JOURNALOFVIROLOGY,June 1983,p.690-702 0022-538X/83/060690-13$02.00/0
Copyright©1983,AmericanSocietyforMicrobiology
Vol. 46, No. 3
In
Vitro
Synthesis of Late Bacteriophage 4)29 RNA
R.DOUGLAS HOLDERANDH. R. WHITELEY*
Departmentof Microbiology and Immunology, UniversityofWashington, Seattle, Washington 98195 Received 14 February 1983/Accepted 4 March 1983
A crude P-100 fraction prepared from Bacillus subtilis 21 min after infection with wild-type phage 4)29 supported the in vitro synthesis of late 4)29 RNA by added RNA polymerase. Synthesis of late RNAwasalso detectedwhen purified
4)29 DNA was transcribed by RNA polymerase in the presence of an S-150
fraction obtained by lysis of 4)29-infected cells in thepresenceof1 MNaCl. Late
4)29RNA was not synthesized when either the P-100 or the S-150 fractionwas
prepared from cultures infected with 4)29 having amutation ingene 4.
Bacillus subtilis phage 4)29 contains linear
double-stranded DNA having a molecular
weight of approximately 12 x 106 (3). The
genome consists of 18 cistrons, and the
func-tions and products ofmostofthe cistrons have
beenidentified(28, 33). Immediately after
infec-tion, thehost RNA polymerase transcribes the
early genes, which account for approximately
40% ofthe coding capacity ofthe genome. As
shown inFig.1,the early RNAisproducedfrom
the"light strand" of429 DNA{i.e., the strand
of lower density in
CsCl-polyuridylic-polygua-nylic acid [poly(UG)] gradients [31]} and is
syn-thesized throughout infection (24, 36, 39). The
lateclassofRNA, whichrepresents the
remain-ing 60% ofthe genome, is transcribedfrom the
"heavy strand" (31, 39). Althoughthe onset of
late transcription occurs at approximately the
sametime thatviral replication begins (36), late
transcription isindependent ofviral DNA
repli-cation (24). Synthesis ofthelate class ofRNA
requires the product ofan early 429 gene,
cis-tron 4 (4, 5, 39); the mechanism by which the gene 4 product regulates transcription is not
known.Thelocations oftheearlyand late genes
have been mappedandcorrelated witha
restric-tion map of thegenome (13, 19, 20, 44).
Transcription ofpurified 4)29 DNAby theB.
subtilis RNA polymerase in vitro yields early RNA (7, 19, 20). The number and location of
early 429promoters have been determined (8),
and the DNA sequences of three early
promot-ers have been compared (32). Synthesis of late
mRNA in vitro has not been reported and the location of the late promoters has not been
ascertained, although two polymerase-binding
siteswerefound in the lateregionof the genome
byelectronmicroscopy ofDNA-RNA polymer-ase complexes (39).
Regulationof viraltranscriptionthrough
mod-ification ofB.subtilisRNApolymerasehas been studied extensively during the development of
bacteriophages SP82 (23, 42) and SPO1 (13).
These phages code for several small polypep-tides which bind to and alterthetranscriptional specificity of the host RNApolymerase. Earlier
investigations (17) of RNApolymerase
extract-edfrom 4)29-infected cells showed that approxi-mately 10% ofthe host polymerase containeda
30,000-dalton (30K) peptide which was not
found inpolymerase from uninfectedcells. The
transcriptional specificity ofthe 30K-containing
polymerase with respect to 4)29 DNA was not
determined. It is known that at least the 3
subunit ofthe host RNApolymerase is required
for viral transcription since synthesis of late
mRNA is inhibited by the addition of rifampin
(24,
37).
In the present paper, we report that late 4)29
RNAwassynthesized invitrobypurified
rifam-pin-resistant RNA polymerase from uninfected
B. subtilis in the presence ofcrude P-100
frac-tionsfrom cellsinfected with wild-type4)29and
treated with rifampin toinhibit the endogenous
polymerase.Wealso report thatsynthesis oflate
mRNA was observed when reaction mixtures
containing purified 429DNAandpurified RNA
polymerase were supplementedwitha
superna-tant fraction (S-150) obtained by lysing
4)29-infected cells in the presence of a high salt
concentration. In both instances, i.e., in the presence of eitheraP-100orthe high-saltS-150
fraction, late mRNA was not produced if the
cellswereinfected withphage havingamutation in gene4.
MATERIALSANDMETHODS
Strains andgrowthofphage-infected B. subtilis. B. subtilis SR22, anasporogenous mutantof B. subtilis
168, obtained fromJ.Spizizen(Departmentof
Micro-biology, UniversityofArizona, Tucson),was usedas
the hostforwild-type 429 and as the nonpermissive host for4)29 sus4. Thesuppressor strain, B. subtilis
Su+44, and the sus4(56) mutant were obtained from B. E. Reilly(SchoolofDentistry, Universityof Min-690
on November 10, 2019 by guest
http://jvi.asm.org/
nesota, Minneapolis). B. subtilisOSB420, a rifampin-resistant mutant of B. subtilis 168, was obtained from L. Brown(Departmentof Microbiology, Oregon State University,Corvallis).
Phage-infected B. subtilis was grown in 750-ml to 10-liter batches of M medium (1% tryptone, 0.5% yeast extract, 1% NaCI). After sterilization, this medium wassupplemented with 0.4 mMMnC12, 5 mM MgSO4, and0.1% glucose added from sterile stock solutions. Cultures were grownat37°C to a concentration of 2 x
108 cells per ml and infected with 5 to 104)29particles percell. After 21 min, sodium azide was added to a 0.01 M final concentration, the temperature was re-duced to 0 to 5°C by the addition of ice, and the cells were harvested bycentrifugation, frozen in a dry ice-ethanolbath, and storedat-70°C. When infected cells were treated with rifampin before harvesting, the inhibitor was added at a final concentration of 10 ,ug/ml
at 18minafter infection, incubation was continued to
21 min, and the cells were collected as described above. Lysates of4)29wereprepared using B. subtilis SR22for wild-type phage and B. subtilisSu+"forthe sus4 mutant, using the M medium described above. Lysis occurred at 36 to 39 min, yielding approximately 101l phage per ml.
Preparation of the P-100 fractionandthe high-salt
S-150fraction.+29-infectedcells which had been treated withrifampin andharvestedat21 minafterinfectionas
described above were usedfor the isolation of both fractions. Except when indicatedotherwise, the fol-lowingoperationswerecarried out at0°C.Toobtain the P-100fraction, frozen cells (ca. 0.2 g [wetweight]) were suspended in 4 ml of buffer A (0.05 M Tris-hydrochloride [pH 7.5], 0.1 M NaCl, 1 mM EDTA, 0.01M2-mercaptoethanol,200 ,ugof
phenylmethylsul-fonyl fluoride perml) supplementedwith30%o
glycer-ol,lysozymewasadded(0.5mlofa5-mg/ml solution),
and thesuspensionwasincubatedfor 25to30minat
100C.Thepreparationwasthencentrifugedfor 20 min
at100,000 xginaBeckman type 40rotor at4°C,the
supernatantwasremovedcarefully, and bufferAwas addedtothesedimenttogiveatotalvolume of 1.5 ml. Thethin, yellowish-brownsediment withaclear
gelat-inouslayer abovewasresuspended by gentle homog-enization with fourorfive strokes ofalooselyfitted
Douncehomogenizerand usedimmediatelyasa
tem-plate for RNA synthesis. In some experiments, the
preparationswerecentrifugedfor10minat10,000 rpm in a Sorvall centrifuge to remove any intact cells before high-speed centrifugation. In most
experi-ments, this preliminary centrifugation was omitted, andif any intact cells were detected after the
high-speedcentrifugation (i.e.,ifagrey-whiteopaque cen-ter of any size could be seen in the yellowish sedi-ment), the preparation was discarded. "Successful lysis" yielded P-100fractions which had anegligible number ofunlysed cells. Direct counts, bymeansofa
Petroff-Hauser counter, showed that there were 2 x 106to 5x10'cells per ml(mostly swollen and atypical
inappearance) inourusualP-100preparationsand ca. 2x 104 viable cellsasdeterminedby plate counts;the original4ml ofsuspensionwascalculatedtohave ca. 5
x 109 cells per ml.
