Modelling host evolutionary responses to infection
Sophie Johns
Supervisors: Michael Jennions & Megan Head
A thesis submitted for the degree of Masters of Philosophy
The Australian National University
Research School of Biology
Ecology & Evolution
Declaration
The research presented in this thesis is my own original work. All of the chapters are co-authored. The authorship order indicates the intellectual input and workload. No part of this thesis has been submitted for any previous degree.
Sophie Louise Johns
Acknowledgements
Completing my Masters thesis has been an unexpected journey. It began with a move to a foreign country and definitely did not go how I expected to but I am thankful I have reach this point with the support of the people around me.
After having Michael Jennions as an honours supervisor I knew I was in safe hands. Mike, you have always encouraged me to push myself and seek expertise. Thank you for taking me as your student despite my unwavering interest in modelling. I never would have been able to make such a leap without your understanding and encouragement.
Megan, thank you for being there for me, even though I was on the other side of the globe for part of my masters. Your dedication, commitment and passion for research is inspiring. I am so grateful that I had someone to openly talk to throughout this whole process.
Sally, I cannot believe I lucked into such a knowledgeable and understanding professor. I came to you with minimal modelling skills and you patiently took me through the process of creating my own model. You helped me think outside the box but also gave me a greater overall understanding of theory. Thank you for teaching me.
Jono, throughout my research you have been incredibly open and ready to help me with any questions I have. I appreciate that you would take the time to explain theoretical concepts and convey them in an easy to understand way.
4
I would also like to thank the people in my personal life who have supported me along this journey. To my partner, Tom, thank you for seeing something in me that I don’t always see in myself. You simultaneously inspire me and lift me up, I could not have done this without you. To my friends Claudia, Katie, Leah, Marcella, Seamus, Jarod, Kevin, Tanner, and all the Fablites, thank you for being my rocks and allowing me to let me hair down. To my family, Ross, Julie, Matthew and Rebecca. Thank you for giving me the opportunity to follow my passions and for always supporting my decisions.
Abstract
Infectious diseases are pervasive, producing strong evolutionary pressure on their hosts. Often epidemiological models focus on pathogen evolution because they have short lifespans relative to their hosts. However, pathogens also impose strong selective pressure on their hosts. With prolonged exposure to pathogens host populations adapt to pathogen’s selection pressure. Host evolution could occur in a variety of ways - from minimising infection costs to disease avoidance. In this thesis I present two theoretical models that examine optimal host evolutionary responses to infectious diseases.
Chapter 1 explores how sexually transmitted infections shape female reproductive investment and, in turn, alters selection on males for infection resistance. I discuss the conditions required for female terminal investment to be favorable, and in populations where females terminally invest, I then consider the ramifications for selection on male immune resistance, since infecting their mate has a reproductive advantage.
6
Thesis outline
The following chapters compose this thesis:
1. Males can evolve lower resistance to sexually transmitted infections to infect their mates and thereby increase their own fitness.
Evolutionary Ecology 2019 33 (2): 149 – 172.
2. Behavioural evolution could rescue Tasmanian devils from disease-induced extinction: a theoretical model.
Contents
Declaration ... 1
Acknowledgements ... 3
Abstract ... 5
Thesis outline ... 6
Introduction …... 9
Chapter 1 ... 26
Appendix 1 ... 45
Chapter 2 ... 52
Appendix 2 ... 80
Introduction
Here I introduce the main concepts and provide some relevant background information to better understand the two chapters within this thesis. I will briefly cover infectious diseases, immunity, epidemiology, and evolutionary rescue. Understanding these four topics will make the chapters easier to understand. Both chapters present mathematical models that have been submitted for publication. The first chapter describes a model of sexually transmitted infections (STIs). It examines if, and then how, females should alter their reproductive effort in response to becoming infected and how any change in female behaviour then impacts selection on males for increased or, more intriguingly, decreased STI resistance. The second chapter examines the effect of devil facial tumour disease (DFTD) on Tasmanian devils, Sarcophilus harrisii. Specifically, I explore the possibility that devils evolve to be less aggressive to reduce the risk of acquiring an infection. The common feature that unites both models is that they examine host evolution in response to infectious diseases.
Infectious diseases
An infectious disease is a transmissible condition that uses an individual’s resources that they might otherwise use for reproduction, subsistence, or competition. Infectious diseases range in severity from those with mild symptoms that clear quickly (e.g. the common cold: Thielmann et al. 2018) to aggressive symptoms and deadly consequences (e.g. ebola: Khan et al. 2017). They are caused by pathogenic microorganisms including bacteria, viruses, fungi, and parasites (Gislason 2015). Due to the large body of literature on human diseases and the relative paucity of literature on animal diseases it is easy to underestimate the breadth of non-human infections.
10
fighting infections which then limits the resources available for host reproduction, reducing host competitive ability due to poorer physical condition, and directly causing host mortality. The importance of infectious disease research is magnified in light of climate change which impacts pathogens in a variety of ways (Wu et al. 2014; Gislason 2015) including range expansion (e.g. Ogden et al. 2006).
Infectious diseases in conservation
Infectious disease pandemics can cause rapid population decline and even threaten species with extinction (Smith, Sax & Lafferty 2006: e.g. Skerratt et al. 2016). Population decline or extinction in a single species can have long reaching effects for an entire ecosystem (e.g. Hollings et al. 2016). Conservation efforts to protect focal populations require sufficient understanding of diseases to implement appropriate control measures. This understanding can partly come from theoretical models (Joseph et al. 2013). For example, Canessa et al. (2018) recommend Batrachochytrium salamandrivorans control measures to prevent Palearctic salamander, Salamandra salamandra, diversity loss. Their model showed that anti-fungal and probiotic treatments must reduce transmission by 90% to be effective, preventing resources being used on ineffective treatments. Additionally, the model revealed local population culls effectively cause local disease extinction and prevent spread to surrounding populations only in the absence of reservoir hosts (non- Palearctic salamander hosts). Another example of research aiding disease management occurs in DFTD which is currently threatening Tasmanian devils with extinction (McCallum et al. 2009). Many control methods have been suggested, including controlled Tasmanian devil culls to reduce disease spread and eradicate the disease (McCallum & Jones 2006). However, Beeton & McCallum (2011) developed a mathematical model which determined that culling would be ineffective. Thankfully, their findings were accepted before any devils were actually culled.
Infectious diseases in domestic animals
Green (2018) data analysis determined that ‘routine’ footbathing of herds has no impact on lameness unless performed during an outbreak. Since footbathing costs time and money this knowledge benefits farm managers. Research into farm practices to prevent diseases must consider economic viability otherwise farm managers will be unlikely to implement these practices into their business. The combination of biological and economic modelling is therefore a powerful way to solve practical problems faced by those working in agriculture.
Infectious diseases threatening human health
Controlling animal diseases has a direct impact on human health as many pathogens can jump the species barrier to humans (zoonotic disease: Olival et al. 2017). There are numerous well-known zoonotic diseases, but the emergence of new diseases is of major concern (Nil-Trebi 2017; Olival et al. 2017). Species that have regular direct contact with humans are of particular danger (e.g. bed bugs: Lai et al. 2016). The devastating impact of emerging zoonotic diseases, and the importance of research into them was clearly seen in the responses to both the HIV (Volberding 2011) and Ebola outbreaks (Mutters et al. 2018).
