Effects of Deletion or Stringent Repression of the H3L
Envelope Gene on Vaccinia Virus Replication
FLA´ VIO G.DAFONSECA,† ELIZABETH J. WOLFFE, ANDREA WEISBERG,ANDBERNARD MOSS*
Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland 20892-0445
Received 22 March 2000/Accepted 17 May 2000
The C-terminal membrane anchor protein encoded by the H3L open reading frame of vaccinia virus is located on the surfaces of intracellular mature virions. To investigate the role of the H3L protein, we constructed deletion (vH3⌬) and inducible (vH3i) null mutants. The H3L protein was not detected in lysates of cells infected with vH3⌬or vH3i in the absence of inducer. Under these conditions, plaques were small and round instead of large and comet shaped, indicative of decreased virus replication or cell-to-cell spread. The mutant phenotype was correlated with reduced yields of infectious intra- and extracellular virus in one-step growth experiments. The defect in vH3i replication could not be attributed to a role of the H3L protein in virus binding, internalization, or any event prior to late gene expression. Electron microscopic examination of cells infected with vH3⌬or vH3i in the absence of inducer revealed that virion assembly was impaired, resulting in a high ratio of immature to mature virus forms with an accumulation of crescent membranes adjacent to granular material and DNA crystalloids. The absence of the H3L protein did not impair the membrane localization of virion surface proteins encoded by the A27L, D8L, and L1R genes. The wrapping of virions and actin tail formation were not specifically blocked, but there was an apparent defect in low-pH-mediated syncytium formation that could be attributed to decreased virus particle production. The phenotypes of the H3L deletion and repression mutants were identical to each other but differed from those produced by null mutations of genes encoding other vaccinia virus membrane components.
Poxviruses are the largest and most complex of all animal viruses. The first visible step in the assembly of vaccinia virus, the prototype poxvirus, is the formation of crescent mem-branes within viral factory areas of the cytoplasm (8). The crescents enclose electron-dense material to become spherical immature virions (IV) that progress into infectious intracellu-lar mature virions (IMV). Some IMV are then wrapped by a double layer of membranes, derived from the trans-Golgi or endosomal cisternae, and migrate to the periphery of the cell where the outermost viral and plasma membranes may fuse (15, 17, 21, 35, 38). Virus spread is mediated largely by extra-cellular particles, which have one more membrane than the IMV, and are called cell-associated extracellular enveloped virions (CEV) if they remain adherent and extracellular envel-oped virions (EEV) if they are detached from the cell surface (2, 4, 5, 24).
Vaccinia virus mutants provide powerful tools for analyzing the sequential steps in membrane formation and morphogen-esis. For example, studies with mutants indicated that (i) the F10L protein kinase is required for formation of the first viral membranes (39, 43); (ii) small vesicles that may be precursors of the viral membranes accumulate in the absence of A17L and A14L expression (28, 29, 40, 47); (iii) D13L expression is needed for coating the viral membranes to form crescents (50); (iv) IV accumulate when expression of L1R is blocked (25); (v) IMV remain largely unwrapped by cisternae when A27R (27), F13L (3), and B5R (12, 45) are not expressed; and (vi) actin
tail formation is dependent on synthesis of proteins encoded by A33R, A34R, and A36R genes (30, 33, 46, 49).
We recently initiated investigations of the role of the H3L protein, an immunodominant component of IMV membranes (18, 37, 51), and presented evidence that it is anchored to the surfaces of IMV by a C-terminal hydrophobic tail that can mediate posttranslational membrane insertion (7). As a con-tinuation of that study we constructed two vaccinia virus mu-tants, one with the H3L open reading frame (ORF) deleted and the other with the H3L ORF stringently repressed, and found that immature viral forms accumulated, with a corre-sponding reduction in infectious virions, under conditions in which H3L was not expressed.
MATERIALS AND METHODS
Cells and viruses.CV-1, RK13, and BS-C-1 cells were grown in Earle’s
mod-ified Eagle’s medium (Quality Biological Inc.) supplemented with 10% fetal calf serum. The vaccinia virus WR strain and the recombinant vaccinia virus vT7LacOI (1) were propagated in HeLa cells as described previously (10), and stocks were maintained at⫺70°C.
Plasmids.A copy of the H3L ORF containingNcoI andBamHI sites at the respective 5⬘and 3⬘ends was generated by PCR. The ATG of theNcoI site was used for translation initiation without altering the coding sequence. PCR was performed using vaccinia virus strain DNA as the template and primers GGG CCATGGCGGCGGCGAAAACTCCTGTTATTGTTGTGCC and GGGGGA TCCTTATTAGATAAATGCGGTAACGAATGTTCCTGTAAGGAACC. Re-striction sites are underlined, and initiation and termination codons are in italics. The PCR product was cut withNcoI andBamHI and inserted into the pVOTE.1 plasmid (44) to form pFFH3L. A 527-bp DNA segment corresponding to the downstream flanking region of the H3L gene, comprising a portion of the H4L ORF, was generated through PCR using vaccinia virus DNA as the template and primers GGGGTCGACCGCCGCCGCCATTTAGTTATTGAAATTAATC and GGGAAGCTTCCTACTCCATCGGTCGAACCAACTG, which contain SalI andHindIII restriction sites at their 5⬘ends, respectively. The DNA fragment was digested and inserted intoSalI andHindIII sites of the pZippy-NEO/GUS plasmid (provided by T. Shors), which contains a copy of the neomycin resistance gene (neo) plus a copy of theEscherichia coli-glucuronidase gene flanked by the multiple cloning sites, to form pFFH4LNG. Another DNA segment of 538 bp, containing the H3L upstream region and comprising a portion of the H2R ORF, was also generated by PCR. Vaccinia virus DNA was used as the template,
* Corresponding author. Mailing address: 4 Center Dr., MSC 0445, National Institutes of Health, Bethesda, MD 20892-0455. Phone: (301) 496-9869. Fax: (301) 480-1147. E-mail: [email protected].
† Present address: Laborato´rio de Vı´rus, Departamento de Micro-biologia, Instituto de Cieˆncias Biolo´gicas da Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brazil.