To testforpossibledegradation ofthe DNAin the P-100fraction, 0.5-mlamounts weredilutedwith 2 mlof proteinaseKsolution(50,ug/ml in0.01 M Tris-hydro-chloride [pH7.51-0.01 M EDTA-0.5% sodium
dode-cyl sulfate[SDS])and incubated for 2 hat30°C. The samples were then extracted with phenol, dialyzed
against 100 volumesof 0.1 MTris-hydrochloride(pH
7.5)-imMEDTA for24hat4°C,treated with1 ILgof
RNaseApermlfor 60 minat22°C,andanalyzed by
electrophoresis through vertical agarose slab gels,
using TBE buffer(0.089 M Tris base,0.089 M boric acid, 0.0025MEDTA).
A high-salt S-150fraction was obtained byoneof
two methods. In thefirst, theprocedure givenabove for isolation of the P-100 fractionwasfollowedexcept thatthetreatmentwithlysozymewasreducedto12to
15 min at 10°C, NaClwas added from a 5 M stock solutiontogiveafinal concentrationof 1 M, and the preparationwasthoroughlymixed, cooledto0°C,and centrifuged for 30 minat 150,000 x g in aBeckman type 40 rotorat 4°C. The supernatant was removed and desalted by passage throughacolumn of Sepha-dexG-25 equilibrated with buffer A, and the fraction having the highest absorbency at 280 nm ("high-salt S-150 fraction") was tested for its ability to stimulate latetranscription fromi029DNA. Inthesecond meth-od, two or more P-100fractions isolated in the
pres-enceof 0.1MNaClweresupplementedwith 5 MNaCl
togiveafinal concentration of 1 M, and the prepara-tions were left on ice for 10min, centrifuged,
com-bined, andprocessedasdescribedfor the high-salt S-150 fraction. The second method also yielded preparations which stimulated the synthesis of late RNA, but they were considerably less effective and less stable than thehigh-salt S-150 fractionobtained by thefirst method.
PreparationofphageDNA.Phagewereprecipitated from 10-liter lysatesby the addition of polyethylene glycol (43), dialyzed for 24 to 48 h against buffer B
(0.01 M Tris-hydrochloride [pH 7.5], 0.1 M NaCl, 0.005 M MgCI2),centrifuged through cesium chloride (31), and dialyzed for 12 to 18 h against buffer B
containingEDTAandSDS atfinal concentrations of 0.01 M and1%, respectively.Afterheatingat60°Cfor 2min,anequal volume ofproteinaseK(100 Lg/rnlin 0.01 M Tris-hydrochloride [pH 7.51-0.01 M EDTA) wasadded,and thepreparations were incubated for4 to6 h at37°C and then extracted with phenol. Phage DNAwasprecipitatedwithethanol, dissolved in 0.01 M Tris-hydrochloride (pH 7.5)-i mM EDTA, and dialyzed against the latter solution for24h.
Digestionof DNAwithrestrictionenzymesand elec-trophoresis. 4)29 DNA was digested with EcoRI or HindIll obtained fromNewEngland Biolabs (Beverly, Mass.)accordingtomethods describedby the
manu-facturer. Fragmentswereseparatedby electrophoresis throughavertical 0.7to1.4% agarose slab gel (12 by
14by 0.6 cm),asindicated, using TBE buffer, at100 V. Staining of gels with ethidium bromide, visualiza-tion under UV light, and photography have been describedpreviously (1).
StrandseparationofEcoRI fragmentsof+29DNA. DNA was digested with EcoRl andcomplexed with poly(UG) byamodification ofthemethodof Goldbach
etal.(14). Specifically,10p.gofdigested DNAin0.1 ml of 0.1 mM EDTA was mixed with 0.06 ml of partially hydrolyzed poly(UG)at aconcentration ofI
mg/ml, and themixturewasthen heatedfor3 min at
100°Ctodenature the DNAandquickly cooledto0°C. Acriticalstepin thisprocedurewasthepreparation of partially hydrolyzed poly(UG). Thiswasachievedby
on November 10, 2019 by guest
http://jvi.asm.org/
692 HOLDER AND WHITELEY
mixing 1 volume of a3-mg/ml solution of the polymer (Miles Laboratories, Inc., Elkhart, Ind.) with 0.1 volume of 0.5 M Na2CO3, incubating for 15 min at 67°C, and then cooling and adding 1 volume of 0.05 M HCl and 1 volume of 0.08 M Tris-hydrochloride (pH 7.7).After the addition of Ficoll and bromphenol blue, samples containing denatured fragments complexed with poly(UG) were subjected to electrophoresis on horizontal0.45%agaroseslab gels, using 0.04 M Tris-acetate (pH 7.7)-i mM EDTA as the buffer (14). Electrophoresis was performed at 4°C at a constant current of45 mAfor 18 to 20h, and the gels were stained withethidium bromide.
Invitro synthesis of RNA. When preparations of the P-100 fraction were used as the template, reaction mixtures (total volume, 0.24 ml) contained 60 ,ul of the P-100fraction, 10 to 20pgofB.subtilis OSB420 RNA polymerase, 0.04 M Tris-hydrochloride (pH 7.9), 0.1 M NaCl, 0.015 MMgCl2, 0.01 M 2-mercaptoethanol, 0.5mgof bovine serum albumin per ml, and 10 ,ug of rifampin per ml. The preparations were maintained at 0°C and mixed thoroughly, and an aliquot of 0.1 ml was withdrawn and added to a 4-,ul aliquot of a nucleotide solution containing 8 mM ATP, GTP, and CTP and4 mMUTPplus1 to2,uCi of[a-32P]UTP(30 to 50mCi/mmol; New England Nuclear Corp., Bos-ton, Mass.).The reaction mixtures were incubated for
10 min at 30°C or at the indicated temperature, and RNA was extracted for hybridization as described previously (41). When purifiedDNAwas the template, the same procedure was used except that the P-100 fraction was replaced with 20 to 30 ,ug of4)29DNA per ml.Methods used to assay theactivity ofRNA
poly-merasehave been described previously (1).
Labeling RNA in vivo. RNA was pulse-labeled at intervals during infection by the addition of4,uCi of [3H]uridine (41.5Ci/mmol; ICN, Irvine, Calif.) per ml and extractedaccordingtopreviouslydescribed meth-ods (41). Alternatively, phage-infected cultures were
grown in a low-phosphate medium (1) and pulse-labeled with 100 ,uCi of 32PO4 (carrier-free; New
England Nuclear) per ml.
Hybridizationof[32PJRNAtorestrictionfragmentsof 4o29 DNA. Fragments weretransferred to nitrocellu-lose filters by the methods of Ketner and Kelly (21), and the nitrocellulose was cut into 3 to 4-mm-wide strips. Eachstripwasincubated with[32P]RNAin3to
4mlof buffer C (0.025MTris-hydrochloride[pH 7.5],
0.825 M NaCl) containing 0.1% SDS in a sealed
polyethylenebag for 18to20 hat67°C.Thestripswere
washedinbuffer C for 20min at60°C, treated with10
pugof RNaseAper ml in 0.3MNaCl-0.03 M sodium citrate for20min at 37°C,and washedat 60°C fora totalof 60 minwiththree changes of buffer C contain-ing 0.1% SDS,0.01 Msodium PP,,0.004 M disodium phosphate, and 1 to 2,g ofATP per ml. Thefilters were dried and exposed to Kodak X-Omat film with an intensifyingscreen at -75°C.
RNA-DNA competition-hybridization. Anti-mRNAs may be synthesized by in vitro transcription of4)29
DNA(19).These RNAsmayself-anneal or hybridize with added -competitor RNAs or both, thus leading to spurious results in competition-hybridization
reac-tions. To minimize these effects, the competition-hybridization procedure (22) was modified to permit presaturation with competitor RNAs (26). In brief, nitrocellulose filters (5-mmdiameter) containing2 to 3
,ug of denatured 4)29 DNA were incubated with 0 to 0.75 mg of unlabeled competitor RNA in 0.3 ml of hybridization buffer (0.01 M Tris-hydrochloride [pH 7.5], 0.33 M NaCl, 0.1% SDS) at 67°C for 16 to 20 h. The filters were washed to remove unhybridized RNA,and labeled RNA was added and incubated with the filters as described above. After washing, the filters were treated for 60min at 20°C with a mixture of
RNases AandTI,washed, and dried, and the radioac-tivity was determined in a scintillation counter, using a toluene-based scintillant.
Miscellaneous methods. RNA polymerase was puri-fiedasdescribedby Achberger and Whiteley (1). The procedure of Roberts (34) was used to purify rho factor from Escherichia coli MRE600; rho activity was as-sayed according to Lowery-Goldhammer and
Richard-son (25). Protein concentrations were determined by the methodof Bradford (6), using gamma globulin as thestandard.