Host immunity
12
variation in resistance both within and between amphibian species reflecting genetic differences in their innate immunity. Frog populations with greater historical exposure to infection have a more robust innate immune response to the fungus (Grogan et al. 2018), implying selection for immune investment. In addition, however, individuals that have cleared the virus develop an acquired immunity reflecting an adaptive immune response (Fu & Waldman 2017). Although adaptive immunity is acquired over an individual’s lifetime there is still the potential for evolutionary selection on its effectiveness and sensitivity to pathogens (i.e. equivalent to selection on reaction norms leading to adaptive phenotypic plasticity). In this thesis, in the first model I investigate the evolution of changes in investment in innate rather than adaptive immunity in response to a sexually transmitted disease. It should be noted, however, that my model also considers the evolution of a plastic shift in investment into innate immunity versus reproduction upon becoming infected (see below ‘Immune-Reproduction Trade-off’).
Disease avoidance
Since diseases are so costly to hosts, there is strong selective pressure for hosts to avoid infection (e.g. Epstein et al. 2016). Disease avoidance can be achieved in a range of ways from the evolution of strong innate immunity to prevent infection, to a robust adaptive immune system to quickly acquire immunity if infected; and from evolving behaviour to avoid initial disease exposure, to the evolution of behaviours to acquire immunity through early disease exposure.
Immune investment
Immune-reproduction trade-off
Immune investment can be costly and since resources are often limited this can result in a trade-off between immune and reproductive investment (Verhulst, Riedstra, & Wiersma 2005; Luong & Polak 2007; Dallas, Holtackers, & Drake 2016). Investment in reproduction could include nutritional resources to produce gametes and sustain embryos (Marzal et al. 2005), traits that facilitate mate attraction (Jacot et al. 2005; Martin & Lopez 2015; McKean & Nunney 2001), and parental care (Bonneaud et al. 2003). An increase in reproductive investment often leads to improved reproductive success as offspring have a greater chance of surviving to sexual maturity and being in superior condition (e.g. Marzal et al. 2005). However, reproductive investment can detract from immune investment resulting in a heightened risk of acquiring an infection upon exposure to a disease-producing pathogen. This could lower overall life-time reproductive success since an infection can reduce reproduction, or even kill the host. All else being equal, individuals should therefore invest more into immunity if there is a higher probability of coming into contact with disease-producing pathogens. Given this scenario there would then be fewer resources available for reproduction, but the individual would be healthier and still capable of reproducing (self-maintenance strategy). Hypothetically, there exists an optimal investment strategy which balances the risk of encountering and acquiring an infection against declines in reproductive success that arise from investment into disease prevention.
Immune investment once infected
14
determine experimentally, but mathematical models can help us to determine likely evolutionary trends and optimal strategies. In my first model I consider the optimal trade-off into immunity versus reproduction by females in response to acquiring a sexually transmitted infection. I then consider how this female response affects male investment into innate immunity.
Behavioural changes
Behaviour is a key aspect of a host that is often under selection to reduce the risk of acquiring a disease (Sarabian, Curtis, & McMullan 2018; Weston, Hauck, & Amlot 2018). Particular behaviours can increase or decrease an individual’s risk of disease exposure (e.g. being less sexually active lowers the risk of acquiring a sexually transmitted infection: Berec, Janouskova, & Theuer 2017). Some behaviours are a direct response to a given stimulus that are shared by all members of a species, but often individuals have a behavioural disposition (behavioural syndrome: Adriaenssens & Johnsson 2013) that affects the extent to which they respond to certain stimuli. If these behavioural propensities are heritable, then selection for disease avoidance can influence behaviour. Intriguingly, optimal disease avoidance strategies have the potential to create sexual conflict due to life-history differences between the sexes, that affect the costs and benefits of specific behaviours (Fischer et al. 2015; Kelly 2017). In my second model I consider that trade-off between a behaviour that reduces disease exposure at the expense of a decreased birth-rate.
Epidemiology
has been used to describe Ebola which includes an extra classification for hosts that have been infected but cannot yet infect others (E, Exposed: Diaz et al. 2018). This occurs when a host first becomes infected with cells but there has been insufficient time for the cells to replicate to the point that they can be transmitted to other hosts. Ignoring the presence of an exposed class can lead to underestimating how many individuals a single host can infect, a term which is called the disease’s reproductive ratio (Wearing, Rohani, & Keeling 2005). Models are often used to predict future disease trends (Huppert & Katriel 2013) and to uncover appropriate control measures. These measures could include immunisation schemes (McCallum 2016: e.g. recommended vaccination strategy in maternal pertussis Fabricius et al. 2018), reducing transmission between hosts (e.g. culling: Beeton & McCallum 2011), and predicting pathogen evolution (e.g. drug-resistance: McBryde et al. 2017) to work out the optimal scheduling and combination of different medical treatments. Surprisingly few models use an epidemiological framework to examine rapid host evolution in response to strong pathogen selective pressure, despite the existence of empirical evidence for evolution over ecological time frames (e.g. evolution in response to chtridiomycosis: Grogan et al. 2018 and DFTD: Epstein et al. 2016). There is a tendency to assume that evolution is too ‘slow acting’ to be relevant to modeling most disease-host interactions.
Density and frequency dependent transmission
16
transmission since individuals are assumed to maintain the same mating rate despite population size. At low population densities hosts actively seek mates maintaining the same rate of disease spread even though the population is small. A lack of positive density-dependent transmission means that frequency-dependent diseases have the potential to cause host extinction. However, there are exceptions to this rule with some STIs displaying density-dependent transmission (Johnson & Geffen 2016; Ryder et al. 2005) and some non-STI, communicable diseases displaying frequency-dependent transmission (e.g. DFTD in Tasmanian devils: McCallum et al. 2009; and sarcoptic mange in red foxes: Devenish-Nelson et al. 2014). In frequency-dependent systems, hosts are assumed to continue interacting even when population numbers are extremely low, perhaps due to a communal feeding source or other social behaviour (including mating) that brings hosts together. Although models typically use one type of transmission it is possible to incorporate both (Ryder et al. 2007). In my first model on STIs I assume that transmission is frequency-dependent. In my second model on Tasmanian devils I assume that transmission is frequency-dependent.
Evolutionary rescue
unlikely (Wal et al. 2013). In nature there are putative cases of evolutionary rescue, but definitive evidence is elusive. One example of rapid evolution seemingly rescuing a population from extinction comes from Franks et al. (2007) who resurrected Brassica rapa seeds produced prior to an extended drought. Plants prior to the drought had genotypes with regular flowering times whereas those after the drought had delayed flowering time. Presumably the climactic pressure caused strong selection on flowering time leading to its rapid evolution. Another example comes from the Tasmanian devil where Epstein et al. (2016) has shown a rapid genomic shift in response to DFTD. These cases demonstrate plausibility of populations being rescued by rapid evolution.