7518
on November 9, 2019 by guest
http://jvi.asm.org/
and the oligonucleotides GGGAGATCTGGCTATAGTAGGCGTACAAGCA GCC and GGGGCGGCCGCCACTATTCCATATTACTAAATCGGAACACCA ATGCGG withBglII andNotI sites at their 5⬘ends, respectively, were used as primers. The DNA product was digested withBglII andNotI and inserted into the pFFH4LNG plasmid, which had been digested with same enzymes to form pFFH2RH4LNG. Ready-to-Go PCR beads (Pharmacia Biotech) and the Expand High Fidelity PCR system (Boehringer Mannheim) were used for PCR. Restriction enzymes were from Gibco-BRL. The cloned segments of all plasmids were checked by nucleotide sequencing.
Recombinant viruses.Procedures for the construction and propagation of recombinant viruses were similar to those previously described (11, 47). vH3i was constructed as follows. CV-1 cells were infected with vT7LacOI at 0.5 PFU per cell and then transfected with pFFH3L in Lipofectamine reagent and the Opti-Mem I reduced medium system (Gibco-BRL Life Technologies) for 5 h. After an additional 48 h of incubation, the cells were harvested and diluted lysates were used to infect BS-C-1 monolayers in the presence of mycophenolic acid. The monolayer was covered with agar, and mycophenolic acid-resistant plaques were visualized with neutral red and picked with a pipette. New BS-C-1 monolayers were infected with individual plaques, and the cycle was repeated for three successive rounds to generate vT7LacOI-H3L. CV-1 cells were then infected with vT7LacOI-H3L and transfected with pFFH2RH4LNG as described above. The lysates were used to infect BS-C-1 monolayers in the presence of 2 mg of Geneticin/ml and 50 to 100M isopropylthiogalactopyranoside (IPTG). Plaques that stained blue with 0.2 mg of 5-bromo-4-chloro-3-indolyl--D-glucuronic acid
(X-Gluc; Clontech, Palo Alto, Calif.)/ml were picked, and stocks of vH3i were amplified in BS-C-1 cells in the absence of IPTG and maintained as aliquots at
⫺70°C.
vH3⌬was constructed by infecting CV-1 cells with wild-type vaccinia virus and
transfecting them with pFFH2RH4LNG. Plaques were formed on BS-C-1 mono-layers in the presence of Geneticin and picked after staining with X-Gluc.
Western blot analysis.BS-C-1 monolayers in six-well plates were infected with vaccinia virus (10 PFU per cell) with or without IPTG. After 24 h, the cells were harvested, collected by centrifugation, and suspended in Laemmli reducing buffer (Sigma). Proteins were separated on 4 to 20% polyacrylamide gradient sodium dodecyl sulfate (SDS) gels (Owl Separation Systems) in a Tris-glycine-SDS buffer system and transferred by electrophoresis to membranes (Immo-bilon-P; Millipore). The membranes were blocked for 1 h in TTBS (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween 20) containing 2.5% (wt/vol) dried nonfat milk. As the primary antibody we used a 1:500 dilution of the H3L peptide rabbit antiserum (7) or a rabbit antiserum generated against purified, infectious vaccinia virus (provided by L. Potash). The membrane was washed and incubated with an anti-rabbit horseradish conjugate (Amersham Life Science). Immune complexes were detected with the Super-Signal chemiluminescent de-tection kit (Pierce) and visualized with X-Omat film (Kodak). Alternatively, anti-rabbit immunoglobulin G alkaline phosphatase conjugate (Promega) was used as the secondary antibody.
Virus attachment to cells.Confluent BS-C-1 cells in six-well plates were cooled on ice for 30 min. Virus from cell lysates was used to infect cells at a multiplicity of 1 PFU per cell. At various times, the cells were washed twice with cold phosphate-buffered saline (PBS), harvested, and collected by centrifugation. The cells were then suspended in modified Eagle’s medium containing 2.5% fetal calf serum, serially diluted, and applied to duplicate fresh BS-C-1 monolayers for an infectious center assay.
One-step virus yield experiments.Confluent BS-C-1 or RK13cells in six-well
plates were infected with either crude or sucrose gradient-purified vaccinia virus (10 PFU per cell). After 1 h of adsorption, the medium was removed, the cells FIG. 1. Representation of wild-type and mutant genomes. (A) Genomes of wild-type and recombinant viruses. Shown are genome segments containing the following ORFs: J2R (encoding thymidine kinase) H2R, H3L, H4L, and A56R (encoding hemagglutinin). In vH3⌬, the H3L ORF is replaced by theneoandgusgenes, which are regulated by vaccinia virus promoters and which provide antibiotic selection and positive identification of plaques. In vH3i, (i) the H3L ORF is replaced by theneo
andgusgenes regulated by vaccinia virus promoters, (ii) the bacteriophage T7 RNA polymerase ORF regulated by theE. coli lacoperator (lacO) and the vaccinia virus P11 late promoter (PL) and theE. coli lacrepressor (lacI) regulated by the vaccinia virus P7.5 early/late promoter (PEL) are inserted into the J2R ORF, and (iii) the
H3L ORF preceded by the encephalomyocarditis virus untranslated leader sequence (EMC) providing cap-independent translation, a modifiedlacO(SLO), and a bacteriophage T7 promoter (PT7) as well as theE. coli gptgene regulated by a vaccinia virus promoter for antibiotic selection are inserted into the A56R ORF. (B)
Expression of the H3L ORF. BS-C-1 cells that were uninfected (UN) or infected with WR, vH3⌬, or vH3i in the presence (⫹I) or absence (⫺I) of IPTG were harvested after 24 h and the lysates were analyzed by SDS-PAGE and Western blotting with antiserum to a peptide encoded by the H3L ORF. The H3L protein was detected by chemiluminescence. The positions and masses of marker proteins are indicated at the left.
VOL. 74, 2000 VACCINIA VIRUS H3L ENVELOPE PROTEIN MUTANTS 7519
on November 9, 2019 by guest
http://jvi.asm.org/
were washed twice in PBS, and fresh medium was added. At various times, the medium was removed and clarified by centrifugation and the cells were washed and harvested. Cells were lysed by three cycles of freezing and thawing, and lysates and clarified medium were sonicated separately. Virus titers were deter-mined by plaque assay on BS-C-1 cells.
