JOURNAL VIROLOGY, Apr. 1989, p. 1514-1524 Vol. 63, No. 4 0022-538X/89/041514-11$02.00/0
Copyright ©1989, American SocietyforMicrobiology
An
Element
of
the
BK
Virus Enhancer
Required
for
DNA
Replication
ALFRED M. DEL VECCHIO, RICHARD A. STEINMAN, ANDROBERT P. RICCIARDI* WistarInstitute ofAnatomyandBiology, 36th at Spruce Street, Philadelphia, Pennsylvania 191044268
Received 28September 1988/Accepted7December 1988
The human papovavirus BK virus contains three 68-base-pair (bp) repeats that act as transcriptional enhancers. Ananalysisofplasmids containingthe BK virusorigin revealed thatsequences within the 68-bp enhancer are required for DNA replication as well as transcription of the early promoter in COS-1 cells. Originswith asingle68-bp repeat replicatedasefficientlyas did thosewith threerepeatswhentransfected into COS-1cells. Replicationdid not occur in the absence ofenhancersequencesandcouldnotbe restoredby distal placement ofenhancers to enhancerless origins. However,as withsimian virus40, replicationin vitro was not dependent on thepresence ofany enhancersequences. Deletionanalysisshowed that replicationof BK virus origins was dependent on thepresenceofthefirst21 bp of theenhancercontiguouswith theA-T-rich stretch ofthe origin. This 21-bp element is referred to as the rep element. Although in combination with rep the remaining 47 bp of the enhancer appear to increasereplication by two-tofivefold, theyalone are notsufficient to support replication. Deletions orinsertions in the enhancerwhich didnotaltertherepelement hadnomajor effectonreplication. Site-directedmutagenesisof theSpl-likesite within therep element,theNF1sitepresent intheenhancer, or the NF1 siteinadjacentlate-sidesequences each reducedtranscription bytwo- tofivefold, but had noeffectonreplication, suggestingthatreplicationandtranscriptioncan beuncoupled.
The human papovavirus BK (BKV) was first isolated in 1971 by Gardner et al. from the urine of a renal transplant recipient on immunosuppressive therapy (18). Approxi-mately80% of the population of the world has antibodies to BKV(17,50).This virus appears to be acquired during early
childhoodand may persistthroughoutlife (6, 39).
Reactiva-tion of the virus byimmunosuppression has been associated with hemorrhagic cystitis (1). Although BKV can transform human cells (reviewed in reference 24) and BKV DNA has
been foundinavarietyof human tumors (13), its relationship tohuman cancers has not been clearly established.
The size and morphology of BKV are similar to those of otherpapovaviruses such as simian virus 40 (SV40), poly-omavirus (Py), and JC virus. BKV has a double-stranded, circular DNA genome (5, 196 base pairs[bp]in the Gardner strain) which is remarkably similar to that of SV40. Nucle-otide sequence homology between thetwo viruses is greater than 80% (49, 57). Both viruses have complex regulatory regions at their origins of replication from which early mRNAs encoding large and small tumor antigens and late mRNAs encoding viral capsid proteins(VP1, VP2,VP3, and Agno) are synthesized by host cell RNA polymerase II in
oppositedirections on opposite strands. Replication of BKV and SV40 DNA requires host cell DNA polymerases and viral T antigen. Replication of BKV and SV40 can be supported by the heterologous T antigen (5, 36, 38), and in
fact, SV40 T antigen has been shown to contact specific nucleotides within the three BKV T-antigen-binding sites
(46).