Conclusion
In this thesis I use mathematical modelling techniques to determine optimal host strategies in two infectious disease systems. The first model involves a sexually transmitted infection where females can alter their investment into reproductive versus innate immunity in response to becoming infected. I identify the conditions required for terminal investment to evolve as opposed to a self-maintenance strategy. I then examine the impact that female terminal investment has on the evolution of male innate immunity. I show that males might reduce their innate immunity because males that infect their partners can have greater reproductive success than healthy males due to female terminal investment. My second model investigates behavioural evolution in Tasmanian devils in response to DFTD. DFTD is spread through devils biting each other creating selection for lower aggression to avoid infection. I ask whether the evolution of increased passivity could provide a mechanism of evolutionary rescue for devils. Both my models increase our understanding of how hosts can evolve in response to diseases.
18
ANTIA, R., REGOES, R.R., KOELLA, J.C. & BERGSTROM, C.T., 2003. The role of evolution in the emergence of infectious diseases. Nature, 426(6967), 658. BEETON, N. & MCCALLUM, H. 2011. Models predict that culling is not a feasible
strategy to prevent extinction of Tasmanian devils from facial tumour disease. Journal of Applied Ecology, 48, 1315-1323.
BEGON, M., BENNETT, M., BOWERS, R. G., FRENCH, N. P., HAZEL, S. M. & TURNER, J. 2002. A clarification of transmission terms in host-microparasite models: numbers, densities and areas. Epidemiology and Infection, 129, 147-153. BELL, G. 2017. Evolutionary Rescue. In: FUTUYMA, D. J. (ed.) Annual Review of
Ecology, Evolution, and Systematics, Vol 48.
BEREC, L., JANOUSKOVA, E. & THEUER, M. 2017. Sexually transmitted infections and mate-finding Allee effects. Theoretical Population Biology, 114, 59-69. BEST, A., WEBB, S., ANTONOVICS, J. & BOOTS, M. 2012. Local transmission
processes and disease-driven host extinctions. Theoretical Ecology, 5, 211-217. BONNEAUD, C., MAZUC, J., GONZALEZ, G., HAUSSY, C., CHASTEL, O.,
FAIVRE, B. & SORCI, G. 2003. Assessing the cost of mounting an immune response. American Naturalist, 161, 367-379.
BRANNELLY, L. A., WEBB, R., SKERRATT, L. F. & BERGER, L. 2016. Amphibians with infectious disease increase their reproductive effort: evidence for the terminal investment hypothesis. Open Biology, 6.
BROWN, V. R., BOWEN, R. A. & BOSCO-LAUTH, A. M. 2018. Zoonotic pathogens from feral swine that pose a significant threat to public health. Transboundary and Emerging Diseases, 65, 649-659.
CALBACHO-ROSA, L., MORENO-GARCIA, M. A., LANZ-MENDOZA, H., PERETTI, A. V. & CORDOBA-AGUILAR, A. 2012. Reproductive activities impair immunocompetence in Physocyclus dugesi (Araneae: Pholcidae). Journal of Arachnology, 40, 18-22.
CANESSA, S., BOZZUTO, C., GRANT, E. H. C., CRUICKSHANK, S. S., FISHER, M. C., KOELLA, J. C., LOTTERS, S., MARTEL, A., PASMANS, F., SCHEELE, B. C., SPITZEN-VAN DER SLUIJS, A., STEINFARTZ, S. & SCHMIDT, B. R. 2018. Decision-making for mitigating wildlife diseases: From theory to practice for an emerging fungal pathogen of amphibians. Journal of Applied Ecology, 55, 1987-1996.
CHRISTIE, M. R. & SEARLE, C. L. 2018. Evolutionary rescue in a host-pathogen system results in coexistence not clearance. Evolutionary Applications, 11, 681-693. DALLAS, T., HOLTACKERS, M. & DRAKE, J. M. 2016. Costs of resistance and
infection by a generalist pathogen. Ecology and Evolution, 6, 1737-1744.
DE CASTRO, F. & BOLKER, B. 2005. Mechanisms of disease-induced extinction. Ecology Letters, 8, 117-126.
DEVENISH-NELSON, E.S., RICHARDS, S.A., HARRIS, S., SOULSBURY, C. & STEPHENS, P.A., 2014. Demonstrating frequency-dependent transmission of sarcoptic mange in red foxes. Biology letters, 10(10), 20140524.
DIAZ, P., CONSTANTINE, P., KALMBACH, K., JONES, E. & PANKAVICH, S. 2018. A modified SEIR model for the spread of Ebola in Western Africa and metrics for resource allocation. Applied Mathematics and Computation, 324, 141-155. EPSTEIN, B., JONES, M., HAMEDE, R., HENDRICKS, S., MCCALLUM, H.,
MURCHISON, E. P., SCHONFELD, B., WIENCH, C., HOHENLOHE, P. & STORFER, A. 2016. Rapid evolutionary response to a transmissible cancer in Tasmanian devils. Nature Communications, 7.
FABRICIUS, G., AISPURO, P. M., BERGERO, P., BOTTERO, D., GABRIELLI, M. & HOZBOR, D. 2018. Pertussis epidemiology in Argentina: TRENDS after the introduction of maternal immunisation. Epidemiology and Infection, 146, 858-866.
FISCHER, J., JUNG, N., ROBINSON, N. & LEHMANN, C. 2015. Sex differences in immune responses to infectious diseases. Infection, 43, 399-403.
FLAJNIK, M. F. 2018. A cold-blooded view of adaptive immunity. Nature Reviews Immunology, 18, 438-453.
FRANKS, S.J., SIM, S. & WEIS, A.E., 2007. Rapid evolution of flowering time by an annual plant in response to a climate fluctuation. Proceedings of the National Academy of Sciences, 104(4), 1278-1282.
FU, M. J. & WALDMAN, B. 2017. Major histocompatibility complex variation and the evolution of resistance to amphibian chytridiomycosis. Immunogenetics, 69, 529-536.
20
GISLASON, M. K. 2015. Climate change, health and infectious disease. Virulence, 6, 535-538.
GOMULKIEWICZ, R. & SHAW, R. G. 2013. Evolutionary rescue beyond the models. Philosophical Transactions of the Royal Society B-Biological Sciences, 368.
GONZALEZ, A., RONCE, O., FERRIERE, R. & HOCHBERG, M. E. 2013. Evolutionary rescue: an emerging focus at the intersection between ecology and evolution. Philosophical Transactions of the Royal Society B-Biological Sciences, 368. GREEN, L. & CLIFTON, R., 2018. Diagnosing and managing footrot in sheep: an
update. In Practice, 40(1), 17-26.
GROGAN, L. F., CASHINS, S. D., SKERRATT, L. F., BERGER, L., MCFADDEN, M. S., HARLOW, P., HUNTER, D. A., SCHEELE, B. & MULVENNA, J. 2018. Evolution of resistance to chytridiomycosis is associated with a robust early immune response. Molecular Ecology, 27, 919-934.
HENNESSY, D. A. 2017. Conceptual models underlying economic analysis of animal health and welfare with the inclusion of three components: people, products and resources. Revue Scientifique Et Technique-Office International Des Epizooties, 36, 77-85. HOLLINGS, T., JONES, M., MOONEY, N. & MCCALLUM, H. 2016. Disease-induced decline of an apex predator drives invasive dominated states and threatens biodiversity. Ecology, 97, 394-405.