RESULTS
Transcriptional map. As indicated in Fig. 1,
early RNA is transcribed primarily from the
EcoRI A andC fragments. One earlytranscript
extendsslightly into theEcoRI Dfragment,and
hybridization to fragment D has been reported
by some investigators (20, 39)but notbyothers
(19). RNAextracted from4)29-infected cells late
ininfectionhybridizestoEcoRIfragmentsB, D,
and E in addition tofragments A and C.
Sym-metricaltranscriptionof boththe B andD
frag-ments can occur(39).The in vitrotranscription
of 4)29 DNA by B. subtilis RNA polymerase
yields several RNA species which hybridize
predominantly to theEcoRI A and Cfragments
(19,20, 39); in vitrotranscriptionmay alsoyield
significant amountsof anti-late mRNA (19).
Strand separation. To study the in vitro
syn-thesis oflate4)29 mRNA, we have determined
the strand specificity of transcripts produced
from a region of the genome containing late
genes-i.e., the EcoRI A and B fragments.
When EcoRI restriction fragmentsof4)29DNA
weredenatured,complexedwithpoly(UG),and
subjected to electrophoresis, the
complemen-tary strands of the two largest fragments each
yielded two ratherdiffuse, but separable,bands
(Fig. 2, lanes a to d). The complementary
strands ofundigested 4)29DNAand thestrands of the EcoRI Cfragmentwere notseparable by
this method, and theDand Efragments usually
comigratedas asingle largeband.
Figure 3 compares autoradiograms obtained
by hybridizing RNAs synthesized during
infec-tion to EcoRI and HindIlI fragments and to
strand-separated EcoRIfragmentsafter
electro-phoresisand transfer ofthefragmentsto
nitro-cellulose membrane filters.Controlexperiments
(Fig. 3, lanes a,b,g, andh)indicated thatearly
and late RNAs hybridized to the EcoRI and HindIII fragments in accordance with the map shown in Fig. 1. Early RNA hybridized to the J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
IN RNA Genetic 12 3 45 6 7 8 &5 9 10 11 12 13 14 15 16 17
1a a Ia a a I I Ia I I I
Map
. , .. -. . . >
early genes A
Hind IllL
(L) 5'. (H)
3'-late genes
B
B G K HMI E J D
a I I ...a .I a
early RNA
late genes earlygene
E D C
A N F C L
early RNA
late RNA
[image:4.488.81.414.68.239.2]0 25 50 75
FIG. 1. Genetic,physical, and transcriptionalmapsof the4)29genome.
EcoRI C fragment and to the slow-migrating band of the A fragment (Fig. 3, lane d). The latter has been designated "AL," since early RNA istranscribed from the light strand of4)29
DNA (31). The parallel experiment with late RNA(Fig. 3, lane e) demonstrated that in addi-tion to AL, the fast-migrating band of the A fragment and the slow-migrating band of the B fragment were transcribed at late times after infection. These bands were designated "AH"
and "BH," respectively, since late RNA is complementary to the heavy strand of 4)29 DNA. These findings and strand assignments
agreewiththose ofSogoetal. (39), who
report-ed that the fast-moving band of all EcoRI frag-ments of 4)29 DNA, except the A fragment, corresponded to the light strand and the
slow-moving bandcorresponded totheheavy strand. Longer exposures of the autoradiograms
pre-sented inFig. 3 also showedhybridization oflate
mRNAtothe EcoRI D/Efragment region (data notshown). Occasionally, RNA extracted from cultures early in infection showed a small amountofhybridization in the BH region (faint-ly visible in lane d of Fig. 3 and in lane b of Fig. 5), butnohybridizationwasfoundtothe EcoRI Bfragment if the strands of the DNA were not
complexed withpoly(UG) and separated before transfer to nitrocellulose (Fig. 3, lane a).
Be-cause of this occasional spurious hybridization ofearly in vivo RNA to BH, hybridization to both AHandBHwasconsideredtobethemost reliable indicator of the presence of late RNA.
The decreased hybridization of late RNA rela-tive to the hybridization of early RNA to the EcoRI C fragment(Fig. 3, lanes a, b, d, and e)
and tothe HindlIl C fragment (Fig. 3, lanes g
andh) will be discussed separately (R. D.
Hold-erand H. R. Whiteley, manuscriptin
prepara-tion).
In vitrosynthesis oflate RNA.P-100fractions
wereprepared from 429-infected cultures 21 min
afterinfection as described above and used as
templates for the synthesis of RNA. Since DNA replication beginsat8 to10min after infection, thepresence oflarge amountsof 4)29 DNAwas
expected. Electrophoresis of complexes after digestion with proteinase K showed two bands
on agarose gels. One band corresponded in
molecularweight to mature 429 DNA, and the other,
havinf
anapproximate molecular weightof 30 x 10, probably contained sheared B. subtilis DNA. The samplesalso contained some
high-molecular-weight DNA which didnot enter thegel. Therewasnoevidenceofdegradationof either the hostorphage DNA during the
incuba-tion of thecomplexes with nucleotides and add-edpolymeraseasjudged by electrophoresis. The
FIG. 2. Strand separation of EcoRI fragments of 4)29 DNA by gel electrophoresis (photograph of a 0.45%agarosegel stainedwithethidiumbromide). (a) EcoRI-digested4)29 DNA; (b) heat-denatured EcoRI-digested +29 DNA; (c) EcoRI fragments of DNA which were heat denatured and complexed with
po-ly(UG) before electrophoresisasdescribedin thetext; (d) purified EcoRI A fragment denatured and com-plexedwithpoly(UG) before electrophoresis; (e) puri-fied EcoRI Bfragment denaturedandcomplexedwith poly(UG)beforeelectrophoresis.
100
VOL.46,1983 693
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.488.294.395.433.580.2]694 HOLDER AND WHITELEY
a
bc
d
ef
g
hi
'g-~A
:i -AL*wA
X
-
-AH
X
BH
DI
BL
0t0:Ujx;0| 0;
D/E
l_
Z
~~~~~~~~~K
L
FIG. 3. Autoradiograms showing the hybridization of early and late in vivo [32P]RNAs to restriction fragments of4)29DNA.4)29-infectedcells were labeled with32PO4,and RNAs were extracted andhybridized to EcoRI- and Hindlll-digested 429DNA asdescribed in the text. (a) Early RNA(pulse-labeled for 2 to 7min) hybridized toEcoRIfragments; (b) late RNA(pulse-labeled for 25 to 30min) hybridized to EcoRl fragments; (c) photograph of ethidium bromide-stained 0.7% agarose gel ofEcoRI fragments; (d) early RNAhybridized to separated strands of EcoRI fragments; (e) late RNA hybridized to separated strands ofEcoRI fragments; (f) photograph of ethidium bromide-stained 0.45%agarose gel of separated strands ofEcoRI fragments; (g) early RNAhybridized toHindlI fragments; (h) late RNA hybridized to HindII fragments; (i) photograph of ethidium bromide-stained 1.2% agarose gel ofHindlIl fragments.
P-100 fractions also contained many proteins,
including RNA polymerase and probably
ribo-somes, since ribosomesweredetected in earlier
experiments with similar preparations(30).
Sedi-mentationofthe P-100preparations in10to30%
glycerol gradients showed thatRNApolymerase
and additional peptides cosedimented with
DNA, suggesting that the preparations
con-tainednucleoprotein complexes.
Because the P-100fractions contained endog-enouspolymerase, transcription wasperformed in the presence ofrifampin by using
rifampin-resistantpolymeraseisolatedfrom uninfected B.
subtilisOSB420.To ensure thatRNAsynthesis
by preparations containing the P-100 fraction
resulted from de novo initiation by the added
rifampin-resistantpolymerase and did not result
merely from elongationoftranscriptsinitiated in
vivo, cultures were treated with rifampin for 3
minbefore harvestingthecells. The data in Fig.
4show thatthistreatmenttotally inhibited RNA
synthesis by the endogenous polymerase pres-entin the P-100fractions.Itshouldbenoted that
all in vitro transcriptions were performed using
RNA polymerase preparations containing the
deltapeptide (2).
