, 13). They are substan-tially higher, however, than the levels observed in monkeys infected withDnef andDvpxDvpr mutant viruses (3, 13).
Virus loads.Cell-associated virus burdens were evaluated by measuring the numbers of PBMC needed to recover SIV by cocultivation in vitro. As seen in Fig. 2, all but one of the inoculated animals became virus positive and maintained a high level of culturable virus during the course of infection. The patterns of virus loads in the animals infected with the mutant viruses were very similar to those infected with SIV mac239. The only exception is, again, animal 360-93, whose viral burdens declined steadily after the initial peak (Fig. 2). The cell-associated virus loads in 13 of the 14 animals in this assay were similar to those observed with wild-type SIVmac239 in previous studies but substantially higher than the loads ob-served with theDnef andDvprDvpx attenuated viruses (3, 13).
Antibody responses.Most inoculated animals produced an-tibodies against SIV as determined by ELISA to antigens in whole lysed virus (Fig. 3). There was no correlation between the strength of the antibody response and the type of inocu-lated virus. However, the level of antibodies in plasma
re-flected at least to some degree the rate of disease progression. Five of the seven animals that progressed rapidly to death mounted little or no free anti-SIV antibody responses. This is similar to the pattern described previously for the 20 to 50% of wild-type virus-infected monkeys that are rapid progressors (3, 12). The strongest antibody response was recorded in 360-93 (DNFkBDSp1234), the animal with the lowest levels of plasma antigenemia and cell-associated virus loads.
Disease progression. One animal (104-94) died and nine others had to be euthanized because of poor health during the first 60 weeks of follow-up. One of these animals, 66-94, died of causes apparently unrelated to SIV infection and was not considered further. The remaining nine animals showed le-sions at necropsy that have commonly been associated with SIV-induced AIDS, including disseminated cytomegalovirus infection, Pneumocysitis carinii pneumonia, Mycobacterium
avium-M. intracellulareinfection, cryptosporidiosis,
lymphosar-coma, and characteristic changes in lymphoid tissue (Table 3). Thymic dysinvolution with loss of corticomedullary distinc-tion and interstitial fibrosis is commonly observed in animals which die of SIV-induced AIDS (Fig. 4). In this study, two animals, 183-94 and 146-94, had thymuses with unusually large cortices, although other signs of dysinvolution were present. 183-94 also had a B-cell lymphoma within spinal cord and thymic interstitium, and intrathymic lymphoid hyperplasia.
[image:4.612.62.555.77.399.2]In situ hybridization for SIV was performed on the thymuses of seven of the animals, spleens of four animals, and lymph nodes of three animals. Numerous SIV nucleic acid-containing cells (hundreds) were observed in each section of every tissue
FIG. 3. Antibody levels in infected rhesus monkeys. Measurements are optical densities in ELISA assays directed to purified, lysed SIVmac.
on November 9, 2019 by guest
http://jvi.asm.org/
except those of 146-94 (Table 3 and Fig. 4). This animal had rare positive cells in the thymus, and more frequent positive cells in the lymph node and spleen, although not to the extent of the other animals. As can be seen in Fig. 4, animal 146-94 had a large thymus with a prominent expanded cortex (A), and only occasional cells containing SIV in the thymus (D) and spleen (G). In contrast, animals 101-94 and 104-94 had severe thymic dysinvolution (B, C) and numerous SIV-positive cells in both thymus (E, F) and spleen (H, I).
Genetic analysis of SIV in infected rhesus monkeys.Total cellular DNA was isolated from PBMC of infected animals 20 weeks after experimental infection or at the latest available time point if the animal died earlier. After PCR amplification of U3 and R regions, multiple clones were derived and se-quenced from most of the infected animals (Table 4). No consistent changes were detected in the DNA derived from the animals infected with viruses containing deletions in the tran-scriptional control elements. All introduced deletions were preserved. Also, no specific reversional events were detected in the animals infected with SIVsubstSp12. However, the mu-tated sequences in SIVsubstSp1234 developed significant changes in both animals infected with this strain (Fig. 5). By 20 weeks after inoculation, parts of the mutagenized fragment were deleted in animals 185-94 and 187-94. The size and the exact location of the deletion differed. However, in both cases the acquired deletion included part of the introduced mutation and did not interfere with essential regions, in some cases stopping just one base short of the TATA box (Table 4 and Fig. 5). Some of these deletions could be detected as early as 4 weeks postinfection.