HUPPERT, A. & KATRIEL, G. 2013. Mathematical modelling and prediction in infectious disease epidemiology. Clinical Microbiology and Infection, 19, 999-1005. JACOT, A., SCHEUBER, H., KURTZ, J. & BRINKHOF, M. W. G. 2005. Juvenile
immune status affects the expression of a sexually selected trait in field crickets. Journal of Evolutionary Biology, 18, 1060-1068.
JOHNSON, L. F. & GEFFEN, N. 2016. A Comparison of Two Mathematical Modeling Frameworks for Evaluating Sexually Transmitted Infection Epidemiology. Sexually Transmitted Diseases, 43, 139-146.
JOSEPH, M. B., MIHALJEVIC, J. R., ARELLANO, A. L., KUENEMAN, J. G., PRESTON, D. L., CROSS, P. C. & JOHNSON, P. T. J. 2013. Taming wildlife disease: bridging the gap between science and management. Journal of Applied Ecology, 50, 702-712.
KEELING, M. J. & ROHANI, P. 2008. Modeling Infectious Diseases in Humans and Animals, Princeton University Press.
crassidens (Orthoptera: Tettigonioidea: Anostostomatidae). Ethology, 123, 785-792. KHAN, F. N., QAZI, S., TANVEER, K. & RAZA, K. 2017. A review on the antagonist Ebola: A prophylactic approach. Biomedicine & Pharmacotherapy, 96, 1513-1526.
LAI, O., HO, D., GLICK, S. & JAGDEO, J. 2016. Bed bugs and possible transmission of human pathogens: a systematic review. Archives of Dermatological Research, 308, 531-538.
LUONG, L. T. & POLAK, M. 2007. Costs of resistance in the Drosophila-macrocheles system: A negative genetic correlation between ectoparasite resistance and reproduction. Evolution, 61, 1391-1402.
LYNCH, M. & LANDE, R. 1993. EVOLUTION AND EXTINCTION IN RESPONSE TO ENVIRONMENTAL-CHANGE.
MARTIN, J. & LOPEZ, P. 2015. Condition-dependent chemosignals in reproductive behavior of lizards. Hormones and Behavior, 68, 14-24.
MARZAL, A., DE LOPE, F., NAVARRO, C. & MOLLER, A. P. 2005. Malarial parasites decrease reproductive success: an experimental study in a passerine bird. Oecologia, 142, 541-545.
MCBRYDE, E. S., MEEHAN, M. T., DOAN, T. N., RAGONNET, R., MARAIS, B., GUERNIER, V. & TRAUER, J. M. 2017. The risk of global epidemic replacement with drug-resistant Mycobacterium tuberculosis strains. International Journal of Infectious Diseases, 56, 14-20.
MCCALLUM, H. 2016. Models for managing wildlife disease. Parasitology, 143, 805-820.
MCCALLUM, H., BARLOW, N. & HONE, J. 2001. How should pathogen transmission be modelled? Trends in Ecology & Evolution, 16, 295-300.
MCCALLUM, H. & JONES, M. 2006. To lose both would look like carelessness: Tasmanian devil facial tumour disease. Plos Biology, 4, 1671-1674.
22
nonself by the innate immune system. Science, 296, 298-300.
MOONEY, H.A. & CLELAND, E.E., 2001. The evolutionary impact of invasive species. Proceedings of the National Academy of Sciences, 98(10), pp.5446-5451.
MUTTERS, N. T., MALEK, V., AGNANDJI, S. T., GUNTHER, F. & TACCONELLI, E. 2018. Evaluation of the scientific impact of the Ebola epidemic: a systematic review. Clinical Microbiology and Infection, 24, 573-576. NII-TREBI, N. I. 2017. Emerging and Neglected Infectious Diseases: Insights,
Advances, and Challenges. Biomed Research International, 1-15.
O'HARA, P. A. 2007. The global spread of AIDS and HIV. Journal of Economic Issues, 41, 459-468.
OGDEN, N.H., MAAROUF, A., BARKER, I.K., BIGRAS-POULIN, M., LINDSAY, L.R., MORSHED, M.G., O'CALLAGHAN, C.J., RAMAY, F., WALTNER-TOEWS, D. & CHARRON, D.F., 2006. Climate change and the potential for range expansion of the Lyme disease vector Ixodes scapularis in Canada. International journal for parasitology, 36(1), pp.63-70.
OLIVAL, K. J., HOSSEINI, P. R., ZAMBRANA-TORRELIO, C., ROSS, N., BOGICH, T. L. & DASZAK, P. 2017. Host and viral traits predict zoonotic spillover from mammals. Nature, 546, 646-+.
OTTO, S.P. & DAY, T., 2007. A biologist's guide to mathematical modeling in ecology and evolution. Princeton University Press.
PALANISAMY, R., BHATT, P., KUMARESAN, V., PASUPULETI, M. & AROCKIARAJ, J. 2018. Innate and adaptive immune molecules of striped murrel Channastriatus. Reviews in Aquaculture, 10, 296-319.
RONGVAUX, A. 2018. Innate immunity and tolerance toward mitochondria. Mitochondrion, 41, 14-20.
RYDER, J. J., MILLER, M. R., WHITE, A., KNELL, R. J. & BOOTS, M. 2007. Host-parasite population dynamics under combined frequency- and density-dependent transmission. Oikos, 116, 2017-2026.
RYDER, J. J., WEBBERLEY, K. M., BOOTS, M. & KNELL, R. J. 2005. Measuring the transmission dynamics of a sexually transmitted disease. Proceedings of the National Academy of Sciences of the United States of America, 102, 15140-15143.
SARABIAN, C., CURTIS, V. & MCMULLAN, R. 2018. Evolution of pathogen and parasite avoidance behaviours. Philosophical Transactions of the Royal Society B-Biological Sciences, 373.
MARANTELLI, G., NEWELL, D. A., PHILIPS, A., MCFADDEN, M., HINES, H. B., SCHEELE, B., BRANNELLY, L. A., SPEARE, R., VERSTEEGEN, S., CASHINS, S. D. & WEST, M. 2016. Priorities for management of chytridiomycosis in Australia: saving frogs from extinction. Wildlife Research, 43, 105-120.
SMITH, K. F., SAX, D. F. & LAFFERTY, K. D. 2006. Evidence for the role of infectious disease in species extinction and endangerment. Conservation Biology, 20, 1349-1357.
THIELMANN, A., GERASIMOVSKA-KITANOVSKA, B., KOSKELA, T. H., MEVSIM, V., WELTERMANN, B. & EUROPEAN GENERAL PRACTICE, R. 2018. Self-care for common colds: A European multicenter survey on the role of subjective discomfort and knowledge about the self-limited course - The COCO study. Plos One, 13.
TURNER, J., BEGON, M. & BOWERS, R. G. 2003. Modelling pathogen transmission: the interrelationship between local and global approaches. Proceedings of the Royal Society B-Biological Sciences, 270, 105-112.
VERHULST, S., RIEDSTRA, B. & WIERSMA, P. 2005. Brood size and immunity costs in zebra finches Taeniopygia guttata. Journal of Avian Biology, 36, 22-30.
VOLBERDING, P. 2011. The impact of HIV research on health outcome and healthcare policy. Annals of Oncology, 22, 50-53.
WAL, E. V., GARANT, D., FESTA-BIANCHET, M. & PELLETIER, F. 2013. Evolutionary rescue in vertebrates: evidence, applications and uncertainty. Philosophical Transactions of the Royal Society B-Biological Sciences, 368.