Two typesof P-100 fractionswerecompared:
thoseobtained from cells infected with wild-type
4)29 and those prepared from nonpermissive
cells infected with 4)29 carrying a nonsense
mutation in cistron 4. Mutations in this cistron
havepreviously been shown toprevent
expres-sion of late genes in vivo (4, 5, 39). RNAs
synthesized from such complexes by
exoge-nously added rifampin-resistant polymerase
were analyzed by hybridization to blots of
strand-separated EcoRI fragments of4)29DNA
asinFig. 3. HybridizationtofragmentsAH and BH was defined as indicating the synthesis of
late mRNA.
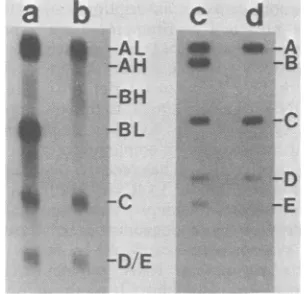
Transcription of the P-100 fraction derived
from cells infected with wild-type 4)29 yielded
RNAs which hybridized strongly to fragments
AL, AH, and BH and weakly to fragment C
(Fig. 5, lane a). The same fragments were
hy-bridizedby RNAs synthesized in vivo 25 to 30 J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
20
-E
1.5-0
0101.0
0.
1-0.
0~
0.-0 2 4 6 8 10 12 Time
(min)
FIG. 4. RNA synthesis from the P-100 fraction in the presenceand absenceof added RNA polymerase. Reaction mixturescontained 150 ,ul of P-100 fraction, 10 g of rifampin per ml, 50 ,ug of rifampin-resistant RNApolymerase(O or A) or no added polymerase(0 orA),buffer, and nucleotides including[32P]UTPin a total volumeof 0.75. The reactions were incubated at
30°C,and atthe indicated times, S0-pl samples were withdrawn and the incorporation of radioactivity into trichloroacetic acid-insoluble material was measured.
(0, 0)P-100fraction isolated from4)29-infectedcells
which had not been treated with rifampin before
harvesting; (A, A)P-100fraction isolated from o029-infected cells which had been treated with rifampin beforeharvesting.
min afterinfection (Fig. 3, lane e) except that
there was a relatively weaker hybridization to
ALin the latterexperiments.Transcription of
P-100obtained fromcellsinfectedwith thegene 4 mutantdid notyield late RNA (Fig. 5, lane b). Hybridization wasfoundpredominantly to
frag-ment AL and more weakly to fragment BH.
SincehybridizationtofragmentAHcouldnotbe
detected and therewas nohybridizationtothe B
fragment if the poly(UG) complexing step was
omitted (data not shown), the hybridization to
BH was considered to be an artifact. The
ab-senceofhybridizationtotheEcoRI Cfragment
in lane b ofFig. 5 will be discussed elsewhere
(HolderandWhiteley, in preparation). For
com-parison,lane cofFig. 5presents the resultsofa
separateexperiment in which purified4)29DNA was used as the template inplace ofthe P-100
fraction. Late RNA was not synthesized from
this template, as evidenced by the absence of
hybridizationtoAHandBH. Hybridizationwas
observedtofragments AL, C,andD/E,
indicat-ing the synthesis of earlyRNA(19, 20).
Hybrid-izationwasalso found tofragment BL,
indicat-ing the synthesis of anti-late RNA. This
observationis discussed in a later section.
Paral-lel experiments in which the S-100fraction ob-tained from cells infected with wild-type 4)29
was used for the synthesis of RNA by rifampin-resistant polymerase yielded only trace amounts
of RNA, probably because the S-100 fraction
contained relatively low concentrations of
DNA. The hybridization of this RNA was not
investigated.
Confirmation that the P-100 fraction from cells
infected with wild-type 4)29 supported the
syn-thesis of late RNA in vitro came from
RNA-DNA competition-hybridization experiments
(Fig. 6). Thedifferences in hybridization in the
presence of early and late competitor RNAs
were similar to those obtained with
pulse-la-beled in vivo late RNA. In contrast, the data
obtained with RNAsproduced from purified4)29
DNAindicate that only earlyRNA was
synthe-sized; the ability of late RNAto competein this
hybridizationwasduetothefact that early RNA
is produced throughout infection (7, 24, 37).
Hybridization ofRNA synthesized from P-100
tosaturatingamountsof 4)29 DNA indicated that
approximately 25% of the transcripts were429
specific (datanotshown),and therelative
inten-sities of the bands in lane a of Fig. 5 and
a
b
c
AL_
---AL
AHd
~-AH
BL-
*-BL
C-'-C
D/E
D,-);E
FIG. 5. Autoradiogram showing the hybridization ofseparated strands ofEcoRIfragments of4)29DNA with RNAs synthesized in vitro. RNA was synthe-sizedas described inthe text during a 10-min incuba-tion at 30°C, using rifampin-resistant RNA polymer-ase, rifampin, and: (a) a P-100 fraction isolated from cells infected with wild-type429,(b)aP-100 fraction isolated from cells infected with a gene 4mutant of
4)29,and(c)purified4)29DNA.Note that two sets of filterswereused in thisexperiment: one set forlanes a andbandthe other setfor lane c.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.488.66.216.60.267.2] [image:6.488.286.408.388.573.2]6% HOLDER AND WHITELEY
L..
o
20-0
0
100
0
2 80
IL A
60
40
20-0
0 0.5 1.0 1.5 2.0 2.5
Competitor RNA
(mg/ml)
FIG. 6. Competition-hybridization experiments in which RNAs synthesized in vivo and in vitro were
used. (A) RNA extractedfrom4)29-infected cells la-beled with 32PO4 at 2 to 7 min after infection and competed with 7-min unlabeled RNA (0) or 30-min unlabeled RNA(0);RNAextracted from4)29-infected cells labeled with32P04at25to30 minafter infection andcompetedwith 7-min unlabeled RNA (A) or 30-minunlabeled RNA(A).(B)[32P]RNAsynthesizedin vitro as described in thetextfrompurified 4)29DNA andcompetedwith 7-min unlabeled RNA(0) or 30-min unlabeled RNA (0); [32P]RNA synthesized in vitro as described in the text from aP-100 fraction isolated from+29-infectedcellsandcompetedwith
7-minunlabeledRNA(A)or30-min unlabeledRNA(A\).
subsequent figures indicate that significant
amountsoflate RNA were synthesized.
Effects oftemperature,dilution,andNaCi.The
capacity oftheP-100fractionisolated from cells
infected with wild-type 4)29 to support the in
vitro synthesis of late RNA was highly
depen-dent upon the temperature at which transcrip-tion was performed. Figure 7 presents dataon the hybridization between strand-separated
EcoRI fragments of4)29 DNA and RNAs
syn-thesized by preparations containing P-100 prepa-rations at 20, 25, 30, 37, and 42°C. Two overall trends were apparent from these
autoradio-grams. First, late RNA was not synthesized at
the two higher temperatures asjudged by hy-bridization to EcoRIfragmentsAH and BH(Fig. 7, lanes d and e), and synthesis of anti-late RNA
was observed (hybridization to fragment BL).
Temperature shift experiments (data not shown) indicated that thefactor orfactorsrequired for late transcription were irreversibly inactivated
in vitro at temperatures higher than 30°C. The
second trend evident inFig.7 was aprogressive
inhibition in the synthesis of early RNA as the temperature was decreased. RNA complemen-tary to the EcoRI C fragment was not synthe-sized at 25°C (Fig. 7, lane b), and at 20°C there
was little detectable transcription from the
EcoRI A fragment (Fig. 7, lane a). Thus, at
lowertemperatures,virtuallyalltranscription by
reaction mixtures containing the P-100 fraction
came from late sequences. Since these assays
were performed with crude preparations, it is
not known whether the apparent temperature
lability of the factor(s) is dueto degradation by
proteases or to some othermechanism.
The efficient synthesis of late RNA in vitro
required relatively high concentrations ofthe
P-100 fraction (50 to 100 ,ul per 240-,ul reaction
mixture; the reaction mixtures also contained
0.5 mg of bovine serumalbumin perml). When
low concentrations wereused(30
RI
per 240-,ulreaction mixture), therewaslittleor no
hybrid-izationto AH andBH, andhybridizationtoBL
was detected, indicating the synthesis of
anti-a
b
c
d
e
-AH
w || X -BH
~:
.dl-BL
-D/E
FIG. 7. Autoradiogram showing the hybridization ofseparated strandsofEcoRI fragments of4)29DNA
withRNAssynthesized at different temperatures from
aP-100fraction.[32P]RNAswereextractedfrom
reac-tion mixtures containing rifampin-resistant
polymer-ase and a P-100 fraction isolated from 4)29-infected
cellsafter10min of incubationat:(a)20,(b) 25, (c) 30, (d) 37,and(e)42°C.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.488.72.219.62.404.2] [image:7.488.272.419.454.600.2]SYNTHESIS 429
late RNA. It maybe speculated that the gene4
product was presentinlimitingamounts orthat
itwasreadily inactivated by dilution orboth.