The proviral sequence of the entire upstream U3 region and the remainder of the nef reading frame was also determined in
all animals infected with DNFkBDSp1234. The aim was to identify changes which could have possibly contributed to the wild-type phenotype of infection in animals 146-94 and 66-94. While individual point substitutions were noted in some clones, none of the changes were consistently present and none had the appearance of being compensatory in nature.
DISCUSSION
SIVmac and HIV-2 appear to differ from HIV-1 in their reliance on NF-kB and Sp1 binding elements for virus expres-sion. Removal of all Sp1 and NF-kB elements from HIV-1 reduces transcription in transient assays to below detectable levels and abolishes any detectable spread of virus in lymphoid cultures (14, 25). Removal of all Sp1 and NF-kB elements from SIVmac results in only a partial reduction in transcription in transient assays and results in little or no reduction in viral replication in lymphoid cultures (9). The sequences principally responsible for this surprising activity in the absence of NF-kB and Sp1 binding elements appear to be located immediately upstream of the NF-kB element in both SIVmac and HIV-2 (1, 9, 16, 17).
[image:5.612.55.558.82.363.2]We have now extended the surprising activity of NF-kB and Sp1 mutants of SIVmac to rhesus monkeys. The loss of either the single NF-kB or the multiple Sp1 elements had little or no effect on the ability of SIVmac239 to replicate in rhesus mon-keys and to cause disease. Virus lacking all NF-kB and Sp1 elements (DNFkBDSp1234), while still capable of causing AIDS, did appear to exhibit diminished replication in vivo. One of the animals infected with this mutant, 360-93, had only a very low level of plasma antigenemia at week 2, and cell-associated virus loads declined dramatically from peak levels TABLE 3. Postmortem findings
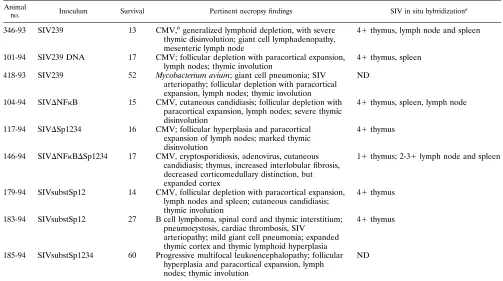
Animal
no. Inoculum Survival Pertinent necropsy findings SIV in situ hybridizationa
346-93 SIV239 13 CMV,bgeneralized lymphoid depletion, with severe
thymic disinvolution; giant cell lymphadenopathy, mesenteric lymph node
41thymus, lymph node and spleen
101-94 SIV239 DNA 17 CMV; follicular depletion with paracortical expansion,
lymph nodes; thymic involution 41thymus, spleen 418-93 SIV239 52 Mycobacterium avium; giant cell pneumonia; SIV
arteriopathy; follicular depletion with paracortical expansion, lymph nodes; thymic involution
ND
104-94 SIVDNFkB 15 CMV, cutaneous candidiasis; follicular depletion with paracortical expansion, lymph nodes; severe thymic disinvolution
41thymus, spleen, lymph node
117-94 SIVDSp1234 16 CMV; follicular hyperplasia and paracortical expansion of lymph nodes; marked thymic disinvolution
41thymus
146-94 SIVDNFkBDSp1234 17 CMV, cryptosporidiosis, adenovirus, cutaneous candidiasis; thymus, increased interlobular fibrosis, decreased corticomedullary distinction, but expanded cortex
11thymus; 2-31lymph node and spleen
179-94 SIVsubstSp12 14 CMV, follicular depletion with paracortical expansion, lymph nodes and spleen; cutaneous candidiasis; thymic involution
41thymus
183-94 SIVsubstSp12 27 B cell lymphoma, spinal cord and thymic interstitium; pneumocystosis, cardiac thrombosis, SIV
arteriopathy; mild giant cell pneumonia; expanded thymic cortex and thymic lymphoid hyperplasia
41thymus
185-94 SIVsubstSp1234 60 Progressive multifocal leukoencephalopathy; follicular hyperplasia and paracortical expansion, lymph nodes; thymic involution
ND
a11, 1 to 10 positive cells per cm2; 21, 10 to 25 positive cells; 31, 25 to 100 positive cells; 41,.100 positives; ND, not performed. bCMV, cytomegalovirus.