Syncytium formation.Confluent BS-C-1 or RK13monolayers were infected
with vaccinia virus (10 PFU per cell). After adsorption, the inoculum was re-moved and replaced by fresh medium. The infected cells were then incubated for 12 h at 37°C. Cells were washed and treated for 2 min at 37°C with PBS containing 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) and 10 mM HEPES, pH 5.0 or 7.4. The fusion buffers were replaced by fresh medium, and the cells were incubated for 1 to 3 h at 37°C and then photographed with a phase-contrast microscope.
Electron microscopy.RK13cells were cultured in 60-mm-diameter dishes and
infected with vaccinia virus (10 PFU per cell). After 23 h, the cells were fixed in 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). Samples were then prepared by osmication, dehydration, and embedding in Epon resin. Thin sec-tions were obtained, placed on grids, and stained with uranyl acetate and Reyn-old’s lead citrate (47). Images were acquired using a Philips CM100 electron microscope. Immunogold labeling was performed as described in the accompa-nying paper (7).
RESULTS
Construction of H3L mutant viruses. A genetic approach was taken to investigate the role of the H3L protein in vaccinia virus replication. Two different types of mutants were con-structed for this purpose: a deletion mutant and an inducible mutant. The deletion mutant, vH3⌬(Fig. 1A), was isolated by infecting cells with vaccinia virus strain WR and then trans-fecting them with a plasmid that containedneoandgus expres-sion cassettes replacing most of the H3L ORF but keeping the adjacent H2R and H4L genes intact. Recombinant virus was selected in the presence of Geneticin, and plaques that stained
blue with X-Gluc were picked repeatedly to obtain a pure clone. As will be shown later, the plaques were smaller than those of parental WR virus. The genotype of the mutant and the absence of contaminating WR were verified by PCR anal-ysis (data not shown).
The second type of mutant, vH3i, has a deleted H3L gene as well as a new inducible one (Fig. 1A). We used a high-strin-gency system (44, 47) in which the recombinant vaccinia virus contained three main components: (i) a continuously ex-pressedEscherichia coli lacrepressor gene (lacI), (ii) a bacte-riophage T7 RNA polymerase gene regulated by a T7 pro-moter and a modifiedE. coli lacoperator (lacO), and (iii) an inducible gene regulated by the T7 promoter and a modified
lacO. In this system, the repressor inhibited two consecutive steps: transcription of the T7 RNA polymerase gene and tran-scription of the inducible gene thereby providing the basis for high stringency. Controlled expression of the inducible gene was achieved by addition of the desired concentration of IPTG. To produce vH3i, we infected cells with the WR-derived virus vT7lacOI (1), already carrying the repressor and bacteriophage T7 RNA polymerase genes. The infected cells were then trans-fected with a plasmid transfer vector (44) containing the H3L ORF regulated by the T7 promoter and modifiedlacO, theE.
coli gpt gene to provide mycophenolic acid resistance, and
flanking sequences derived from the nonessential A56R ORF. The recombinant virus was plaque purified in the presence of mycophenolic acid, and the presence of the inserted gene was verified by PCR analysis. This intermediate-stage virus still contained the original H3L ORF as well as the new inducible one. The plasmid containing the neo and gusgene cassettes replacing most of the H3L gene, used above to delete H3L from WR, was then transfected into cells that had been in-fected with the intermediate virus to form vH3i, which has a single H3L ORF (Fig. 1A). This step was carried out in the presence of IPTG to ensure expression of the inducible copy of the H3L gene during isolation. After repeated plaque purifi-cation, deletion of the original H3L gene was verified by PCR. Stocks of vH3i to be used for experiments were made in the absence of IPTG so that the virions would lack the H3L pro-tein as well as traces of inducer.
The mutant virus stocks were checked for H3L gene expres-sion using an antibody to an H3L-derived peptide (7). H3L protein synthesis could not be detected by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting of lysates from BS-C-1 cells infected for 24 h with vH3⌬or vH3i in the absence of IPTG (Fig. 1B). However, the H3L protein was made in cells infected with vH3i in the presence of 100 M IPTG (Fig. 1B).
Plaque phenotypes of vH3⌬and vH3i.The ability to isolate a virus with a deleted H3L gene indicated that the H3L gene was not essential for replication. Nevertheless, vH3⌬plaques could be distinguished from those of WR by their smaller size on BS-C-1 monolayers (Fig. 2A). A size difference between vH3i plaques that formed in the absence and presence of IPTG was also noted (Fig. 2A). Even in the presence of IPTG, however, vH3i plaques were smaller than those of WR, reflect-ing the slower replication of the parental vT7lacOI virus. The most striking difference in plaque phenotype was found with RK13cells, which are known to release large amounts of ex-tracellular vaccinia virus resulting in elongated or comet-shaped plaques (23). The WR strain of vaccinia virus formed comet-shaped plaques by 48 h (Fig. 2B), albeit ones smaller than those formed by some other vaccinia virus strains. In contrast, vH3⌬produced only small round plaques during the same period (Fig. 2B). Moreover, vH3i produced comet-shaped plaques in the presence of IPTG and round ones in the
FIG. 2. Plaque phenotypes of mutant viruses. (A) BS-C-1 cell monolayers were inoculated with WR, vH3⌬, or vH3i in the presence (⫹I) or absence (⫺I) of IPTG and stained with crystal violet after 48 h. (B) Same as panel A except
that RK13cells were used.
on November 9, 2019 by guest
http://jvi.asm.org/
absence of the inducer (Fig. 2B). The difference was not ab-solute, however, because vH3⌬ and vH3i in the absence of IPTG produced small comets at 72 to 96 h. By that time, however, RK13cell monolayers infected with wild-type virus or vH3i in the presence of IPTG were totally degraded by the spreading infection (data not shown).