Althoughtheoverall arrangement of elements within the regulatory regions of SV40 and BKV is similar, there are critical differences. The regulatory region of both viruses from the early to late side begins with start sites for early transcription, three T-antigen-binding sites at the origin of
replication, an A-T-rich stretch, a set of transcriptional enhancers, and late transcription start sites. BKV and SV40
*Correspondingauthor.
diverge greatly with respect to their enhancer elements. SV40contains three 21-bptandem repeats situated between the A-T-rich stretch and the two 72-bp transcriptional en-hancers (52, 54, 58). Each21-bprepeatcontains twocopies of a G-C "box" sequence which forms the recognition
sequencefor thetranscriptionfactorSpl (14). One function of the 21-bp repeats is to augment transcription from both early and late promoters (2, 28). The72-bpenhancers binda number of different factors (i.e., AP-1, AP-2, and AP-3) involved intranscription(27,55). In contrast to SV40, BKV has aunique setof three 68-bp tandem repeats: the middle repeat hasadeletion of 18 nucleotides (nt) (16, 49).Similarto those of SV40, the BKV enhancers stimulate transcription
from the early promoter (11, 45) and can stimulate
transcrip-tion from heterologous promoters as well (11). It is likely thattheenhancerelementsplayacritical role indetermining the host range of these viruses (29, 43).
Papovaviral enhancers can augmentreplicationaswellas transcription. Replication in vivofrom the SV40 origin still occurs in the absence of either the 72-bp enhancers or the
21-bprepeatsbut isdramaticallyreduced when bothof these cis-acting elementsareeliminated (7, 22, 25, 34).Stimulation ofreplication bythe21-bprepeatsorthe72-bpenhancersor both occurs independently of their orientation, but is lost whentheseelementsareplaced distal to the A-T-richstretch (22, 32). However, neither the 21-bp nor the 72-bp repeats arerequiredtosupport SV40DNAreplication in vitro(34).
Replication ofPy is dependenton its enhancer element (8, 40, 53). A combination oftwo of the four nonhomologous subdomains withinthe Pyenhancer can supportreplication, suggesting that they are functionally redundant (53). More-over, either the SV40 enhancer or the immunoglobulin enhancer cansubstitute for the Py enhancer inreplication (4, 8). AlthoughMuller et al. (40)have shownthatPy enhancer sequences lose their ability to support replication if
posi-tioned further than 400 bp from the origin, others have suggestedthatstimulation ofPyreplication bySV40 enhanc-ers cantake placeover much greaterdistances (8).
1514
on November 10, 2019 by guest
http://jvi.asm.org/
Inthisstudywereveal thatthe BKVenhancer isrequired for the replication of plasmids containing a BKV origin in COS-1 cells. A BKV origin plasmid with a single 68-bp
enhancer replicates as well as a plasmid containing three enhancers. Deletion analysis showed that replication re-quires that the first 21 bp of the enhancer becontiguouswith theA-T-rich stretch. Distal placement of theBKVenhancers failedto restore replication. Ananalysis ofpoint mutations
within the enhancer suggests that the replication and
tran-scription functions ofthe BKV enhancer canbe separated.
MATERIALS AND METHODS
Plasmidconstructions. Plasmids p33-1 andp34-2, contain-ing the entire genomesoftheGardnerandDunlop strains of BKV, respectively, cloned into theBamHI site of pBR322
were used as parental constructs. The HindlIl C origin fragments of Gardner and Dunlop were cloned into the HindIII site of puc 19 to generate
pBKori(Gardner)
and pBKori(Dunlop), respectively (see Fig. 1). PlasmidpBKcorBKen+L(D) was constructed by digesting pBKori
(Dunlop) with Bsu361 and religating the large fragment. PlasmidpBKcorBKen wasobtainedbyfilling inthe endsof
the Hinfl fragment (BKV, nt 3284 to
3519),
using Klenow,andcloning into theSmiaI site ofpuc 19. Inthisconstructthe
late side of the enhancer is next to the KpnI site of the polylinker; plasmid pBKcorBKen(R) contains the identical fragment cloned into puc 19 in the reverse orientation.
pBKcorBKen, containing a single enhancer, served as the
parental plasmid for all deletion and insertion constructs
described below.