The addition of0.05 to 0.15 M NaCl had no
effect on the synthesis of late RNA, although
some inhibitionwasnoted at aconcentration of
0.2MNaCI. Thisfinding is of interest because of
the effect of NaCI in limiting the formation of
nonspecific complexes between RNA
polymer-aseand DNA. Detailed studiesonthebinding of
B.subtilisRNApolymerase to phage SP82DNA
(1)have shownthat in thepresenceof0.1 to0.2
M NaCl, B. subtilis RNA polymerase
prepara-tionswhichcontainthedeltapeptideform
com-plexes onlywithstrongearly phage
promoters-i.e., there is little or no nonspecific complex
formation. The formation of complexesby
SP82-modified forms of RNA polymerase with middle
and late SP82 promoters was even more
sensi-tive to increasing concentrations of NaCl than
the interaction of unmodified polymerase with
earlySP82promoters. If theinteractionof
poly-merase with late 4)29 promoters resembles the
interaction of thisenzymewith SP82promoters,
then the observation that late 4)29 RNA was
produced in the presence of 0.15 M NaCl
sug-gests that this synthesis did not result from a
nonspecificinteraction betweenpolymerase and 4)29DNA.
Location of the promoter(s) for late
transcrip-tion.Asnotedabove,thelocation and number of
late promoters on 4)29 DNA have not been
determined. Tolocalize the site(s)atwhich late
transcription was initiated in vitro in the
pres-ence of the P-100 fraction, transcription was
terminated after increasing intervals of
incuba-tion and theRNAs werethenhybridizedtoblots
of HindIII restriction fragments of 4)29 DNA
(Fig. 8, lanes a to f). Lanes g and h ofFig. 3
show, respectively, the hybridization of RNAs
pulse-labeledatearly and late times of infection.
RNAs synthesized in vivo 2 to 8 min after
infectionhybridized toHindlllfragments B,C,
G, H, and K, whereas RNAs produce 25 to30
minafter infectionhybridized tothe same
frag-ments asearly RNAplusHindIIIfragments A,
D, E, F, I, and J(Fig. 3, lane g).Hybridization
to the two smallest fragments, HindIII-L and
-M, was not detected in any experiments; a similarlackofhybridization to smallrestriction
fragmentswasnotedoriginally bySouthern(40).
RNAssynthesized in vitro duringthefirst30 s
of incubation in the presence ofthe P-100
frac-tion were complementary to the Hindlll B, C,
H,and Ifragments(Fig. 8, lanea).With
increas-ing times of incubation (Fig.8, lanes b to f),
additional regions of the genome were
tran-scribed ina sequence which is consistent with
the transcriptional map shown inFig. 1, ifone
allows for the fact that hybridization to
frag-a
b
c
d
e
f
-A
& w -B -D
* 0*
*-E
-F
*
4*
* **-G
-
I--J
L-FIG. 8. Autoradiogram showing the hybridization
of HindIIIfragmentsof+29DNAwith RNAs
synthe-sized afterincreasingintervalsofincubation froma P-100fraction.[32P]RNAswereextractedfrom reaction mixturescontainingrifampin-resistant polymeraseand
aP-100fraction isolated from429-infected cells and incubated at30°Cfor: (a) 30 s,(b)60 s,(c)90 s,(d)2 min, (e)4min, and (f)8min.
mentsLandM was notdetected. The results of
the experiment presented in Fig. 8 suggest that
early RNA synthesis in vitro is initiated on the
HindlIl B, C, H, and I fragmentsandproceeds
from rightto lefton the physical map. In vitro
transcription of the late regions was evidently
initiated in theregionnearorwithin the
HindlIl-H-I region, and the sequential appearance of RNAcomplementary tofragments E,J(faintly hybridized), D, A, and F indicates that the
direction oftranscriptionwasfromlefttoright.
Although the HindlIl I fragment was the first
exclusivelylatefragmenttowhichhybridization
was detected, the exact position of the late
promoter could not be determined since the H
and Ifragmentsareseparated by theMfragment
which didnothybridize. Itisevident, however,
fromFig.8 thattranscription comparabletothat
ofthe H-Iregionwas notinitiated inthemiddle
of the lateregionoratthe Bi and B2sites(39).
The latter two sites, at positions 59.4 and 79.3
fromtheleft end ofthe genome, wereidentified
asbinding sites forB. subtilisRNApolymerase
by electron microscopic examination of
poly-merase-DNAcomplexes.
Elution of thefactor(s)from the P-100 fraction. Treatmentof the P-100fraction with 1 MNaCl,
followed by centrifugation or lysis of
phage-VOL. 1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.488.280.413.72.288.2]698 HOLDER AND WHITELEY infected cells in the presen
apparently released the fact
transcription of late sequence
sedimentedby high-speed cei
a to e of Fig. 9 indicate th
increasing amounts ofthe hi
tion (after decreasingthe con
to 0.1 M) to transcription r
purified
4)29
DNAand purifiewereemployednot onlystimu
of late RNA (hybridization ti
alsoprogressivelyreducedth
RNA (hybridizationto AL ai
RNA (hybridization to
BL)
discussed below, RNAs corr
EcoRI D and E fragments
producedbyreadthrough fro
in EcoRI-C. The P-150 fract
the high-salt treatment was
effective in stimulating late
was a P-100fraction (data not
same experiments were per1
infected with4)29 carrying a
in cistron 4, the high-salt '
(Fig. 9, lane g) did not stimul;
late sequences in vitro, and :
complementaryto theCand
not detected. The lack ofsyl
fragment in this experiment:
a
bcde
S
..:**
*
p
FIG. 9. Autoradiogram showi ofseparatedstrands of EcoRI fra withRNAssynthesizedinthepr S-150fraction isolated from cellh type4)29 andfrom cellsinfectedi
of4)29.[32P]RNAsweresynthesiz
as described in the text in reac
volume,0.1ml)containing3 ,ug( rifampin-resistantRNApolymera perml,buffer, nucleotides,and ti of thehigh-salt S-150fractionfro] wild-type 429:(a)none,(b)6 ,u1,(
32 p1, (f) none, and (g) 32 ,t1 fraction isolated from cells infe mutant of 4)29.
ice of 1 M NaCl, obtained in two earlier experiments (Fig. 3,
tor(s) required for lanesb, e,and h,andFig. 5, laneb).
:s from the material Sincethe4)29DNA used in theabove
experi-ntrifugation. Lanes ments was purified using proteinase K
treat-iat the addition of ment,thetemplate lacked the
4)29
gene3proteinigh-salt S-150 frac- (16, 35). Synthesis oflateRNA in vitro,
there-icentrationof NaCl fore, doesnotdepend on thisprotein. Previous
reactions in which studies have demonstrated that the gene 3
prod-d RNA polymerase uct doesnotaffect either RNApolymerase
bind-ildated the synthesis ing (39) or in vitro synthesis of early RNA (8,
o AH and BH) but 20).
esynthesisof early The followingpreliminary observations have
ndC) and anti-late beenmade withrespecttotheproperties of the
). Presumably, as high-salt S-150 preparation. (i) Electrophoresis
iplementary to the onSDS-polyacrylamide gelsand onagarosegels
and to BL were showedthat thesefractionscontained acomplex
imaninitiation site mixtureofpeptides and small amountsof
nucle-tion resulting from ic acids (some rRNA and trace amounts of
significantly less DNA). (ii) As stated earlier, theS-150 fractions
transcription than prepared by either of two methods did not
shown). When the provide a template for added RNA
polymer-formed using cells ase-i.e., the addition ofpurified429 DNAwas
nonsense mutation required for RNA synthesis and late
transcrip-S-150 preparations tion. (iii)Norifampin-resistant incorporation of
atetranscription of [3H]UTP into RNA could be detected in the
synthesis ofRNAs presence of4)29 DNA when the high-salt S-150
D/E fragmentswas fraction was used as the source of RNA
poly-nthesis from the C merase in the standard RNA assay procedure
agrees with results (the concentration of NaCi in the eluate was
reducedto 0.1 Mbefore assay).(iv)Theaddition
of the high-salt S-150 fraction obtained from
cells infected with wild-type 4)29 or with the
t
g^
cistron 4 mutant resulted in a two- to threefoldt ~ stimulation in the activity ofthe
rifampin-resis--AL tant RNA polymerase. This increase wasnoted
-A H inassaysinwhich two DNA
templates
(4)29
and-BH
SP82
DNAs) were employed and probably canbe attributedtothe release ofthe sigma subunit
-BL from endogenous RNA polymerase. (v)
Treat-mentwith 1 MNaClwasrequiredtoreleasethe
active factor(s) from P-100 fractions. (vi) The
-C
cfactor(s)
responsible
forsynthesis
of late RNAwasrapidlyinactivated bydilution andexposure
totemperaturesgreaterthan 30°C. (vii)To date,
-
D/E
attemptstofractionate the factor(s) bysedimen-tation in glycerol gradients orby
chromatogra-ing the hybridization phy at 40C on DEAE-cellulose,
phosphocellu-Lgments of+29 DNA
lose,
Biorex70,
and DNA-cellulose have beenesence of a high-salt unsuccessful.