Comparison of virus yields under one-step growth condi-tions. A small-plaque phenotype could result from inhibition of virus production or spread. To investigate this, BS-C-1 cells were infected with 10 PFU of purified virus per cell and the intra- and extracellular virus yields were determined as a func-tion of time. In BS-C-1 cells, replicafunc-tion of both WR and vH3⌬ was detected between 6 and 12 h after infection. However, there was 10-fold more intracellular WR than vH3⌬at 12 h and about 7.5-fold more at 24 h (Fig. 3A). A similar difference was found by comparing levels of vH3i replication in the pres-ence and abspres-ence of IPTG (Fig. 3B). Although typically only small amounts of vaccinia virus are released from BS-C-1 cells, there was about fivefold more released from cells infected with WR than from cells infected with vH3⌬ (Fig. 3C) and also about fivefold more released from cells infected with vH3i in the presence of IPTG than in the absence of IPTG (Fig. 3D). Similar results were obtained when unpurified virus was used for infection, indicating that the results were not due to insta-bility of mutant viruses in sucrose.
The results obtained with RK13 cells were comparable to those obtained with BS-C-1 cells. Viruses expressing the H3L protein, WR and vH3i in the presence of IPTG, produced 7- to 10-times-higher intracellular titers than viruses not expressing H3L protein, vH3⌬and vH3i in the absence of IPTG (Fig. 3E
and F). A similar difference in virus yield was found at 24 and 48 h, indicating that the absence of H3L protein does not merely delay the formation of infectious virus. Although the yields of extracellular vaccinia virus were higher in RK13cells than in BS-C-1 cells, there was still about a fivefold advantage for a virus expressing H3L (Fig. 3G and H). We concluded that there was an overall diminution in production of infectious virus by H3L mutants rather than a specific defect in formation of extracellular particles.
[image:4.612.58.550.72.372.2]Binding of virus to cells.The difference in yields of vH3i in the presence and absence of IPTG could not be explained by an effect on virus binding or entry because the same virus stock prepared in the absence of IPTG was used under both condi-tions. However, the lower yield of vH3⌬than of WR might be partially explained by such defects. To investigate binding, BS-C-1 cells were incubated on ice with 1 PFU of either un-purified WR or vH3⌬/cell. At intervals, the cells were washed, harvested, and dispersed. An infectious-center assay was then carried out by diluting the cells and plating them on fresh BS-C-1 monolayers. Under these conditions, the rate of bind-ing of vH3⌬was nearly the same as that of WR (Fig. 4). Similar results were obtained when the cells that had bound virus were frozen, thawed, and sonicated before plaquing (data not shown). It is important to emphasize that this assay compared the binding of infectious vH3⌬and WR rather than physical particles. As will be noted later, the yield of IMV is greatly reduced when H3L is not expressed, and consequently purified vH3⌬may have a higher percentage of immature forms as well as other contaminants than WR.
FIG. 3. One-step growth curves. BS-C-1 cells (A to D) and RK13cells (E to H) were infected with purified WR, vH3⌬, or vH3i in the presence (⫹I) or absence
(⫺I) of IPTG as indicated. At 2, 6, 12, 24, and 48 h, the medium (C, D, G, and H) and cells (A, B, E, and F) were harvested separately and infectious virus titers were determined by plaque assay.
VOL. 74, 2000 VACCINIA VIRUS H3L ENVELOPE PROTEIN MUTANTS 7521
on November 9, 2019 by guest
http://jvi.asm.org/
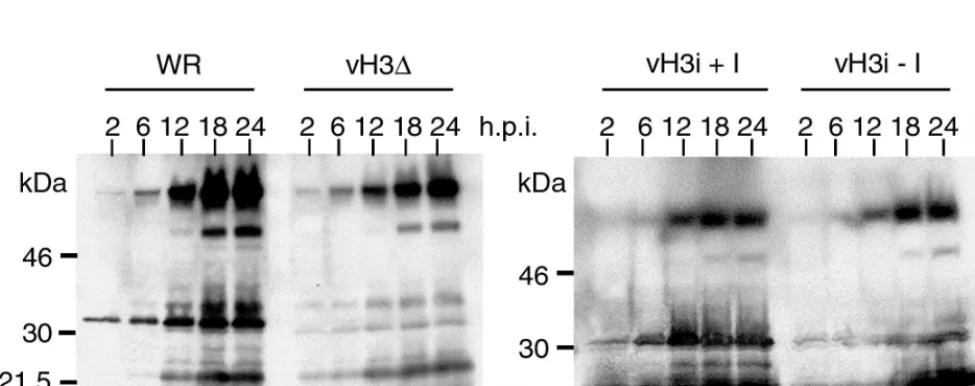
Viral protein synthesis.To investigate the role of the H3L protein in postbinding steps, cells were infected with 10 PFU of WR, vH3⌬, or vH3i in the presence or absence of IPTG and then harvested at various times. The cell lysates were analyzed by SDS-PAGE and Western blotting using an antiserum that was prepared against purified virions and which reacts strongly with late structural proteins including H3L. By 12 h, similar patterns were present in all cases (Fig. 5). The major difference between the blots of cells infected with viruses expressing and not expressing the H3L ORF was that the latter showed de-creased labeling intensity in the region of the gel expected to contain the H3L protein. The slightly lower overall intensity of viral protein bands in cells infected with vH3⌬than in cells
infected with WR could be due small differences in the rate of entry of H3L⫹ and H3L⫺ virus. This was not a factor in
comparing results for vH3i in the presence and absence of IPTG as the same virus stock was used in both cases. We concluded that the H3L protein was not crucial for steps in virus replication leading up to late protein synthesis.