Aseries ofplasmids with deletionsonthe late sideofthe
enhancer were generated from pBKcorBKen (see Fig. 3). Deletion plasmids pA47L,
pA43L,
pA38L,
pA22L, and pA19L were created bydeleting
from the NcoI, BsmI, or Bsu36I site in the BKV enhancer to the NdeI site in the vector,followed by eithermungbean nucleasetreatment orfilling in with Klenow. Another deletion ofthe enhancer,
pA56L, was created by cloning the Hinfl-AluI fragment
(BKV nt 3284 to 3465) into the SmaI site ofpuc 19. For
convenience,
nt3456inthe enhancerwaschanged
fromAto Tby site-directed mutagenesis (31), transforming aHaeIII site intoaStul site.Digestion
withStul,
whichseparatesthe enhancer from the A-T-richstretch,
was used to generatepBKcor, which is devoid of any of the
68-bp
enhancer sequences;pBKcor contains BKVnt3284to 3453cloned intheSmaIsite ofpuc19.A26-bp deletion fromthe
early
side of the enhancer wasconstructedusing
thisengineered
StuI site; pA26Edeletes BKVnt3454to3480. Aseries ofinternaldeletions
and insertions in the enhancer ofpBKcorBKen
weregeneratedutilizing unique restriction sites NcoI, BsmI, andBsu36I in the enhancer. Plasmid pAN-M(-) deletes nt 3475 to 3504; pAI-N(-) deletes nt 3476 to
3482;
pAI-Bdeletes nt 3480 to 3486;
pAI-M(-)
deletes nt 3501 to 3503.Plasmid pI-N(+) inserts CATG between nt 3479.and 3480; pAB-M inserts 51 bp between nt 3484 and
3504;
pI-M(+)
deletes nt3501to 3508 and inserts a G.
Plasmids containing distally placed enhancer elements were constructed from replication-defective plasmid
pMLA56L,
which containsthesameorigin
aspA56L cloned intopMLtet (35). The HaeIIIfragment containing
the BKV enhancers servedastheinsertin allconstructs (seeFig. 7).
In pBKen(A) the enhancers were inserted into theBamHI
site, placing
them 372 bp from their naturalposition.
InpBKen(E) the enhancers reside 3.7 kilobases from their natural location. In
pBKenTag(E)
the enhancers abut theearly boundary
ofT-antigen-binding
siteIattheStuIsite,
94bp
from their naturalposition.
PlasmidpMLBKcorBKen
+L(Gardner)
contains a BKVorigin
with asingle,
intact68-bp
repeat and late-side sequences from the Gardnerstrain.
Plasmid
pBKcorBKen+L(G)CAT
wasusedasthe paren-tal construct for site-directedmutagenesis
ofthe enhancer andwasmade frompBKCAT-1 (5) by
digesting completely
with Bsu36Iand
religating
thelarge
fragment.
All constructs were
propagated
inEscherichia
coli XL-1(3)
andpurified by
tworounds ofequilibrium
centrifugation
oncesium chloride
gradients.
Allconstructs wereverifiedby
dideoxy-chain
terminationsequencing
(47)
ofthe BKV in-serts,using
thedouble-strandedplasmid
DNAas atemplate
(30).
Cells and transfections. COS-1 cells
(20)
were grown inDulbeccomodified
Eagle
medium with7.5% fetal calfserumand
gentamicin.
Cells under passage 10 were seeded 24 hbefore transfectionata
density
of6 x105
cellsper 100-mmdish. Cells werecotransfected with 5 ,ugofthetest
plasmid
and5 ,ug ofaninternal standard
plasmid (pBR325
orpRSV
P-globin) by
calciumphosphate
precipitation,
followedby
a60-s15%
glycerol
shock(21).
The inclusion ofanonreplicat-ing
internalstandardplasmid
wasused to correctfor differ-ences in transfectionefficiency.
DNA extractions and Southern
blotting.
At 48 h aftertransfection,
cells were washed threetimes and DNA wasextracted
by
the method ofHirt(23).