s infected with wild- In view of the earlier observation (17) that a with a gene 4 mutant small part of the RNApolymerase isolated from
red at 30'C for10min 429-infected cells contained a 30K peptide, it
Ation mixtures (total was ofinteresttodetermine whetherthe
synthe-of
429DNA,4pLg
of sisoflate4)29
RNAcouldbecorrelated withtheefollowginof
armompnts
presence of thispeptide
in thehigh-salt
S-150m cells infected with
preparation.
Electrophoretic comparisons of'c) 121Ll,(d) 20p1,(e) such fractions obtained fromcells infected with
of a high-salt S-150 wild-type and gene 4 mutantphages showed no
cted with a gene 4 significant differences in theamountsof peptides
in the 25,000- to 35,000-dalton range (R. D. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.488.58.230.394.536.2]IN VITRO SYNTHESIS OF LATE 4)29 RNA 699
Holder, Ph.D.thesis,University of Washington,
Seattle, 1981). Furthermore, when extracts of
cells infected with wild-type 4)29 were
precip-itated with antibodytothe coresubunits ofRNA
polymerase and the antibody complexes were
analyzed by electrophoresis on
SDS-polyacryl-amide gels, no additional peptides were found
associated with the core subunits. However,
when thesameantibody preparationswereused
in parallel experiments with extracts of
SP82-infected cells, the SP82-coded peptides were
found in association with thecoresubunits(D. L.
Linemeyer, Ph.D. thesis, University of
Wash-ington, Seattle 1977). These observations,
cou-pledwith thefact thatareproducible isolation of
a 30K-containing form ofRNApolymerase has
notbeenachieved, suggestthat the 30Kpeptide
is not involved in transcription of late genes.
Alternatively, the 30K peptide may havea low
affinity for thecoresubunits and therefore isnot
precipitatedby antibodiestothecore orretained
with the core subunits duringpurification.
In vitrosynthesisofanti-lateRNA. Inspection
of the restriction map of 4)29 DNA (Fig. 1)
indicates that the in vitro synthesis of anti-late
RNA(RNAwhichhybridizestoBL;Fig.5, lane
c and Fig. 7, lanes d and e) results from read
through of transcription beyond a termination
site near the junction of the EcoRI C and D
fragments. Davison et al. (8) have shown that
although RNA polymerase will terminate
tran-scription atthis site invitro, termination is not
efficient and RNA ofhigh molecular weight is
produced by read through beyond this site.
Perhaps such readthroughmay occurinvivoas
well, since hybridization of early RNA to the
EcoRI D fragment has been demonstrated (20,
39). Inciarteetal. (19) reported that the addition
ofacrude extractofB. subtilis decreased
tran-scription through this site and suggested that a
termination factor may be involved. We
at-tempted to isolate a B. subtilis rho factor by
using methods described for the isolation (34)
andassay(25) ofE.coli rho but didnotsucceed (Holder, Ph.D. thesis); subsequently, Hwang
and Doi(18)isolatedarho factor fromB.subtilis
by usingother methods.
However,wefound that the addition of theE.
coli rho factor decreased the incorporation of
[3H]UTP
into RNAbyB.subtilisRNApolymer-asebyapproximately 30%when
4)29
DNA wasthe template and that E. coli rho had noeffect
when polydeoxyadenylic
acid-polydeoxythymi-dylic acid was the template (data not shown).
Figure 10presents dataon the hybridization of
RNAs synthesized in the presence and absence
of E. coli rho. The addition of this protein
inhibited the synthesis of anti-late RNA-i.e.,
RNAthathybridizedto BL(Fig.10,lanesaand
b). Lanes c and d ofFig. 10 provide evidence
a
b
I
.-AL.. w:
-AH-i-BH
-BL
Q
-c d
S-AA
-B
-L. ._.-c
_w _ -Ew
[image:10.488.274.426.77.224.2]A
D/-D
EFIG. 10. Autoradiograms showing the hybridiza-tion of EcoRI fragments of +29 DNA with RNAs synthesized from +29 DNAin the presence and
ab-sence ofEcoRI rho factor. (32P]RNAs were synthe-sized asdescribed in the legendtoFig. 8 in the absence (lanesaand c) or presence(lanes b and d) of 3 Fgof
EcoRIrhofactor. Lanes aand b show the hybridiza-tion to separated strands of EcoRI fragments, and lanes c and d show the hybridization toEcoRI frag-mentsof4)29DNA.
that rho prevented synthesis from the E
frag-ment aswellasfrom theBfragment.E.coli
rho-induced termination occurred, therefore, near
thejunction of the EcoRI D and E fragments.
However,hybridizationofearlyand late invivo
RNAs(Fig. 3) demonstrated thatearly
transcrip-tion did notextendinto the EcoRI-Dregion; in
our experiments, the B, D, and E fragments
represent exclusively late regions. Therefore, although the E. coli rho factor was able to
terminate in vitrotranscription of4)29 DNA by
B. subtilis RNA polymerase and prevent the
synthesisofatleastapartof the anti-lateRNA, it seemsthatthis factor doesnot recognizethe
termination site utilized in vivoby4)29-infected
B. subtilis. It should be noted also that the
pattern ofhybridization in lane c ofFig. 10 is
identicaltothatobserved with late in vivo RNA
(Fig. 3, lane b). Use ofseparated strands,
how-ever, allows discrimination between late and
anti-lateRNAandclearlyshows that
hybridiza-tion of the B fragment in lane c of Fig. 10
represents anti-late transcription.
DISCUSSION
The present investigation demonstrated that
bothearlyand late
4)29
RNAsweresynthesizedinvitrobyRNApolymerase fromDNA
provid-edbyacrudeP-100 fraction obtained from
4)29-infected B. subtilis at a late stage ofinfection.
Rifampin was added tothe culturesjust before
extraction oftheP-100 fractiontoinhibit
endog-46, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
700 HOLDER AND WHITELEY
enouspolymerase; transcription was performed
in the presence of rifampin, using a purified
rifampin-resistant RNA polymerase. The
pat-ternofRNAsynthesis bythis system, as
detect-ed by hybridization to restriction fragments of
4)29DNA,was verysimilartothat obtained with
RNA synthesized in vivo late in infection. A
similar in vitro systememployinga crude lysate
of T4-infectedE.coli has recentlybeen reported
to synthesize middle T4RNA (9, 10). Asinthe
present studies, rifampin was used to block
transcription by endogenous polymerase, and
RNA synthesis was catalyzed by added
rifam-pin-resistant E. coli RNA polymerase in the
presence of this inhibitor. It was proposed that
the binding of rifampin-inhibitedpolymerase to
immediate early promoters prevented chain
elongation to the middlegenes, thereby
permit-ting detection of middlegene expression under
the control of the motgene product. It is not
known what effect, ifany, thebinding of
rifam-pin-inhibited polymerasetoearlypromotershad
onthe4)29in vitrosystem.Possibly, such
bind-ingmayhave decreasedtranscription from early
promoters, thus allowing amore active
synthe-sisof late RNA.
Underoptimal conditions oftemperature and
concentration of the P-100 fraction,there was no
synthesis of anti-late 4)29mRNAinourin vitro
reactions. Read through of a termination site
nearthe EcoRI C and Dfragments to produce
anti-late mRNA was observed when purified
4)29DNAwasusedas atemplate in placeof the
P-100 fraction. Addition of theE. coli rhofactor
prevented synthesis of some of the anti-late
RNA, butterminationdidnot occur atprecisely
the same site astermination in 4)29-infectedB.
subtilis. Possibly, a B. subtilis rho factor (18)
functions both in vivo and in the in vitrosystem
toprevent read through.