WEARING, H. J., ROHANI, P. & KEELING, M. J. 2005. Appropriate models for the management of infectious diseases. Plos Medicine, 2, 621-627.
WEI, X., LIN, W. & HENNESSY, D.A., 2015. Biosecurity and disease management in China’s animal agriculture sector. Food Policy, 54, pp.52-64.
WESTON, D., HAUCK, K. & AMLOT, R. 2018. Infection prevention behaviour and infectious disease modelling: a review of the literature and recommendations for the future. Bmc Public Health, 18.
24
26
Chapter 1
Males can evolve lower resistance to sexually transmitted infections to infect
their mates and thereby increase their fitness
Vol.:(0123456789) Evolutionary Ecology (2019) 33:149–172
https://doi.org/10.1007/s10682-019-09976-1
1 3
ORIGINAL PAPERMales can evolve lower resistance to sexually transmitted infections to infect their mates and thereby increase their own fitness
Sophie Johns1 · Jonathan M. Henshaw1,2 · Michael D. Jennions1 · Megan L. Head1
Received: 22 November 2018 / Accepted: 19 February 2019 / Published online: 25 February 2019 © Springer Nature Switzerland AG 2019
Abstract
Sexually transmitted infections (STIs) often lower their host’s future reproductive success by inducing sterility. Females can minimise the reproductive cost of infection by plasti-cally increasing their current reproductive effort (i.e. terminal investment) before they become sterile. In polyandrous systems, long-term female survival or fecundity is often irrelevant to male fitness. Mating with an infected, terminally investing female potentially yields greater fitness gains for males than mating with an uninfected female. Males might consequently benefit from infecting females with an STI. We construct mathematical mod-els of the evolutionary consequences of a sterilising STI. We show that females should terminally invest in response to an STI when immune investment is relatively ineffective at delaying STI-induced sterility. Cost-effective immune responses may conversely select for reduced reproductive effort after infection (‘terminal divestment’). Crucially, we then show that female terminal investment can select for lower STI resistance in males. This selection is driven by fitness gains to males that acquire the STI and subsequently infect their mates, which offset any costs of infection (e.g. male sterility). This type of adaptive mate harm generates sexual conflict over the optimal level of resistance to STIs. It could partly explain why immune reactions to new infections are weaker in males than females of many species.
Keywords Life history · Model · Sexual conflict · Sexual dimorphism · Sexually antagonistic · Terminal investment
Sophie Johns, Jonathan M. Henshaw: Joint first authors.
* Jonathan M. Henshaw [email protected]
1 Division of Ecology and Evolution, Research School of Biology, The Australian National University, Canberra, ACT 2601, Australia
28
150 Evolutionary Ecology (2019) 33:149–172
1 3
IntroductionSex differences in the harmful effects of sexually transmitted infections (STIs) can lead to intra-locus sexual conflict over disease resistance, where optimal immune investment is higher for the sex that suffers greater STI harm (Forman et al. 2012). More intriguing, however, is the idea that sexual conflict over immunity can arise because males benefit from transferring an STI to their mates (see Haaland et al. 2017, who discuss this idea in the framework of differential allocation). Specifically, this could occur if STI-infected females increase investment into their current offspring as an adaptive response to their reduced long-term fitness prospects (a form of ‘terminal investment’: Clutton-Brock 1984; note that, despite the name, terminal investment need not lead to the death of the female). This female response provides a fitness benefit to the sires of her current offspring. Con-sequently, males might be under selection to reduce their own immune resistance in order to acquire an STI and then infect their mates. This unusual mechanism would create sexual conflict over disease resistance, which might help to explain why males of many species have weaker immune responses to initial infections (Tschirren et al. 2003; Cordoba-Agui-lar et al. 2006) and higher infection rates (Strandberg and Tucker 1974; Zuk and McKean 1996) than females (although patterns of sexual dimorphism in immunity are heterogene-ous across species: Kelly et al. 2018; see also Cousineau and Alizon 2014; Gipson and Hall 2016).
Sexual conflict and mate harm
The evolutionary interests of male and female mating partners are never perfectly aligned (Chapman et al. 2003). Reproductive investment is a prime battleground for sexual con-flict, with each sex generally preferring that their mates invest more in offspring. In many species, this conflict leads to inter-locus sexual conflict and selection for direct manipula-tion of partner behavior (Perry and Rowe 2015). For example, male Drosophila influence female egg laying rate via seminal fluid proteins (Rice 1996; Wolfner 1997, 2002; Yapici et al. 2008; Wilburn and Swanson 2016). Male traits that elevate egg production (Chapman et al. 1998; Lessells 2005; Bonduriansky 2014) are often accompanied by female traits to resist such manipulation (Wigby and Chapman 2004; Nandy et al. 2013). Parental invest-ment decisions can also be manipulated indirectly by changing the conditions under which they are made. For instance, some studies suggest that males could evolve to harm their partners’ future fitness prospects to induce an increase in current egg production (Lessells 2005; Bonduriansky 2014).
Terminal investment: going out with a bang
151 Evolutionary Ecology (2019) 33:149–172
1 3
Terminal investment is often discussed in the context of senescence (Creighton et al. 2009), but many other factors can reduce future fitness prospects, including an increase in preda-tion risk (Tyson et al. 2010), lower food availability (Kruuk and Parish 1982), and infection by pathogens (Knell and Webberley 2004). For example, female pea aphids Acyrthosiphon pisum injected with Escherichia coli produce more offspring than control females (Altin-cicek et al. 2008).STIs as a manipulatable trigger for terminal investment
Female terminal investment potentially provides a mechanism that males can exploit to increase female investment in shared offspring. Males need only manipulate the ‘cue’ that triggers greater female investment. We see three reasons why STIs might be a plausible mechanism of adaptive mate harm, especially relative to other infectious diseases. First, STIs often cause sterility (Lockhart et al. 1996; Antonovics et al. 2011; Gimenes et al. 2014), which ends a host’s reproductive lifespan and may consequently select for termi-nal investment. Indeed, termitermi-nally investing females have been shown to increase both egg quantity (Strandberg and Tucker 1974; Simmons and Rodgers 1994; Snook and Markow 2002; Rittschof et al. 2013; Staudacher et al. 2015) and quality (Bowers et al. 2012). Sec-ond, unlike many other forms of mate harm, such as physical injury, delayed sterility is unlikely to lower the fitness of the offspring of a current mate (Hurst et al. 1995). Conse-quently, a male may not suffer a fitness loss from infecting his mate. Third, many STIs are detrimental to females but cause minimal direct harm to males.
Terminal ‘divestment’: when it’s better to just hang on in there
In contrast to our arguments above, in some cases infection may trigger an increase in immune defense, at the expense of reproductive investment (Ilmonen et al. 2000). We call such a strategy ‘terminal divestment’ if it leads to reduced overall investment in current offspring: i.e., the opposite of terminal investment. Since STIs are often life-long infec-tions, additional immune investment might be insufficient to clear an infection, but it could delay sterility and extend an individual’s reproductive lifespan. If selection favours reduced female reproductive investment upon acquiring an STI, this will increase the costs of infec-tion for males, and thereby select for greater male STI resistance.