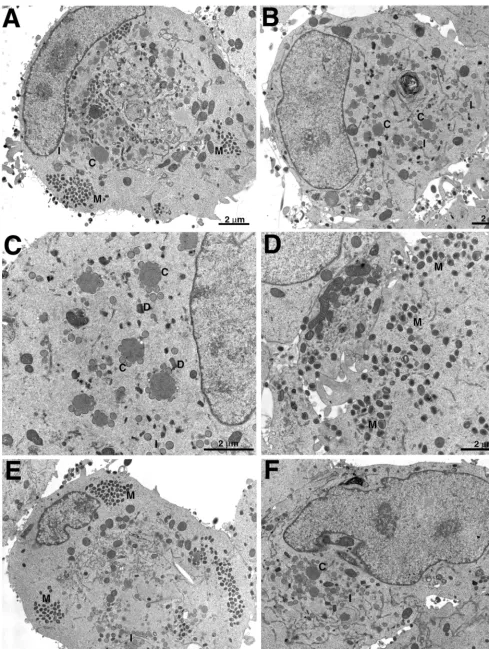
Electron microscopy of infected cells.To further investigate the stage at which virus replication was diminished as a con-sequence of H3L deletion or repression, we used electron microscopy to examine thin sections of RK13cells infected with wild-type virus, vH3⌬, or vH3i in the presence or absence of IPTG. After 23 h, cells infected with WR contained large clusters of mature virions and extracellular particles as well as some immature forms (Fig. 6A). In contrast, typical fields of cells infected with vH3⌬ contained predominantly crescents adjacent to large masses of granular material and immature forms of virus (Fig. 6B and C). DNA crystalloids, a hallmark of decreased morphogenesis (14), were also found when the H3L protein was not made (Fig. 6C). Although immature forms were predominant in cells infected with vH3⌬, mature forms were occasionally seen and some cells contained clusters of IMV (Fig. 6D). The wrapping of IMV was not specifically blocked because intracellular enveloped virions (IEV) can be seen in Fig. 6D and at a higher magnification in a subsequent figure. The picture was similar in cells infected with vH3i: in the presence of IPTG, typical fields showed clusters of mature virions as well as some immature forms (Fig. 6E), whereas the latter were predominant in the absence of IPTG (Fig. 6F). To confirm our visual impression, the viral forms in random fields were enumerated. In WR-infected cells, the intracellular ma-ture forms comprising IMV and IEV constituted 67% of the total, the CEV and EEV made up 25%, and the crescents and IV totaled only 9% (Table 1). Similar numbers were obtained for cells infected with vH3i in the presence of inducer (Table 1). In cells infected with vH3⌬or vH3i in the absence of IPTG, however, the crescents and IV constituted 61 to 70% of the total with correspondingly lower numbers of intra- and extra-cellular mature forms (Table 1).
[image:5.612.60.286.72.270.2]Incorporation of other membrane proteins into IV and IMV in the absence of H3L expression. Because the H3L protein
FIG. 4. Binding of infectious WR and vH3⌬ to cells. Replicate wells of chilled BS-C-1 cells were inoculated with 1 PFU of unpurified WR or vH3⌬per cell and maintained on ice. At 1, 10, 20, 30, 45, and 60 min, cells were washed with cold PBS, harvested, dispersed, and plated on fresh BS-C-1 monolayers at 37°C. After 48 h, the cells were stained with crystal violet and the plaques were enumerated.
FIG. 5. Synthesis of viral proteins. BS-C-1 cells were infected with WR, vH3⌬, or vH3i in the presence (⫹I) or absence (⫺I) of IPTG. At 2, 6, 12, 18, and 24 h after infection, cells were harvested and total proteins were subjected to SDS-PAGE. The resolved proteins were transferred to membranes, probed with antiserum made in rabbits infected with purified vaccinia virus, and detected by chemiluminescence. The positions and masses of marker proteins are shown on the left.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.58.546.505.698.2]FIG. 6. Electron microscopy of ultrathin sections of infected cells. RK13cells were infected with WR (A), vH3⌬(B to D), or vH3i in the presence (E) or absence
(F) of IPTG and incubated for 23 h. Abbreviations: C, crescents; I, IV; M, mature virions; D, DNA crystalloids. Magnification is indicated by bars.
7523
on November 9, 2019 by guest
http://jvi.asm.org/
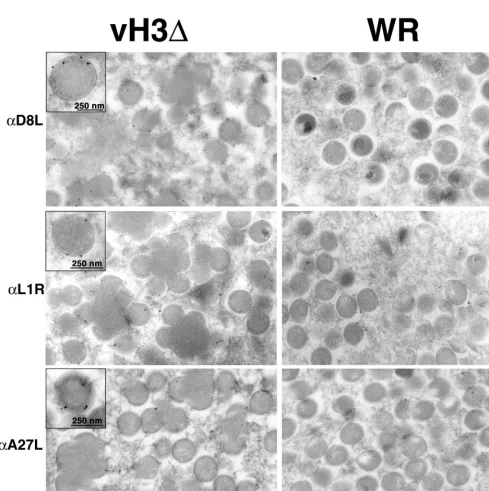
can insert directly into membranes via its hydrophobic C ter-minus, we considered that it might have a role in the associa-tion of other viral membrane proteins. The A27L, L1R, and D8L proteins also have a surface location, and the latter two resemble H3L with regard to their C-terminal hydrophobic domains. The A27L protein is largely hydrophobic and does not have a typical transmembrane structure. To test this hy-pothesis, we infected cells with vH3⌬or WR and then incu-bated cryosections with an antibody to one of the three pro-teins followed by protein A-gold. Both the A27L (36) and the L1R (48) proteins were previously reported to label immature
[image:7.612.60.549.208.699.2]FIG. 7. Detection of D8L, L1R, and A27L proteins on membranes of IV formed in cells infected with vH3⌬or WR. Infected cells were cryosectioned and incubated with antibody (␣) to D8L, L1R, or A27L followed by protein A-gold. Insets show high magnification of IV selected for good visualization of gold grains overlying the membrane.