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 4. Thymic histology (A to C) and SIV in situ hybridization (D to I) of infected animals. 146-94 had a large thymus with prominent expanded cortex (A) and only occasional cells containing SIV nucleic acid in the thymus (D) and spleen (G). In contrast, there was severe thymic disinvolution in animals 101-94 (B) and 104-94 (C), with numerous SIV-positive cells in both thymus (E, F) and spleen (H, I). (A to C, bar5150mm; D to I, bar540mm).
TABLE 4. Proviral sequences in PBMC DNA of animals infected with SIVmac239 mutants 20 weeks postinoculation Animal
no. Viral strain sequencedClones Changes in mutagenizedfragment Additional changes in U3 and R regions (location, no. of clones in which theywere present, amino acid change)a
104-94 SIVDNFkB 3 Original deletion retained G3D(9567, 1, downstream out of frame) 117-94 SIVDSp1234 4 Original deletion retained A3T (9576, 4, Y3F)
66-94 SIVDNFkBDSp1234 5 Original deletion retained A3G (9488, 1, R3G); A3G (9651, 1, H3R); C3T (10113, 1, n/c) 360-93 SIVDNFkBDSp1234 2 Original deletion retained G3A (10086, 1, n/c)
146-94 SIVDNFkBDSp1234 4 Original deletion retained T3G (9550, 3, D3E), G3D(9685, 1, downstream out of frame) 179-94 SIVsubstSp12 4 Original substitution retained A3G (9492, 1, H3R); G3T (9495, 1, R3I); G3A (9647, 1,
E3K); G3A (10116, 1, n/c), 19 bp3D(9762-80, 1, downstream out of frame)
183-94 SIVsubstSp12 4 Original substitution retained C3T (9585, 1, T3I); G3A (9749, 1, A3T); G3A (9806, 1, V3I); C3T (9990, 1, n/c); T3G (9971, 1, n/c); G3A (10001, 1, n/c) 185-94 SIVsubstSp1234 3 Part of the substituted
fragment (9 bp) deleted; T at position 9931
substituted for G
C3T (9532, 1, silent): G3A (9620, 1, V3I); G3A (9864, 1, R3H)
187-94 SIVsubstSp1234 5 Various parts of the substituted fragment deleted (7 bp in two clones, 32 bp in three clones)
C3T (9532, 4, silent); G3A (9544, 1, W3stop); T3A (9973, 1, n/c); G3A (10004, 1, n/c); G3A (10015, 1, n/c); C3T (10078, 1, n/c); G3A (10150, 1, n/c)
aD, deletion; n/c, noncoding region. Silent, no amino acid change.3, change of nucleotide or amino acid. Amino acid changes are in thenefreading frame. Nucleotide numbers refer to those described by Regier and Desrosiers (22).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.60.557.507.708.2]during the course of infection. We have seen this pattern rarely if at all with the parental SIVmac239. Animal 146-94 died with AIDS following infection withDNFkBDSp1234, but the levels of SIV expression in tissues were the lowest of all animals examined. Thus, the NF-kB and Sp1 binding elements proba-bly do contribute to the level of SIV replication in rhesus monkeys but the contribution is not major as it is with, for example, thenefgene (13).
Our results suggest that the spatial distribution of enhancer elements may be important for maintaining optimal levels of virus replication, as has been noted previously in transient assays (21). While the deletion mutants did not acquire any compensatory changes, the substSp1234 mutant quite consis-tently acquired deletions in the point-mutated region. This likely reflects a need to bring the remaining enhancer elements in closer proximity to the TATA box. It is also possible, how-ever, that these sequences are selected against for other rea-sons.
Our results also allow some speculation on the possible role of NF-kB and Sp1 elements in replication in particular cell types and tissues and in tissue-specific disease manifestations (1, 7, 9, 20). The behavior of the mutant viruses studied in this report in lymphoid cell cultures was more predictive of the in vivo outcome than the replication of these viruses in primary alveolar macrophage cultures. Mutation of the Sp1 elements had little or no effect on SIV replication in lymphoid cell cultures but had severe effects on SIV replication in primary macrophage cultures (9). The wild-type virus loads seen with Sp1-mutant viruses in vivo are thus consistent with lymphoid cells being the major source of virus production in vivo, as suggested by in situ hybridization and immunohistochemical measurements. It is curious that none of the animals with severely mutated Sp1 elements developed a primary lentiviral encephalitis or granulomatous pneumonia, lesions that are usually seen in about 40% of monkeys that die from SIVmac239 infection (26). These are pathologic manifestations associated with extensive virus replication in tissue macrophages. Thus, while mutation of Sp1 elements may have minimally affected overall levels of virus production in vivo and the progression to disease, it may have affected virus replication in tissue macro-phages and with it the associated tissue-specific disease mani-festations. Animal 146-94, infected withDNFkBDSp1234, died with AIDS but curiously did not exhibit the severe thymic involution usually seen in animals which die of SIV-induced AIDS; 146-94 exhibited only very low levels of SIV sequences in its thymus. Additional studies will be needed to determine whether theDNFkBDSp1234 mutation has a specific effect on virus replication in thymocytes.