Our modelling approach
Terminal investment and divestment by females generate sex differences in the costs and benefits of STI infection. This should drive evolution of sex-specific patterns of immunity and resistance to STIs. When females terminally invest, infected (but fertile) males gain an immediate fitness benefit from transmitting the STI to their partners. Consequently, even if an STI is harmful to net male fitness, the potential to increase their mate’s reproduc-tive investment could reduce the fitness costs of infection relareproduc-tive to that experienced by a female, and therefore lead to sexual conflict over the optimal level of resistance.
30
152 Evolutionary Ecology (2019) 33:149–172
1 3
resistance and reproductive investment in both sexes. Our goal was not to explore all pos-sible factors that could moderate these effects, but rather to confirm that the basic verbal argument is sound, and to establish a framework for more detailed models that could be parameterized and tested using empirical data.
Mate harm via STI infection
We first provide a simple continuous-time model of female reproductive effort under STI infection. The model serves both to recap previous sexual conflict theory (e.g. Lessells 2005; Johnstone and Keller 2000) and to set the stage for a full epidemiological model in the next section. We show that females infected with an STI should invest more in their current offspring than uninfected females, and that males may consequently benefit by infecting their mates. We assume that the STI leads to delayed female sterility, but does not directly affect mortality, nor the ability to produce offspring prior to sterilization (see “Discussion” section). However, changes in female reproductive effort after infection may affect both her offspring production and mortality. We initially ignore any detrimental effects of the STI on males: these costs are examined in our full epidemiological model (next section).
We assume that mature females follow the following breeding cycle: (1) mate with a sin-gle male; (2) fertilise eggs immediately after mating; (3) invest in eggs or offspring as they Table 1 Summary of variables and parameters
Unmarked symbols pertain to females, whereas the corresponding symbols for males are marked with a sperm-like tilde (like this: ã ). Subscripts U , I , or S indicate that the symbol applies to uninfected, infected-but-fertile, or sterile individuals, respectively
Symbol Description w Lifetime fitness
x Reproductive effort
y Immune effort
𝜇 Mortality rate
𝜇min Minimum mortality rate (for individuals that invest neither in reproduction nor immunity)
C Steepness with which mortality increases with increasing total effort x+y
p Probability of becoming infected at any particular mating (when partner infection status is
unknown)
𝛼 Probability of becoming infected when mating with an infected partner
𝛼max Maximum value for 𝛼 , occurring when an individual does not invest in STI resistance (i.e. yU=0) A Effectiveness of immune effort yU in preventing STI acquisition
𝛽 Rate at which an infected individual becomes sterile
𝛽max Maximum value for 𝛽 , occurring when an individual does not invest in immunity after infection (i.e. yI=0)
B Effectiveness of immune effort yI in delaying the onset of sterility
U , I , S Proportion of uninfected, infected-but-fertile, and sterile females in the population
R Rate at which newly mature females are recruited into the population
rM Rate at which any female mates with males of infection status M
̃
[image:31.595.105.438.121.358.2]153 Evolutionary Ecology (2019) 33:149–172
1 3
develop. Once a female’s offspring are fully developed, she remates immediately and the breeding cycle is repeated until she either dies or becomes sterile (note that the onset of steril-ity has no effect on already-fertilised offspring). Sterility (for infected females) and mortality may occur at any stage during the breeding cycle. For simplicity, we assume that females mate exactly once per batch of eggs, which are always fertilised by her last mate (i.e. no sperm stor-age or multiple paternity). Note that such ‘serial monogamy’ is predicted as an evolutionary result of STI infection under some circumstances (McLeod and Day 2017). We also assume that no offspring survive if the mother dies before they are fully developed. This means that a female’s short-term survival is in her mate’s evolutionary interest, but her long-term prospects are irrelevant to males.The fitness value of a set of offspring is proportional to a female’s reproductive effort x after her most recent mating, which may depend on her infection status (variables and param-eters are summarised in Table 1). Our assumption of substantial post-mating investment in off -spring is plausible if, for example, eggs are provisioned after mating (Staudacher et al. 2015; Giehr et al. 2017) or there is post-zygotic parental care (Hanssen 2006; Bowers et al. 2015; Amininasab et al. 2017). A female’s instantaneous rate of mortality 𝜇(x) increases with her reproductive effort during her current reproductive bout. We assume that offspring develop-ment takes an average of one unit time; this means that all time measuredevelop-ments are given in units of the mean offspring development time. For mathematical convenience we assume that the development times of individual broods follow an exponential distribution.
Let us write xU for the reproductive effort of an uninfected female and xI for that of an
infected female. The mortality rates of these females are respectively 𝜇U=𝜇(xU) and
𝜇I =𝜇(xI) . We first consider the average number of times an unmated female will mate with-out becoming infected. Suppose that at each mating, her probability of becoming infected is p . After her first mating, the female remains uninfected with probability 1−p . Since she experi-ences mortality at a constant rate of 𝜇U and her offspring mature at a rate of 1, she survives until her offspring mature with probability 1
1+𝜇U . She then remates immediately, after which
she remains uninfected with probability 1−p . Elaborating this pattern, the expected number of matings after which the female remains uninfected is given by a geometric series:
By similar logic, the probability that the female survives to become infected is given by
p (
𝜇U+1 𝜇U+p )
.
Once infected, we assume that females risk becoming sterile. Sterility does not occur immediately upon infection. Rather, the onset of sterility occurs at a constant rate of 𝛽 per unit
time, regardless of a female’s behavior (we lift the latter restriction in the full model). The STI does not directly affect a female’s mortality or offspring production, although changes in her reproductive effort could influence both traits. After infection, a female’s expected number of matings is 1+ 1
𝜇I+𝛽 , including the mating in which she became infected. Consequently, the
expected number of matings while infected but still fertile is:
(1)
nU= (1−p) [
1+ 1−p 1+ 𝜇U +
( 1−p
1+ 𝜇U
)2
+ ⋯
]
= (1−p) (
𝜇U+ 1
𝜇U+ p )
(2)
nI=p
(
𝜇U+1
𝜇U+p
)(
1+ 1
𝜇I+𝛽
32
154 Evolutionary Ecology (2019) 33:149–172
1 3
The expected fitness gain from mating equals the female’s reproductive effort times the
probability of surviving to produce offspring. This is vU= xU
1+𝜇U for uninfected females and
vI= xI
1+𝜇I for infected females. A female’s expected lifetime fitness is then:
For a given choice of the parameters, optimal reproductive effort ( xU and xI ) is found by
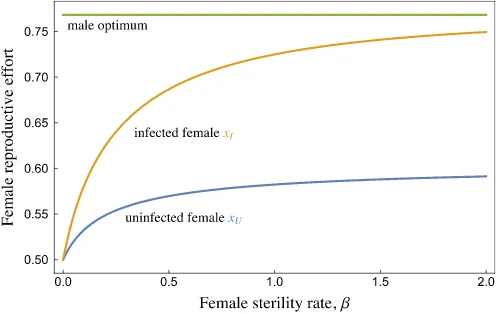
maximizing this expression numerically (Fig. 1).