TABLE 1. Distribution of immature and mature virus particles
Virusb Totala % of particles that are:
Crescents IV IMV CEV EEV
WR 1,100 3.1 5.7 66.5 19.7 5.0
vH3⌬ 671 17.3 43.6 21.5 11.2 6.4
vH3i⫹I 732 6.4 8.6 61.0 20.3 3.7
vH3i⫺I 743 23.9 45.7 22.8 5.2 2.4
aTotal number of crescents, IV, IMV, CEV, and EEV counted.
bvH3i⫹I, vH3i in the presence of IPTG; vH3i⫺I, vH3i in the absence of
IPTG.
on November 9, 2019 by guest
http://jvi.asm.org/
virus forms to a lesser extent than mature ones, and we found a similar situation with regard to D8L (Fig. 7 and 8). However, in cells infected with vH3⌬, the labeling of IV appeared to be increased, particularly for D8L, although this was not quanti-fied (Fig. 7). If the association of the surface proteins with viral membranes occurred at the same rate in cells infected with vH3⌬and WR, the greater labeling of vH3⌬IV could simply reflect their greater age due to the maturation block. Although there were fewer clusters of mature virions in cells infected with vH3⌬than in cells infected with WR, they were similarly labeled with antibodies to L1R, A27L, and D8L proteins (Fig. 8). As seen in Fig. 8, many of the virions are enclosed within cisternal membranes indicating that they are IEV.
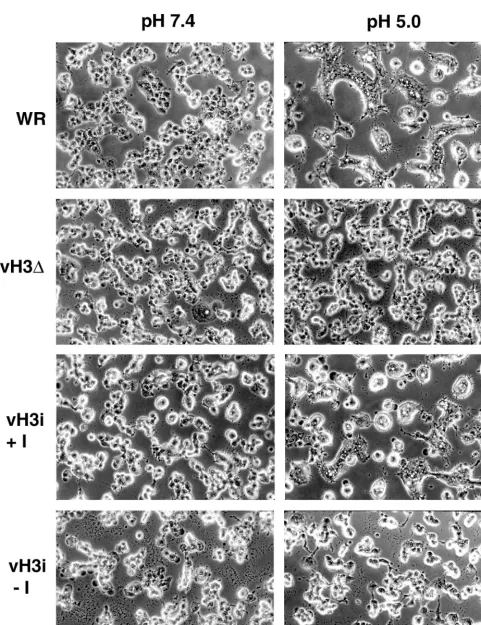
[image:8.612.56.553.68.554.2]Low-pH-induced fusion.Brief low-pH treatment of cells in-fected with vaccinia virus leads to formation of syncytia (9, 13). There appear to be at least two requirements for fusion: ex-pression of the A27L protein (13, 42) and formation of extra-cellular virus particles (3, 45). We anticipated some decrease in fusion due to decreased amounts of mature virions. To exam-ine this directly, BS-C-1 cells were infected at a multiplicity of 10 with WR, vH3⌬, or vH3i in the presence or absence of IPTG. After a 12-h incubation, the cells were treated briefly with fusion buffer (pH 5.0) or control buffer (pH 7.4) and incubated for 2 h more. Fusion did not occur when the pH 7.4 buffer was used (Fig. 9). Extensive syncytia were present when cells were infected with WR or vH3i in the presence of IPTG
FIG. 8. Detection of D8L, L1R, and A27L proteins on virion membranes of cells infected with vH3⌬or WR. Immunogold labeling was as described in the legend to Fig. 7. Insets show mature virions selected for good visualization of gold grains overlying the membrane.
VOL. 74, 2000 VACCINIA VIRUS H3L ENVELOPE PROTEIN MUTANTS 7525
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 9. Low-pH-induced fusion of infected cells. BS-C-1 cells were infected with WR, vH3⌬, or vH3i in the presence (⫹I) or absence (⫺I) of IPTG. At 12 h after infection, the cells were immersed briefly in buffer at pH 5.0 or 7.4. The medium was replaced, and the incubation continued for an additional 3 h. The cells were photographed with a phase-contrast microscope.
on November 9, 2019 by guest
http://jvi.asm.org/
and treated with pH 5.0 buffer (Fig. 9). In contrast, syncytia were not observed under the same conditions when cells were infected with vH3⌬or vH3i in the absence of IPTG (Fig. 9). Similar results were also obtained with infected RK13 cells (data not shown). The absence of detectable syncytia in cells infected with H3L mutants suggests that the amount of CEV is insufficient to mediate this process or that the H3L protein has an additional role in fusion.
DISCUSSION
For the present study, we constructed a recombinant vac-cinia virus with an inducible H3L gene and then deleted the original one giving a virus (vH3i) that produced smaller plaques in the absence than in the presence of IPTG. Although we used a high-stringency repression system and could not detect synthesis of the H3L protein in the absence of inducer, the possibility of slight but biologically significant leakage of gene expression that allowed small-plaque formation could not be ruled out. Therefore, we also constructed a simple H3L deletion mutant, vH3⌬. The ability to isolate this mutant es-tablished that the H3L gene was not absolutely required for infectivity, though again the plaques were much smaller and the titers of mutant virus stocks were 10-fold lower than those of wild-type virus. The availability of both types of mutants proved extremely helpful. With the deletion mutant, we could be confident that there was absolutely no H3L gene expression but could not entirely rule out negative effects on neighboring genes due to the insertion or expression of the selection marker in the H3L locus. In addition, differences in the titers or levels of purity of mutant and wild-type viruses could have significant consequences. This was a special concern because of the very low yield of vH3⌬compared to that of wild-type virus. The main advantage of the inducible mutant was that the effects of H3L gene expression could be studied using a single virus stock by adding IPTG to one set of infected cells and none to another. For most experiments, both types of mutants were used and consistent results were obtained.
The defect in plaque formation was not as severe as that occurring with some other mutant viruses that are defective in production of extracellular virus particles or actin tails (3, 12, 20, 30, 32, 45, 49). Moreover, single-step growth experiments indicated that the reduction in extracellular virus formation could be a secondary consequence of the decrease of up to 1 log unit in the formation of infectious intracellular virus. Elec-tron microscopy revealed that mature intracellular and extra-cellular virus particles could form in the absence of H3L gene expression. However, the numbers of mature particles were greatly reduced, and crescent membranes bordering large granular masses and IV predominated. The relatively small amount of extracellular virus could explain our inability to detect low-pH-mediated fusion of cells. Another possibility is that the H3L protein facilitates this process, which is thought to mimic acid-induced fusion occurring during entry of EEV through an endosomal pathway (41). Actin tails form on IEV and are important for efficient cell-to-cell virus spread (6, 30, 31, 49). Actin tails were visualized by immunofluorescence microscopy in cells infected with vH3⌬or vH3i in the absence of inducer, indicating that there was no specific block at this step (F. G. da Fonseca, unpublished data).