DNA was extractedonce with
phenol
and twice with chloroform and ethanolprecipitated.
DNA was treated with DNase-freeRNase(20
,ug),
and halfofthesample
wasdigested
withDpnI
(10
U)
and an
appropriate
restriction enzyme that linearized the transfectedplasmid;
the otherhalfwasdigested
withMboI(10 U)
and PstI. To ensurecomplete
digestion,
reactions wereincubated for 16to20hat37°C.
Digests
wereelectro-phoresed
through
0.8% agarosegels
and transferred ontonitrocellulose
by
the methodofSouthern(51).
TheHindIII
Cfragment
ofBKV(Dunlop)
was used asthe nick-translatedprobe
onbothDpnI
and MboI blots.Additionally,
theMboIblots were
simultaneously
probed
with either theEcoRV-SaIlI
fragment
ofpBR325
or with theHindIII-BgIH
B frag-ment ofpRSV
P-globin.
Blots werehybridized
in 50% formamide-1x Denhardt solution-4x SSC(lx
SSC is0.15MNaCI
plus
0.015 M sodiumcitrate)-0.2% sodiumdodecyl
sulfate,
washedat65°C
in0.2% sodiumdodecyl
sulfate-0.2xSSC,
andvisualizedby
autoradiography
at-80°C.
Intensi-ties ofreplicative
bands werequantitated
by
scanning
den-sitometry
and normalized to theintensity
of the internal standard bands.In vitro
replication
assay conditions. In vitroreplication
assays were
performed
as describedpreviously (33, 34).
HeLa cells grown in
suspension
were used to make cellextracts
(5
,ug ofprotein
per,ul,
determinedby
Bio-Radassay).
Reactions(30
,ul)
included 2 ,ug ofimmunoaffinity-purified
SV40
large
Tantigen (Molecular
Biological
Re-sourcesInc.),
0.1 p,g ofDNAtemplate,
1pul
of[a-32P]dCTP
(specific activity,
3,000
Ci/mmol),
and 80 ,ugofextract. After incubation at37°C
for 4h,
DNA was isolated. Halfof thesample
wascodigested
for 6 h with 10 UofDpnI
and 10 UofEcoRI,
and the other halfwasleftundigested.
Bothsamples
wererun on1.5%agarose
gels
and thenair driedonNuBond film(FMC
Corp.)
at37°C
andautoradiographed
at-80°C.
The linearDpnI-resistant
bands were excised from thegel
and
quantitated
by
counting
in scintillationfluid.CAT assays.
Chloramphenicol
acetyltransferase
(CAT)
activity
was determined aspreviously
described(21).
Inon November 10, 2019 by guest
http://jvi.asm.org/
I 11 II I
MAf(
H11dI 50 1 68 I. .1
I If III
Hindll IATI 68 l 50 l 68
HZIII
d2b
HindIII
pBKcorBKen+L(D)
I 11 II
Hind III FA *'
I
| Ill Hint
FA
I;F
-68-Hint
pBKcorBKen
pBKcor Hnt
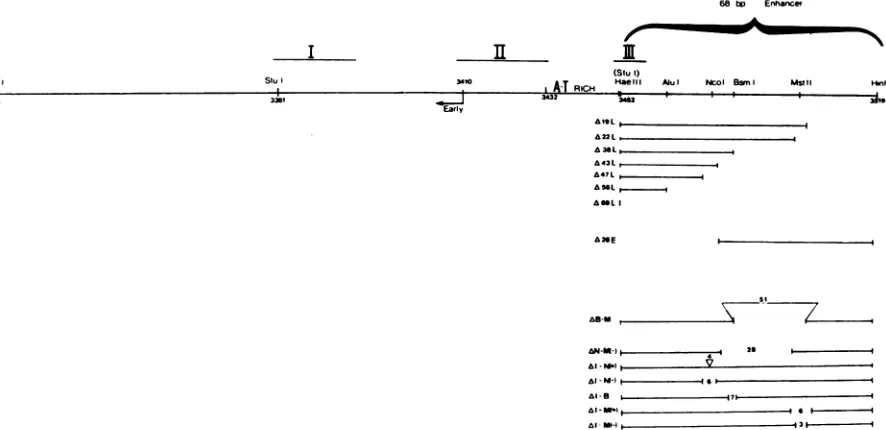
FIG. 1. Diagram of BKV origins fromprototypeviralstrains andsingle-enhancerderivatives. ShownaretheGardner[pBKori(Gardner)]
and Dunlop [pBKori(Dunlop] origins containingthe early (E) transcriptionstart site,T-antigen-binding sites (I, II, andIII),an A-T-rich
stretch, three enhancerrepeatsof68, 50, and68bp,and late(L) transcriptionstartsites. The Gardner strain containsa42-bpstretchadjacent
tothelate side of enhancers that isnot presentinDunlop. This stretch includes the30-bpelementc,which augments early transcription. Single enhancerorigins that either include late-sidesequences[pBKcorBKen+L(D)]ordelete them(pBKcorBKen)werederived from the
Dunlop strain. Also shown is the enhancerless BKVorigin plasmid pBKcor.
each reaction an equal amount of protein from the trans-fected cellextractswas used. Following thin-layer chroma-tography, acetylated forms of ['4C]chloramphenicol were
quantitated by scintillation counting.