Now consider a male who mates with the focal female. His fitness gain Δw̃ from mating
is equal to the female’s reproductive effort x multiplied by the probability 1
1+𝜇(x) that she
survives to produce offspring:
As above, this expression can be maximised numerically to obtain the female behavior that
maximises male fitness. All else being equal, a male should prefer to mate with an
unin-fected female over an inunin-fected female if
If the reverse inequality holds, then males should prefer to mate with an infected female
(all else being equal). Note that males do not want their mates to invest maximally in off
-spring, because we assume that offspring only survive if their mother remains alive until
they mature (Lessells 2005).
The risk of sterility due to STI infection reduces the reproductive value of all females, but more so for those that are already infected (i.e. uninfected females have a probability, (3)
w(xU,xI)=nUvU+nIvI
(4) Δw̃(x) = x
1+ 𝜇(x)
(5) xU
1+𝜇U >
xI
1+𝜇I
Fig. 1 Optimal reproductive effort of infected females (yellow, xI ) and uninfected females (blue, xU ) increases with female sterility rate ( 𝛽 , i.e. the rate at which infected females become sterile). Males always prefer greater reproductive effort by their mating partners (green) than females provide; but infected females approach the male optimum at very high sterility rates. This outcome is based on a simplified model where female infection and sterility rates are fixed and do not depend on their investment decisions (see ‘Mate harm via STI infection’ section). Shown with mortality rate function 𝜇(x) = 0.1+ 0.1x
1−x and a probability
[image:33.595.145.394.427.584.2]155 Evolutionary Ecology (2019) 33:149–172
1 3
but not a certainty, of being infected). Our model predicts that infected females should increase reproductive effort if the risk of sterility 𝛽 is high (‘terminal investment’). Their reproductive effort approaches the optimum for their male partners as 𝛽 increases, reflect-ing the shift of resources towards current, over future, reproduction (Fig. 1). Uninfected females also increase their reproductive effort as 𝛽 increases, but more modestly. Males consequently gain greater fitness from mating with females that are infected. This creates an incentive for males to infect their own partners.Epidemiological model
The above model, while illustrative, is simplified in two important ways. First, reproductive effort may trade off against investment in immune function (Hosken 2001; McNamara and Simmons 2017; Keller et al. 2018). Stronger immunity decreases the likelihood of acquir-ing an STI and lowers the rate at which an acquired STI results in sterilization. If females plastically increase their immune effort in response to STI infection, they might reduce (rather than increase) their reproductive effort in order to free up resources (i.e. terminal divestment). If so, males would lose fitness by infecting their mates.
Second, we have ignored direct effects of the STI on males. In our simplified model, the STI only affected a male’s fitness by changing the reproductive output of his mates. Under this assumption, female terminal investment following STI infection should select for males with no resistance to the STI. On the other hand, female terminal divestment might select for male resistance to avoid acquiring the STI and infecting his mates. If, how-ever, the STI causes sterility in males, then there is a direct reason to invest in resistance. A male’s optimal strategy will then balance the risk of sterility against the costs of immune effort, and the gains or losses of infecting his mates.
Equilibrium strategies for reproductive and immune effort depend on several interact-ing factors. The prevalence of the STI in the population, as well as its sex-specific effects, determine the optimal behavior of each sex. However, this behavior also feeds back to determine STI prevalence (Ashby and Gupta 2013). To predict the coevolution of these traits, we require a full game-theoretic model that incorporates the epidemiology of the STI.
We now consider the evolution of reproductive effort and immunity in both sexes in a population with an endemic STI. All individuals are either uninfected ( U ), infected but still fertile ( I ), or sterile ( S ). Sterile individuals can still mate with and infect others, but they do
not produce viable offspring. We assume that the STI is cryptic so that it is not possible to seek out or otherwise favour mates with a particular infection status. We first construct an epidemiological model to derive the stable demographic structure of a population, assum-ing that rates of mortality, STI transmission and sterility onset are fixed for each sex. We then describe how reproductive and immune effort are assumed to affect STI transmission and mortality. Lastly, we calculate sex-specific behavior and demographic structure in a population at evolutionary equilibrium.
Epidemiology: make-up of a demographically stable population
We begin by calculating the proportion of each type of individual in a population at demo-graphic equilibrium (i.e. we find the proportions U , I and S for females and Ũ , ̃I and S̃ for
34
156 Evolutionary Ecology (2019) 33:149–172
1 3
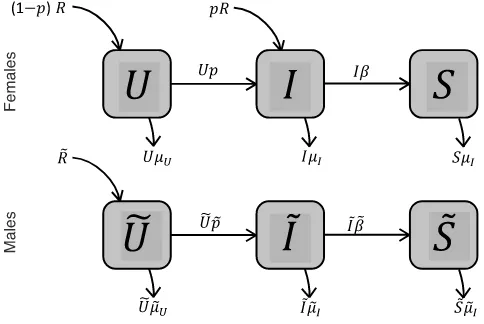
much more quickly than evolution, so that the sex-specific rates of mortality, STI transmission and sterility onset can be treated as fixed. All demographic processes (recruitment, mating, sterility, and mortality) occur continuously at constant rates (Fig. 2).
We assume that population size has reached a stable equilibrium, so that newly mature individuals enter the population at the same rate as older individuals die (Úbeda and Jansen
2016). The total rate of recruitment ( R for females and R̃ for males) is then:
Note that we do not consider population size dynamics explicitly in our model. We assume an even sex ratio at maturity (i.e. R=R̃ ), but sex differences in mortality can nonetheless
lead to biased adult sex ratios.
We first derive the rates of change in the proportion of males and females of each infection status (Fig. 2), and then equate these to zero to find the stable demographic structure. Three processes affect the proportion of uninfected females U . First, virgin females arrive in the pop-ulation at a rate of R . With probability p , these females are infected during their first mating; the remaining (1−p)R females join the uninfected population (note that p is not a constant but depends on both the female’s immune investment and the proportion of infected males in the population: see below). Second, non-virgin uninfected females become infected at a total rate of pU . Third, uninfected females die at a rate of 𝜇UU . The total rate of change in U is then:
Similarly, newly infected females arrive at a total rate of p(R+ U) . Infected females become
sterile at a rate of 𝛽I and die at a rate of 𝜇II . The rate of change in I is consequently: (6) R + R̃= 𝜇UU+ 𝜇I(I+ S) + 𝜇̃UŨ+ 𝜇̃I(̃I+ S̃)
(7) dU
dt = (1−p)R−pU−𝜇UU
Fig. 2 Epidemiological flow-diagram with uninfected (U for females, Ũ for males), infected ( I for females, ̃
[image:35.595.146.388.405.567.2]157 Evolutionary Ecology (2019) 33:149–172
1 3
Last, sterile females arrive at a rate of 𝛽I and die at a rate of 𝜇IS (remember that sterile individuals share the same strategies, hence mortality rates, as infected ones). This gives us:
The equations for males are similar, except that males do not generally mate immediately on maturity, as their mating opportunities are limited by female availability (see below). We write p̃ for the rate at which uninfected males become infected, and 𝛽̃ for the rate of sterility onset in infected males (note that p̃ depends on both male immune investment and the proportion of infected females: see below). The rates of change in males of each type are then given by:
To find the composition of the population at equilibrium, we set the deriva-tives in Eqs. (7)–(12) to zero and solve numerically, subject to the constraint that U+I+S+Ũ +̃I+S̃=1 . For some parameter values, there is no equilibrium where the
STI persists (i.e. the only equilibrium is with U+Ũ =1).