Finally, the high virus loads and disease progression seen with theDNFkBDSp1234 virus provide indirect evidence for the importance of the novel enhancer element just upstream of the NF-kB binding sequence. Further work is needed to char-acterize the nature of this enhancer element and its relative contribution to virus replication in rhesus monkeys.
ACKNOWLEDGMENTS
We thank Prabhat Sehgal for veterinary care and clinical procedures and Dean Regier for DNA sequencing and helpful advice. We also thank Heather Knight and Dan Shvetz for technical assistance and Joan Lane, Laura Chalifoux, Ron Veazey, Vito Sasseville, and Fred Doddy for necropsies.
This work was supported by PHS grants AI35365, AI25328, and RR00168.
REFERENCES
1.Clark, N. M., M. C. Hannibal, and D. M. Markovitz.1995. The peri-kB site mediates human immunodeficiency virus type 2 enhancer activation in monocytes but not in T cells. J. Virol.69:4854–4862.
2.Garcia, J. A., F. K. Wu, R. Mitsuyasu, and R. B. Gaynor.1987. Interactions of cellular proteins involved in the transcriptional regulation of the human immunodeficiency virus. EMBO J.6:3761–3770.
3.Gibbs, J. S., A. A. Lackner, S. M. Lang, M. A. Simon, P. K. Sehgal, M. D. Daniel, and R. C. Desrosiers.1995. Progression to AIDS in the absence of a gene forvprorvpx. J. Virol.69:2378–2383.
4.Guyader, M., M. Emerman, P. Sonigo, F. Clavel, L. Montagnier, and M. Alizon.1987. Genome organization and transactivation of the human immu-nodeficiency virus type 2. Nature236:662–669.
5.Hannibal, M. C., D. M. Markovitz, N. Clark, and G. J. Nabel.1993. Differ-ential activation of human immunodeficiency virus type 1 and 2 transcription by specific T-cell activation signals. J. Virol.67:5035–5040.
6.Harrich, D., J. Garcia, F. Wu, R. Mitsuyasu, J. Gonzalez, and R. Gaynor. 1989. Role of SP1-binding domains in in vivo transcriptional regulation of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 63:2585–2591.
7.Hilfinger, J. M., N. Clark, M. Smith, K. Robinson, and D. M. Markovitz. 1993. Differential regulation of the human immunodeficiency virus type 2 enhancer in monocytes at various stages of differentiation. J. Virol.67:4448– 4453.
8.Ilyinskii, P. O., M. D. Daniel, M. A. Simon, A. A. Lackner, and R. C. Desrosiers.1994. The role of upstream U3 sequences in the pathogenesis of simian immunodeficiency virus-induced AIDS in rhesus monkeys. J. Virol. 68:5933–5944.
9.Ilyinskii, P. O., and R. C. Desrosiers.1996. Efficient transcription and rep-lication of simian immunodeficiency virus in the absence of NF-kB and Sp1 binding elements. J. Virol.70:3118–3126.
10. Jones, K. A., J. T. Kadonaga, P. A. Luciw, and R. Tjian.1986. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Sci-ence232:755–759.
11. Kawakami, K., C. Scheidereit, and R. G. Roeder.1988. Identification and purification of a human immunoglobulin-enhancer-binding protein (NF-kB) that activates transcription from a human immunodeficiency virus type 1 promoterin vitro. Proc. Natl. Acad. Sci. USA85:4700–4704.
12. Kestler, H., T. Kodama, D. Ringler, M. Marthas, N. Pedersen, A. Lackner, D. Regier, P. Sehgal, M. Daniel, N. King, and R. Desrosiers.1990. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science248:1109–1112.
13. Kestler, H. W., III, D. J. Ringler, K. Mori, D. L. Panicali, P. K. Sehgal, M. D. Daniel, and R. C. Desrosiers.1991. Importance of thenefgene for mainte-nance of high virus loads and for development of AIDS. Cell65:651–662. 14. Leonard, J., C. Parrott, A. J. Buckler-White, W. Turner, E. K. Ross, M. A.