Site-directed mutagenesis. Oligonucleotide site-directed mutagenesis was performed by themethod of Kunkel (31).
Oligonucleotides (18-mers) were obtained from the Wistar Institute DNAsynthesisfacility. Mutations were confirmed
byDNA sequencing (47).
RESULTS
Multipleenhancers and late-sidesequences aredispensable
in replication. We and others have previously shown that plasmids containing the complete BKV origin replicate in transfected cell lines which provide 13KVorSV40 T antigen
and that the BKV early promoter is functional in such plasmids (5, 36). To define cis elements involved in the replication ofBKV, a series of origin constructs (Fig. 1)
were tested for their ability to replicate in COS-1 cells, an
African Green monkey kidney cell line which has been transformed by replication-defective SV40 and supplies endogenous SV40Tantigen(20). Replicationwasmonitored
48 haftertransfection by observing theappearanceof either
MboIorDpnIendonuclease-generated bands.Inputplasmid DNAismethylated, making it sensitivetoDpnIbut resistant to the isoschizomer MboI. Conversely, plasmid DNA that hasundergone replication in mammalian cells is nonmethy-lated, making it resistanttoDpnIand sensitivetoMboI. To quantitatively adjust for differences in transfection efficiency
among samples, the intensity of the replicative bands was
normalizedtothat of the nonreplicative internal standard. Replication of plasmids containing the complete origins from the prototype Gardner and Dunlop strains ofBKV is demonstrated by the DpnI-resistant and MboI-generated
bands in Fig. 2 [pBKori(Gardner) and pBKori(Dunlop)]. Both plasmids contain three T-antigen-binding sites, the three enhancerrepeats of 68,50, and 68 bp, andastretchof
sequencesonthe late side of the last enhancerrepeat. They differonly in that the Gardner strain containsanadditional
42-bp stretch thatjuxtaposesthethird enhanceron thelate
side(49, 57) (Fig. 1). This stretch,notpresentin theDunlop strain,waspreviouslyshowntocontainanelementdenoted "c," which augments early transcription (11). Plasmid pBKcorBKen+L(D), which contains a single 68-bp
en-hancerandlate-sidesequencesfrom theDunlopstrain (Fig. 1), replicated as well as the Dunlop and Gardnerplasmids containing three enhancers (Fig. 2). Since late-side se-quencescaninfluencetranscription (e.g., regionelementcof
theGardner strain), it was importantto know whetherany
late-side sequencescould also influencereplication. Plasmid pBKcorBKen, which contains a single 68-bp enhancer but
nolate-side sequences (Fig. 1), replicatedinCOS-1 cellsas
wellas the Gardner andDunlop plasmids (Fig. 2). Replica-tion was not altered when the orientation of this single enhancer origin was reversed in plasmid pBKcorBKen(R) (Fig. 2) orwhen differentvectorswereused (notshown). It
is noted that when pBKcorBKen and all of itsderivatives
were digested with eitherMboI after transfection orDpnI beforetransfection,one or moreminor bandscorresponding to partial digestion productswere seen. This was observed
reproducibly with different plasmid preparations and
ex-tended times of digestion. Thus, replication of a plasmid
containingasingleenhancer andnolate-sidesequencesisas
efficient as one containing three enhancers with late-side
sequences.