Previous studies have shown that theproduct
ofcistron 4 isrequired for the synthesis of the
lateclassof429RNAin vivo(4, 5, 13, 39). We
found that late RNAwasnotsynthesizedin vitro
when theP-100fractionwasderivedfromasus4
mutant,suggestingthatlatetranscriptionin vitro
requiresanactivegene 4protein. Earlier studies
(5) suggested that peptide LM 3B may be the
gene 4product;morerecently(11,29)gene 4 has
been cloned and sequenced, and a peptide of
12,500daltons hasbeen identifiedasitsproduct.
BystoppingtranscriptionfromtheP-100
frac-tion afterincreasingintervals of incubation and
then hybridizing the RNAs to blots of HindIII
fragments of+29DNA, welocalizedtheregion
from whichin vitro latetranscriptionwas
initiat-ed. Theseexperiments indicated that late RNA
synthesis beginsintheHindIII-H-Iregion ofthe
genomeand thattranscription proceeds
sequen-tially from left to right, consistent with the in
vivo transcriptional map presented in Fig. 1.
Electronmicroscopy of polymerase-DNA
com-plexes (39) revealed thata polymerase binding
site (A3)waslocated in this region and that there
were twoadditionalbinding sites(B1 and B2)in
the HindIII A and F fragments, respectively.
Although the results of the present studies do
notruleoutthepossibility that therewas
ineffi-cientorweak initiation from the B1 and B2 sites
concomitantly with stronger or more efficient
initiation from the HindIII-H-I region, the
sim-plestexplanation forourresults isinitiationnear
theA3 binding site followed by rightward
tran-scription.
BytreatingtheP-100fraction with1 M NaCl
orbylysingphage-infected cells in thepresence
of 1 M NaCl, the factor(s) required for late
transcriptionwasreleased into thesupernatant.
In vitro synthesis of late RNA was observed
whensuchsupernatantfractions (high-salt S-150
fraction)werepreparedfrom cells infected with
wild-type 429 and added to transcription
reac-tions containing purified 4)29 DNA and RNA
polymerase from uninfected cells. When the
high-salt S-150 fractionwasobtained from
non-permissive cells infected with 4)29 carrying a
nonsense mutation in cistron 4, late transcrip-tionwas notdetected.
Several possible mechanisms could be
pro-posedto accountfor theeffect ofthehigh-salt
S-150fractions in promoting the synthesis of late
RNA by unmodified RNA polymerase from
pu-rified 429 DNA. One possibility is that these
fractions contain thegene 4product and that this
protein is similar to the phage-coded peptides
which replacesigma in themodification ofRNA
polymeraseinSP82-orSPOt-infectedB.subtilis
(13,23, 42).Thus, it could be imaginedthat the
gene 4 product binds to thefree core subunits
whicharepresent in mostpolymerase
prepara-tionsorthat itdisplaces thesigmasubunit from
theholoenzyme, thereby providingthe
specific-ityrequired fortherecognition of late promoter
sequences. However, if this mechanism
oper-atesin thetranscriptionof4)29 DNA, the in vitro
properties ofthefactor(s)aresignificantly
differ-ent from those of the SP82-coded
specificity-determining peptides. The latterbindsufficiently
tightly to the core subunits to be retained
throughoutthepurificationofRNApolymerase,
theyareprecipitated withthecoreby antibodies
tothe core,andthey donotdisplayany unusual
sensitivitytotemperatureordilution(12, 13, 23,
42).
Anotherpossibility isthat the gene4product
functions in antitermination. An antiterminator
mechanismfor latetranscriptionof
4)29
hasbeensuggested (39) based on the observation that
when large amounts of early RNA and late
chloramphenicolRNAwereused,hybridization
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
wasdetectedtothe heavy strand of the EcoRIA
fragment but not to the heavy strand of the
EcoRIBfragment. Sogoetal. (39)proposedthat the EcoRI A fragment may contain a late pro-moter and thattranscription ofthe B fragment
requires an antiterminator protein which is not
producedearly in infection or in the presence of
chloramphenicol.
The lability of the factor(s) which stimulates
the in vitro synthesis of late 4)29 RNA is of
particularinterest. Wefound that thecapacityof
the P-100fractiontosupportthesynthesis of late
RNA decreased as the temperature of
incuba-tion of thereaction mixtures wasincreased and
thatattemperaturesabove30°C, littleor nolate
RNA was produced. Thefactor(s) required for
late in vitrosynthesiswasirreversibly
inactivat-ed at moderate temperatures and by dilution.
Moreover, asthe temperature ofsynthesis was
decreased, synthesis of earlyRNA wasinhibited
so that at 20°C virtually all RNA synthesized
from the P-100 fraction represented
transcrip-tion from late sequences. It appears, therefore,
thatatlow temperatures, B. subtilis RNA
poly-merase may initiate synthesis in vitro at a late
promoter(s)but is unable todo so at early )29
promoters. If the same factor(s) is involved in
late transcription in vivo, it must either be
stabilizedinsomewayorbesynthesized contin-uously, since raising the temperature to 42 to
45°C decreases the burst size ofwild-type 4)29
by only 50% (38). It is known that the
antiter-minator N protein is unstable in vivo at 37°C
(15). Apparently, continuous synthesis is
re-quiredtomaintain Ninlambda-infectedE.coli, since in the presence of chloramphenicol the proteinhasahalf-life ofonly2min (15).
Mangel andChamberlin (27) have shown that
theinteraction betweenE.coliRNApolymerase
and phage T7promoters is temperature
depen-dentand thatpolymerase is unabletoformopen
promotercomplexesattemperatureslower than
15 to20°C. Similar observations have been made
withB.subtilisRNApolymerase andearly SP82
promoters except that the delta-less form of
RNApolymerasewasabletoinitiatesomeRNA
synthesis at temperatures below 20°C (2). The finding that delta-containing B. subtilis RNA
polymerase is unable to transcribe early 4)29
sequences at20°C probablyreflects the inherent
inability of polymerase to separate the DNA
strands atlow temperatures. Since polymerase
was able to initiate synthesis of late RNA at
20°C, however,possiblythe,29 gene4product,
a basic protein (11, 29), can act as a DNA
"melting protein" and assist in separating the
DNA strands to form open complexes at late
promotersites. Asimilar rolehas beenproposed
forthe mot geneproduct, which controls middle
T4RNAsynthesis in vitro (9,10).Alternatively,
it may be speculated that the gene 4 product
interacts with RNApolymerase to permit
bind-ingatlate promoter sites.
ACKNOWLEDGMENTS
This research was supported by Public Health Service grants GM-20784 andGM-26100 from the National Institute of GeneralMedical Sciences. H.R.W. is a recipient of Research Career Award K6-GM-442 from the National Institute of General Medical Sciences.
LITERATURE CITED
1. Achberger, E. C., and H. R.Whiteley. 1980. The interac-tion of Escherichia coli core RNA polymerase with speci-ficity-determining subunits derived from unmodified and SP82-modified Bacillussubtilis RNApolymerase. J. Biol. Chem. 255:11957-11964.
2. Achberger, E. C., and H. R. Whiteley. 1981.The role of the delta peptide of theBacillus subtilis RNA polymerase in promoterselection. J. Biol. Chem. 256:7424-7432. 3. Anderson, D. L., D. D. Hickman, and B. E.Reilly. 1966.
Structure of Bacillus subtilisbacteriophage +29and the length of +29 deoxyribonucleic acid. J. Bacteriol. 91:2081-2089.
4. Anderson, D. L., and B. E. Reilly. 1974. Analysis of bacteriophage +29 gene function: protein synthesis in suppressor-sensitivemutantinfection of Bacillus subtilis. J.Virol. 13:211-221.
5. Anderson, D. L., and B. E.Reilly.1976.Analysis ofgene function in Bacillus subtilis+29,p. 254-274. In D. Schles-singer (ed.),Microbiology-1976. AmericanSociety for Microbiology,Washington,D.C.
6. Bradford, M. M. 1976. Arapid and sensitive method for quantitation ofmicrogram quantities of proteinutilizing the principle of protein dye binding. Anal. Biochem. 72:245-254.
7. Carrascosa, J. L., F. Jimenez,E.Vinuela,andM.Salas. 1975. Synthesis in vitro of429-specific early proteins directed by phage DNA. Eur. J.Biochem.51:587-591. 8. Davison,B.L., C.L.Murray,andJ. C. Rabinowitz. 1980.