Martin, and A. B. Rabson.1989. The NF-kB binding sites in the human immunodeficiency virus type 1 long terminal repeat are not required for virus infectivity. J. Virol.63:4919–4924.
15. Maciaszek, J. W., D. A. Talmage, and G. A. Viglianti.1994. Synergistic activation of simian immunodeficiency virus and human immunodeficiency virus type 1 transcription by retinoic acid and phorbol ester through an NF-kB-independent mechanism. J. Virol.68:6598–6604.
[image:7.612.71.545.68.131.2]16. Markovitz, D. M., M. Hannibal, V. L. Perez, C. Gauntt, T. M. Folks, and G. J. Nabel.1990. Differential regulation of human immunodeficiency vi-ruses (HIVs): a specific regulatory element in HIV-2 responds to stimulation of the T-cell antigen receptor. Proc. Natl. Acad. Sci. USA87:9099–9102. FIG. 5. Sequence rearrangements in the viral LTR recovered from animals infected with SIVsubstSp1234. Dots are sequence identities; dashes are deletions. Number of clones with this sequence is shown in parentheses.
on November 9, 2019 by guest
http://jvi.asm.org/
17. Markovitz, D. M., M. J. Smith, J. Hilfinger, M. C. Hannibal, B. Petryniak, and G. J. Nabel.1992. Activation of the human immunodeficiency virus type 2 enhancer is dependent on purine box andkB regulatory elements. J. Virol. 66:5479–5484.
18. Nabel, G., and D. Baltimore.1987. An inducible transcription factor acti-vates expression of human immunodeficiency virus in T cells. Nature326: 711–712.
19. Naidu, Y. M., H. W. Kestler III, Y. Li, C. V. Butler, D. P. Silva, D. K. Schmidt, C. D. Troup, P. K. Sehgal, P. Sonigo, M. D. Daniel, and R. C. Desrosiers.1988. Characterization of infectious molecular clones of simian immunodeficiency virus (SIVmac) and human immunodeficiency virus type 2: persistent infection of rhesus monkeys with molecularly cloned SIVmac. J. Virol.62:4691–4696.
20. Parrott, C., T. Seidner, E. Duh, J. Leonard, T. S. Theodore, A. Buckler-White, M. A. Martin, and A. B. Rabson.1991. Variable role of the long terminal repeat Sp1-binding sites in human immunodeficiency virus replica-tion in T lymphocytes. J. Virol.65:1414–1419.
21. Perkins, N. D., N. L. Edwards, C. S. Duckett, A. B. Agranoff, R. M. Schmid, and G. J. Nabel.1994. A cooperative interaction between NF-kB and Sp1 is required for HIV-1 enhancer activation. EMBO J.12:3551–3558. 22. Regier, D. A., and R. C. Desrosiers.1990. The complete nucleotide sequence
of a pathogenic molecular clone of simian immunodeficiency virus. AIDS Res. Hum. Retroviruses6:1221–1231.
23. Renjifo, B., N. A. Speck, S. Winandy, N. Hopkins, and Y. Li.1990.cis-Acting elements in the U3 region of a simian immunodeficiency virus. J. Virol. 64:3130–3134.
24. Rosen, C. A., J. G. Sodroski, and W. A. Haseltine.1985. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell41:813–823.
25. Ross, E. K., A. J. Buckler-White, A. B. Rabson, G. Englund, and M. A. Martin.1991. Contribution of NF-kB and Sp1 binding motifs to the repli-cative capacity of human immunodeficiency virus type 1: distinct patterns of viral growth are determined by T-cell types. J. Virol.65:4350–4358. 26. Simon, M. A., L. V. Chalifoux, and D. J. Ringler.1992. Pathologic features
of SIV-induced disease and the association of macrophage infection with disease evolution. AIDS Res. Hum. Retroviruses8:327–337.
27. Winandy, S., B. Renjifo, Y. Li, and N. Hopkins.1992. Nuclear factors that bind two regions important to transcriptional activity of the simian immu-nodeficiency virus long terminal repeat. J. Virol.66:5216–5223.
28. Wykrzykowska, J. J., D. R. Pauley, A. A. Lackner, and M. A. Simon.1996. Evaluation of anti-human antibodies for immunohistochemistry on archival nonhuman primate tissues. J. Med. Primatol.25:71–77.