Therepelementof the BKVenhancer isrequiredforDNA replication. To examine the involvement of the enhancer
sequencesinreplication,aseriesofdeletionswereproduced onthe late side of thesingleenhancer ofpBKcorBKen (Fig.
pBKori(Gardner)
pBKori(Dunlop)
Hind III
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.85.532.76.303.2]Mbo
IQ a +
c o c: c W
mm c co
R N aL a a m
J-0
C -a +
3l
-C0 ci
cO { a
Ac8
m m mL g o co o
ci 0 0
vj y Yc Y Sl
m m m m m
N R CL aL
40
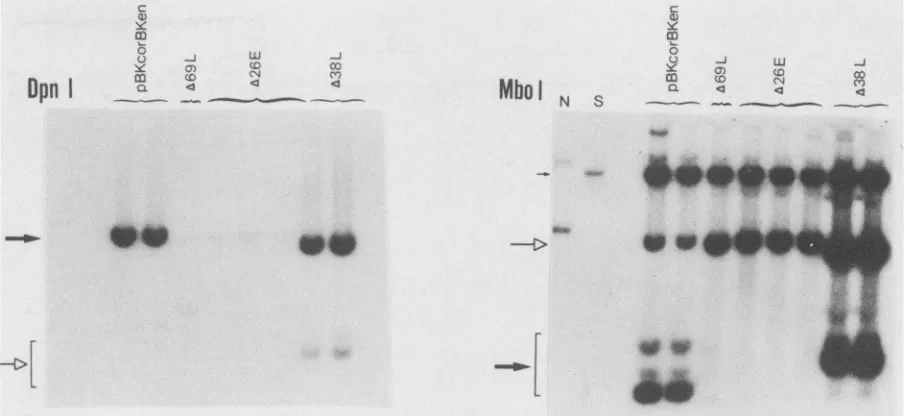
FIG. 2. Replication of plasmids containing multiple- and single-enhancer repeats. BKV plasmids andanonreplicative internal standard
plasmid (pRSV 3-globin) were transfected into COS cells, and Hirt DNAwas extracted after 48 h. Half of the Hirt DNA from each
transfectionwascodigested with DpnI and BamHI and the other halfwasdigested with MboI and PstI and analyzed by Southern blotting.
Both DpnI and MboI filterswereprobedwith the HindIll C origin fragment of BKV of the Gardner strain;additionally, the MboI filterwas
coprobed with theEcoRV-SaIlfragment of pRSV 3-globin. Bandsindicative of replicationaredenoted byaboldarrow,unreplicated input
plasmid bandsare indicated byan open arrow,and internal standard DNA is indicated by athinarrow. To better visualize the internal standard bands in the MboI blot, the film was overexposed. Lanes with marker DNAs representing the positions of replicative (R) and
nonreplicative (N) bands areshown.
3) and tested in COS-1 cells. Sequential deletion of the enhancer resulted inadecreaseinreplication (Fig. 4 and 5).