The formation of crescents and immature virus particles in the absence of H3L gene expression was consistent with anti-body binding experiments, which indicated that the H3L pro-tein is normally associated predominantly with particles at a later stage of maturation (7). Therefore, unlike the A17L and the A14L gene products (26, 29, 40, 47), the H3L protein is not
needed for the initial steps in virus membrane formation but apparently facilitates later steps in morphogenesis. It is inter-esting to compare the effects described here with those of null mutations of other IMV surface proteins. Repression of L1R expression also results in the accumulation of IV but there seems to be a more complete block in formation of IMV (25); repression of A27L expression has no effect on IMV formation but prevents IMV transport and wrapping with modified Golgi membranes to form IEV and EEV (27, 34); deletion of D8L has no effect on morphogenesis, but the IMV have a lower infectivity (16, 22).
In the accompanying paper (7), we show that the H3L pro-tein can insert posttranslationally into membranes via a C-terminal hydrophobic domain leaving the major portion of the protein cytoplasmic. We considered that this cytoplasmic do-main might interact with other membrane proteins, namely, D8L, A27L or L1R, and recruit them to viral membranes. However, these three proteins were found associated with membranes of IV and mature forms in the absence of H3L.
The major defect of H3L null mutants appeared to be im-paired virus maturation. This was unequivocally demonstrated by infecting cells under one-step growth conditions with the same H3L protein-deficient preparation of vH3i in the pres-ence and abspres-ence of IPTG. We could conclude that the phe-notype was due entirely to differences in de novo synthesis of the H3L protein and not to any role of virion-associated H3L protein in the binding of virus to cells, internalization, or other events prior to DNA replication and late gene expression. A quite different experimental situation existed when infections with vH3⌬and wild-type vaccinia viruses were compared be-cause the H3L protein is absent from the IMV membrane in the former and present in the latter. However, the similarity of the phenotypes of vH3⌬ and vH3i in the absence of IPTG suggested that the major defect was the same in both cases. In addition, there was little difference in the binding of infectious vH3⌬ and wild-type virus to cells. If vH3⌬ is less infectious than wild-type virus, however, we may have obscured a partial defect in binding by the design of our experiment. Unfortu-nately, comparing the binding of physical particles was prob-lematic because preparations of the mutant virus were less pure than those of wild-type virus and likely contained larger number of immature forms due to the severe maturation block and the consequent low virus yield. Relevant to this discussion is a report that appeared during the final stage of preparation of our manuscripts. Lin et al. (19) found that deletion of the H3L ORF interfered with virus maturation and reduced virus infectivity and virulence. In addition, they reported that an antibody to the H3L protein was neutralizing and that a solu-ble, truncated form of the protein bound to heparan sulfate and interfered with the binding of H3L protein to cells. There-fore, the H3L protein may have roles in both virus maturation and entry.
ACKNOWLEDGMENTS
We thank Christine White, Joanna Shisler, Linda Wyatt, and Tati-ana Senkevich for protocols, suggestions, and discussion of the data and Norman Cooper for cells and viruses. Erna Kroon was instrumen-tal in establishing the interaction between the Laborato´rio de Vı´rus, ICB, UFMG, Brazil, and the Laboratory of Viral Diseases.
Flavio G. da Fonseca is a graduate student at the Curso de Po´s Graduac¸a˜o em Microbiologia, Universidade Federal de Minas Gerais, Brazil, and was supported by the Fundac¸a˜o Coordenac¸a˜o de Aper-feic¸oamento de Pessoal de Nı´vel Superior and by a stipend from the National Institute of Allergy and Infectious Diseases.
REFERENCES
1.Alexander, W. A., B. Moss, and T. R. Fuerst.1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA
VOL. 74, 2000 VACCINIA VIRUS H3L ENVELOPE PROTEIN MUTANTS 7527
on November 9, 2019 by guest
http://jvi.asm.org/
polymerase and theEscherichia coli lacrepressor. J. Virol.66:2934–2942. 2.Appleyard, G., A. J. Hapel, and E. A. Boulter.1971. An antigenic difference
between intracellular and extracellular rabbitpox virus. J. Gen. Virol.13:9– 17.
3.Blasco, R., and B. Moss.1991. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-dalton outer envelope protein. J. Virol.65:5910–5920. 4.Blasco, R., and B. Moss.1992. Role of cell-associated enveloped vaccinia
virus in cell-to-cell spread. J. Virol.66:4170–4179.
5.Boulter, E. A., and G. Appleyard.1973. Differences between extracellular and intracellular forms of poxvirus and their implications. Prog. Med. Virol.
16:86–108.
6.Cudmore, S., P. Cossart, G. Griffiths, and M. Way.1995. Actin-based mo-tility of vaccinia virus. Nature378:636–638.
7.da Fonseca, F. G., A. Weisberg, E. J. Wolffe, and B. Moss.2000. Character-ization of the vaccinia virus H3L envelope protein: topology and post-trans-lational membrane insertion via the C-terminal hydrophobic tail.74:7508– 7517.
8.Dales, S., and L. Siminovitch.1961. The development of vaccinia virus in Earle’s L strain cells as examined by electron microscopy. J. Biophys. Bio-chem. Cytol.10:475–503.
9.Doms, R. W., R. Blumenthal, and B. Moss.1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol.64:
4884–4892.
10.Earl, P. L., N. Cooper, S. Wyatt, B. Moss, and M. W. Carroll.1998. Prepa-ration of cell cultures and vaccinia virus stocks, p. 16.16.1–16.16.3.InF. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley & Sons, New York, N.Y.
11. Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll.1998. Generation of recombinant vaccinia viruses, p. 16.17.1–16.17.19. InF. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley & Sons, New York, N.Y.
12. Engelstad, M., and G. L. Smith.1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology194:627–637.
13. Gong, S. C., C. F. Lai, and M. Esteban.1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology178:81–91.
14. Grimley, P. M., E. N. Rosenblum, S. J. Mims, and B. Moss.1970. Interrup-tion by rifampin of an early stage in vaccinia virus morphogenesis: accumu-lation of membranes which are precursors of virus envelopes. J. Virol.6:519– 533.