Specificityof promoter site utilizationin vitroby bacterial RNApolymerases onBacillus phage429DNA: transcrip-tion mapping with exonuclease III. J. Biol. Chem. 255:8819-8830.
9. deFranciscis, V., and E. Brody. 1982. Invitro system for middle T4 RNA. I. Studies with Escherichia coli RNA polymerase.J.Biol. Chem. 257:4087-4096.
10. deFranciscis, V., R.Favre, M. Uzan, J. Leautey, and E. Brody. 1982. In vitro system for middle T4 RNA. II. Studies with T4-modified RNA polymerase. J. Biol. Chem. 257:4097-4101.
11. Escarnds, C.,and M. Salas.1982.Nucleotide sequence of the early genes 3 and 4of bacteriophage 4)29. Nucleic Acids Res. 10:5785-5798.
12. Fox,T.D.,andJ. Pero. 1974.NewphageSPOl-induced polypeptidesassociated with BacillussubtilisRNA
poly-merase.Proc. Natl. Acad.Sci.U.S.A.71:2761-2765. 13. Geiduschek,E.P., and J. Ito. 1982.Regulatory
mecha-nisms in thedevelopment oflytic bacteriophagesin Bacil-lus subtilis,p. 203-245. In D. Dubnau(ed.), Molecular biologyof bacillus. Academic Press, Inc.,NewYork. 14. Goldbach, R. W., R. F. Evers, and P. Borst. 1978.
Electrophoretic strand separation of long DNAs with poly (U,G)inagarosegels. Nucleic AcidsRes.5:2743-2754. 15. Greenblatt, J.1973.Regulation of the expression of theN
gene of bacteriophage lambda. Proc. Natl. Acad. Sci. U.S.A.70:421-424.
16. Harding,N.E., J.Ito, and G. S. David. 1978. Identifica-tionoftheproteinfirmlybound to the endsof bacterio-phage4)29DNA.Virology84:279-292.
17. Holland, M.,andH. R.Whiteley.1973. RNApolymerase fromBacillusamyloliquifaciens infected with+29 bacte-riophage.Proc. Natl. Acad.Sci. U.S.A. 70:2234-2237.
on November 10, 2019 by guest
http://jvi.asm.org/
702 HOLDER AND WHITELEY
18. Hwang, J., and R. H. Doi. 1980.Transcription termination factor rho from Bacillus subtilis. Eur. J. Biochem. 104:313-320.
19. Inciarte, M. R., E.Vifiuela, and M. Salas. 1976. Transcrip-tion in vitro of+29DNA and EcoRI fragments by Bacillus subtilis RNA polymerase. Eur. J. Biochem. 71:77-83. 20. Kawamura, F., and J. Ito. 1977. Transcription of the
genome ofbacteriophage+29: isolation and mapping of the major early mRNAsynthesized in vivo and in vitro. J. Virol.23:562-577.
21. Ketner, G.,and T. J. Kelly. 1976. Integrated simian virus 40 sequences in transformed cell DNA: analysis using restrictionendonucleases. Proc. Natl. Acad. Sci. U.S.A. 73:1102-1106.
22. Lawrie, J. M., G. B. Spiegelman, and H. R. Whiteley. 1976. DNA strandspecificity oftemporal RNA classes producedduring infection of Bacillus subtilisbySP82. J. Virol. 19:359-373.
23. Lawrie, J. M., G. B. Spiegelman, and H. R. Whiteley. 1977.Localization oftranscripts producedin vivoandin vitro on phageSP82 genome.Gene 2:251-262. 24. Loskutoff, D. J., J. J. Pene, and D. P. Andrews. 1973.
Geneexpressionduringthedevelopmentof Bacillus subti-lis bacteriophage +29. I.Analysis ofviral-specific
tran-scriptionbydeoxyribonucleic acid-ribonucleic acid
com-petitionhybridization. J.Virol. 11:78-86.
25. Lowery-Goldhammer, C., and J. Richardson. 1979. An
RNA-dependent nucleoside triphosphate phosphorylase (ATP-ase)associated with rho termination factor. Proc. Natl. Acad.Sci. U.S.A. 71:2003-2007.
26. Lucas, J., and H. Ginsberg. 1971. Synthesis of virus-specific ribonucleic acid in KB cells infected with type 2 adenovirus. J. Virol. 8:203-213.
27. Mangel,W. F.,andM.J. Chamberlin. 1974. Studies of ribonucleic acid chain initiationbyEscherichiacoli ribo-nucleic acid polymerase bound to T7deoxyribonucleic acid.I.Anassayfor therateandextentofribonucleic acid chain initiation. J. Biol. Chem. 249:2995-3001. 28. Mellado, R. P., F. Moreno, E.Vinuela, M. Salas,B.E.
Reilly, and D. L. Anderson. 1976. Genetic analysis of bacteriophage +29 ofBacillus subtilis: integration and mapping ofreferencemutantsoftwocollections. J. Virol. 19:495-500.
29. Mellado,R.P.,and M. Salas.1982.Highlevelsynthesisin Escherichia coli of the Bacillus subtilisphage 4)29proteins p3 and p4 under the control ofphagelambdaPLpromoter. Nucleic Acids Res.10:5773-5784.
30. Mizuno,S.,andH.R.Whiteley.1968.Nuclear fraction of Bacillussubtilisas atemplatefor ribonucleic acid synthe-sis. J. Bacteriol. 95:1221-1237.
31. Mosharrafa, E., C. Schachtele, B.E. Reilly, and D. L.
Anderson. 1970. Complementary strands of bacteriophage +29 deoxyribonucleic acid: preparative separation and transcriptionstudies. J. Virol. 6:855-864.
32. Murray,C. L., and J. C. Rabinowitz. 1982. Nucleotide sequences of transcription and translation initiation re-gions in Bacillus phage4)29early genes. J. Biol. Chem. 257:1053-1062.
33. Reilly,B. E., R. A. Nelson, and D. L. Anderson. 1977. Morphogenesisof bacteriophage4)29of Bacillus subtilis: mapping and functional analysis of the head fiber gene. J. Virol. 24:363-377.
34. Roberts, J. W.1969.Termination factor for RNA synthe-sis. Nature(London) 224:1168-1175.
35. Salas, M., R. P. Mellado, E. Vinluela, and J. M. Sogo. 1978.Characterization of a protein covalently linked to the 5'termini of the DNA of Bacillus subtilis phage4)29.J. Mol. Biol. 119:269-291.
36. Schachtele, C. F., C. V. DeSain, and D. L. Anderson. 1973. Transcription during thedevelopment of bacteriophage 4)29: definition of "early" and "late" 4)29 ribonucleic acid. J. Virol. 11:9-16.
37. Schachtele, C. F., C. V. DeSain, L. A. Hawley, and D. L. Anderson. 1972.Transcription during the development of bacteriophage4)29:production of host- and 4)29-specific ribonucleic acid. J. Virol. 10:1170-1178.
38. Schachtele, C. F., R. W.Oman,and D. L.Anderson. 1970. Effect of elevated temperature on deoxyribonucleic acid synthesisinbacteriophage +29-infected Bacillus amyloli-quifaciens. J. Virol. 6:430-437.
39. Sogo, J. M., M. R. Inciarte, J. Corral, E.Vinuela,and M. Salas. 1979.RNApolymerase binding sites and transcrip-tion map of the DNA of Bacillus subtilis phage429.J. Mol.Biol. 127:411-436.
40. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separated by gel electrophoresis. J. Mol.Biol. 98:503-517.
41. Spiegelman, G. B., and H. R. Whiteley. 1974. In vivo and in vitro transcription by RNA polymerase from SP82-infected Bacillus subtilis. J.Biol. Chem. 249:1483-1489. 42. Splegelman,G.B., and H. R. Whiteley. 1978.
Bacterio-phage SP82 induced modifications of Bacillus subtilis RNApolymerase result in the recognition of additional RNAsynthesis initiation sites on phage DNA. Biochem. Biophys. Res. Commun. 81:1058-1065.
43. Yamamoto, K., and B. Alberts. 1970.Rapid bacteriophage sedimentation in the presence ofpolyethylene glycol and itsapplicationtolarge-scale viruspurification. Virology 40:732-744.
44. Yoshikawa,H., and J. Ito. 1982. Nucleotide sequence of themajor early region ofbacteriophage429.Gene 17:323-335.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/