Strategies: reproductive and immune effort
We now consider how immunity, mortality and mating rates are determined by sex-specific investment strategies. Each individual allocates resources to both reproduction ( x for females and x̃ for males) and immune function ( y and ỹ ), both of which impose survival costs. For females, the total fitness value of a brood of offspring is proportional to her reproductive effort x (see earlier). For males, the expected mating rate is proportional to x̃ (see below). Males do not affect the fitness value of a brood, except indirectly by infecting their mating partner. An individual’s investment strategy may depend on its infection status. There are consequently 8 strategy variables: xU , yU , xI and yI for females and x̃U , ỹU , x̃I and ̃yI for males.
The behaviour of sterile individuals is not under direct selection, as they produce no off -spring. We consequently assume that they use the same strategies as infected-but-fertile indi-viduals. Note, however, that sterile individuals still interact with fertile individuals via mating and mortality (which affects the rate of new recruitment). The unselected behavior of sterile individuals therefore affects the ESS strategies for both uninfected and infected individuals.
Immune effort has two possible benefits. First, for uninfected individuals, it decreases the probability ( 𝛼 or 𝛼̃ ) of acquiring the STI when mating with an infected partner. The (8) dI
dt = p(R + U)−𝛽I−𝜇II
(9)
dS
dt =𝛽I−𝜇IS
(10) dŨ
dt = ̃
R−p̃Ũ −𝜇̃UŨ
(11) d̃I
dt =p̃
̃
U−𝛽 ̃̃I−𝜇̃ĨI
(12)
dS̃
36
158 Evolutionary Ecology (2019) 33:149–172
1 3
risk of infection upon mating with an infected individual is described by the following relationships:
The parameters 𝛼max and 𝛼̃max determine the infection probability for individuals who
invest nothing in immune function. The slope parameters A and à determine how eff ec-tively infection risk is reduced by immune effort. The exponential function prevents infec-tion probability from going to zero, which would result in STI extinction.
Second, once infected, immune effort determines the rate ( 𝛽 or 𝛽̃ ) at which the STI leads to sterility:
The parameters 𝛽max and 𝛽̃max are the maximum rates at which infected individuals
become sterile, whereas B and B̃ determine how effectively sterility is delayed by immune
effort. The model allows for sex differences in both the infectivity and virulence of the STI [Eqs. (13) and (14) respectively], even when males and females invest equally in immune function.
We assume that both reproductive and immune effort are costly and increase an indi-vidual’s instantaneous rate of mortality 𝜇 . We model mortality as an accelerating function of the total investment x+y in reproduction and immunity:
The minimum mortality 𝜇min applies to individuals who invest in neither immunity nor reproduction. The slope C determines how steeply mortality increases with total effort. The total investment x+y must be less than one, because x+y=1 implies instant death. Consequently, we can think of 1−x−y as the resources invested in survival (e.g. somatic maintenance or predator avoidance). We write, for example, 𝜇U=𝜇(xU+yU) for the mor-tality rate of an uninfected female,𝜇̃I = 𝜇(x̃I+ ỹI ) for an infected male, and so on.
Female mating rates are not determined by their investment strategies: all females mate immediately upon reaching maturity and then re-mate immediately each time their offspring mature. The total rate of matings in the population is consequently (R + U+ I+ S)P , where P is the equilibrium female population size (note that the average number of matings per indi-vidual per unit time is greater than U+I+S=1 , because a female’s initial mating must also be accounted for). For males, on the other hand, mating rate is determined by their reproduc-tive effort relative to other males in the population, which may depend on their infection sta-tus ( U , I or S , noting that U includes both virgin and non-virgin uninfected males). The share of matings obtained by males of a given infection status consequently depends on both the frequency and the average reproductive effort of these males. The probability that any given mating is with a male of status M (where M=U , I or S ) is given by:
Note that rU+rI+rS=1 . The probability that a female becomes infected from any given mating is p=𝛼(rI+rS).
Similarly, a male of infection status M mates with females of status F at a rate of: (13) 𝛼(yU)=𝛼max exp
(
−AyU), 𝛼̃(ỹU)=𝛼̃maxexp (
−ÃỹU)
(14)
𝛽(yI)=𝛽max exp (−ByI )
, 𝛽̃(ỹI)=𝛽̃maxexp(−B̃ỹI )
(15) 𝜇(x+ y) = 𝜇min+ C(x+ y)
1−(x+ y)
(16) rM= x̃M
̃ M
̃
159 Evolutionary Ecology (2019) 33:149–172
1 3
Here we allow F=R,U,I,S , so that ̃rM,R represents the rate of matings with newly mature virgin females, and r̃M,U represents the rate of matings with non-virgin uninfected females. Note that the Fisher condition is fulfilled because rMF=r̃M,FM̃ for all types M and F (i.e.the total mating rate of type F females with type M males equals that of type M males with type F females, which is a logical necessity: Houston and McNamara 2005;
Jen-nions and Fromhage 2017). The rate at which an uninfected male becomes infected is ̃
p=𝛼̃(r̃U,I+r̃U,S).
Reproductive and immune effort in a population at evolutionary equilibrium
In the previous section, we derived the demographic make-up of a stable population, assuming that all individuals play the same fixed (although sex-specific) strategies for reproductive and immune effort. We can now calculate the fitness of mutant individuals that diverge from the population strategies (details in “Appendix”). We assume that demo-graphic change happens much faster than evolutionary change, so that we can calculate fitness against the background of a demographically stable population.
We write population strategies in vector form as X=(xU,yU,xI,yI) for females and ̃
X=(x̃U,ỹU,x̃I,ỹI) for males. Female or male mutants play strategies X∗=(x∗
U,y
∗
U,x
∗
I,y
∗
I )
or X̃∗=(x̃∗
U,ỹ
∗
U,x̃
∗
I,ỹ
∗
I )
respectively. We write w(X∗) for the fitness of a mutant female playing X∗ in a population where all other females play X and all males play X̃ . For male mutants, we define w̃(X̃∗) analagously. The selection differentials for males and females are then approximately proportional to (Taylor 1996):
This relies on the simplifying assumption that additive genetic variance is approximately equal for all traits. We found evolutionarily equilibria by starting with arbitrary initial strat-egies X0 and X̃0 and following the selection trajectories defined by S(X) and S̃(X̃) until the strategies converged to an equilibrium. This was done by iterating the equation:
with Δ a small positive constant (we found Δ=0.01 suitable). In other words, in each time step, the strategies move a small step in the direction of selection, with the step size equal to Δ times the intensity of selection. Stable population structure was found using
Eqs. (7)–(12) for each iteration. Different choices of initial strategies always led to the same equilibria, except for choices that led to STI extinction.
(17) ̃
rM,F= (
̃ xM
̃
xUŨ +̃xI(̃I+S̃)
)
F
(18) S(X) = 𝜕w(X
∗)
𝜕X∗ | | | |X∗=X
, S̃(X̃)= 𝜕 ̃w (
̃ X∗)
𝜕 ̃X∗ | | | | |X̃∗=X̃
(19) (
Xt+1 ̃ Xt+1
)
=
(
Xt+ΔS(Xt)
̃