15. Hiller, G., and K. Weber.1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol.
55:651–659.
16. Hsiao, J. C., C. S. Chung, and W. Chang.1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol.73:8750–8761.
17. Ichihashi, Y., S. Matsumoto, and S. Dales.1971. Biogenesis of poxviruses: role of A-type inclusions and host cell membranes in virus dissemination. Virology46:507–532.
18. Jensen, O. N., T. Houthaeve, A. Shevchenko, S. Cudmore, T. Ashford, M. Mann, G. Griffiths, and J. K. Locker.1996. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electro-phoresis. J. Virol.70:7485–7497.
19. Lin, C. L., C. S. Chung, H. G. Heine, and W. Chang.2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol.74:3353–3365.
20. McIntosh, A. A., and G. L. Smith.1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol.70:272–281. 21. Morgan, C. 1976. Vaccinia virus reexamined: development and release.
Virology73:43–58.
22. Niles, E. G., and J. Seto.1988. Vaccinia virus gene D8 encodes a virion transmembrane protein. J. Virol.62:3772–3778.
23. Payne, L. G.1979. Identification of the vaccinia hemagglutinin polypeptide from a cell system yielding large amounts of extracellular enveloped virus. J. Virol.31:147–155.
24. Payne, L. G.1980. Significance of extracellular virus in the in vitro and in vivo dissemination of vaccinia virus. J. Gen. Virol.50:89–100.
25. Ravanello, M. P., and D. E. Hruby.1994. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virion assembly. J. Virol.68:6401–6410.
26. Rodrı´guez, D., M. Esteban, and J. R. Rodrı´guez.1995. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J. Virol.
69:4640–4648.
27. Rodriguez, J. F., and G. L. Smith.1990. IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi
mem-brane and egress. Nucleic Acids Res.18:5347–5351.
28. Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez.
1997. Characterization of early stages in vaccinia virus membrane biogenesis: implications of the 21-kilodalton protein and a newly identified 15-kilodalton envelope protein. J. Virol.71:1821–1833.
29. Rodriguez, J. R., C. Risco, J. L. Carrascosa, M. Esteban, and D. Rodriguez.
1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol.72:1287–1296. 30. Roper, R., E. J. Wolffe, A. Weisberg, and B. Moss.1998. The envelope
protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Vi-rol.72:4192–4204.
31. Rottger, S., F. Frischknecht, I. Reckmann, G. L. Smith, and M. Way.1999. Interactions between vaccinia virus IEV membrane proteins and their roles in IEV assembly and actin tail formation. J. Virol.73:2863–2875. 32. Sanderson, C. M., F. Frischknecht, M. Way, M. Hollinshead, and G. L.
Smith.1998. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol.
79:1415–1425.
33. Sanderson, C. M., F. Frischknecht, M. Way, M. Hollinshead, and G. L. Smith.1998. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol.
79:1415–1425.
34. Sanderson, C. M., M. Hollinshead, and G. L. Smith.2000. The vaccinia virus A27L protein is needed for the microtubule-dependent transport of intra-cellular mature virus particles. J. Gen. Virol.81:47–58.
35. Schmelz, M., B. Sodeik, M. Ericsson, E. J. Wolffe, H. Shida, G. Hiller, and G. Griffiths.1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J. Virol.68:130–147.
36. Sodeik, B., S. Cudmore, M. Ericsson, M. Esteban, E. G. Niles, and G. Griffiths.1995. Assembly of vaccinia virus: incorporation of p14 and p32 into the membrane of the intracellular mature virus. J. Virol.69:3560–3574. 37. Takahashi, T., M. Oie, and Y. Ichihashi.1994. N-terminal amino acid
se-quences of vaccinia virus structural proteins. Virology202:844–852. 38. Tooze, J., M. Hollinshead, B. Reis, K. Radsak, and H. Kern.1993. Progeny
vaccinia and human cytomegalovirus particles utilize early endosomal cister-nae for their envelopes. Eur. J. Cell Biol.60:163–178.
39. Traktman, P., A. Caligiuri, S. A. Jesty, and U. Sankar.1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of morphogenesis. J. Virol.69:6581–6587.
40. Traktman, P., K. Liu, J. DeMasi, R. Rollins, S. Jesty, and B. Unger.2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol.74:3682–3695.
41. Vanderplasschen, A., M. Hollinshead, and G. L. Smith.1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol.79:877–887.
42. Vazquez, M. I., and M. Esteban.1999. Identification of functional domains in the 14-kilodalton envelope protein (A27L) of vaccinia virus. J. Virol.
73:9098–9109.
43. Wang, S., and S. Shuman.1995. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase 2. J. Virol.69:6376–6388.
44. Ward, G. A., C. K. Stover, B. Moss, and T. R. Fuerst.1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc. Natl. Acad. Sci. USA92:6773–6777. 45. Wolffe, E. J., S. N. Isaacs, and B. Moss.1993. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extra-cellular virus envelope formation and dissemination. J. Virol.67:4732–4741. 46. Wolffe, E. J., E. Katz, A. Weisberg, and B. Moss.1997. The A34R glycop-rotein gene is required for induction of specialized actin-containing mi-crovilli and efficient cell-to-cell transmission of vaccinia virus. J. Virol.71:
3904–3915.
47. Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss.1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol.70:2797– 2808.
48. Wolffe, E. J., S. Vijaya, and B. Moss.1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology211:53–63.
49. Wolffe, E. J., A. S. Weisberg, and B. Moss.1998. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-contain-ing microvilli and cell-to-cell virus spread. Virology244:20–26.
50. Zhang, Y., and B. Moss.1992. Immature viral envelope formation is inter-rupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology187:643–653. 51. Zinoviev, V. V., N. A. Tchikaev, O. Chertov, and E. G. Malygin.1994.
Identification of the gene encoding vaccinia virus immunodominant protein p35. Gene147:209–214.