Whencompared with thesingle-enhancerconstruct pBKcor-BKen, replication of plasmids
p419L,
pA22L, and pA38L, which eliminate 19, 22, and 38 bp from the late side of the enhancer,wasreproducibly reduced by 40to50%(Fig. 4 and 5). It is noted that the difference in the sizes of the MboI-generated bands in Fig. 4 is due to slight size differencesin the plasmids as well as the loss of an MboI site in theconstruction of the deletions.Replication of plasmids pA43L andpA47L, which delete 43 and 47 bp of the enhancer,was
reduced in bothcasesby 50to80%(Fig. 4). Neither pA56L, which deletes 56 bp of the enhancer, northe enhancerless
plasmid pA69L (same as pBKcor) exhibited any detectable
replication (Fig. 4 and 5). To more precisely map specific sequence requirements for replication, plasmid pA26E,
which deletes 26bp from the early side of the enhancer,was
constructed (Fig. 3). pA26E exhibited no apparent
replica-68bP Enhancer
I
Stu
I
(StuI) ,, RICH Haelli AJuI
A-TRICF1 Ncol Bsni mstli Hnfl
Eat0
A38L6
A43L, A47L
A"L
A26E
5M
AB- IISI ,_<
6N-10-i 4 28
Al-Nia V
At-MN)AI-B i 17 6 * -l
Al-P()1 06 i
FIG. 3. Diagram oforiginswith deletions and insertions inthe BKV enhancer. All constructs werederived from the parental plasmid pBKcorBKen (top)and containT-antigen-bindingsitesI, II,andIII,andtheA-T-rich stretch. Deletionsof theenhancer madefrom theearly side(AE), from the lateside(AL),orinternally (Al)areshown. Deletion and insertion sizesaregivenin basepairs.
Dpn I
--Op-L
H.nf
3264 331
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.137.471.78.264.2] [image:4.612.84.527.473.688.2]1518 DEL VECCHIO ET AL.
c
0
X -J -J _j - -4 -4
m C(Cn N CO N
a Co L) e e N
N R <.d 4 c 4c 4
Dpn
Im
U -j -4 -4 -4
0 -a N CO
n < 4 4
-. 'ww W
:, * ,4X4 .# * *
I..:
....
J.s
*_.N.
:41,..._.
loR A_.l
_mi
_ _ *
[A
:.~~~~~~~~~..,.0
,;.z.
[image:5.612.83.551.74.261.2]*~~~~~~~~~~~~~~~
--
!
FIG. 4. Replication of BKV origin plasmids with deletions and insertions in the BKV enhancer. Constructs depicted in Fig. 3 were
transfected into COS-1 cells, and Hirt DNAwasanalyzedasdescribed in thelegendtoFig. 2. Marker bands representing the positions of replicative (R) and nonreplicative (N) bandsareshown. Arrowsarethesame asinFig. 2.
tion, similar to the enhancerless plasmid pA69L (Fig. 5). Together these results demonstrate that thefirst21bpofthe
enhancercontain an element essential forBKV replication.
We refer to these 21 bp on the early side of the BKV enhanceras the "rep" element.
Constructs shown inFig. 3, which deleted large internal regions [pAN-M(-)] or small regions [pAI-M(-), pMI-B,
pAlI-N(-), and pAI-M(+)]or createdlarge insertions
(pAB-M)or small insertions [pAI-N(+)]in the enhancer, reduced replication by only 5 to 10% (Fig. 4 and 6). These findings confirmed the fact that a critical function for replication is
containedwithintherepelementandthatalterationsoutside
this element have no major effect onreplication.
Addition-ally, these results show that stereospecific alignment
be-Dpn
I
c '3) CO
CO
-J w
CD (0 co
(3 N
4 < 4e
tween different sections of the BKV enhancer does not
appeartobe crucial forreplication.
These results demonstrate that a single, complete BKV
enhancer is required for optimal replication of the BKV origin in COS-1 cells and that at least the first 21 bp of the enhancerarerequiredforreplicationtooccur.Althoughthe remaining47bpof theenhancerappeartoincreasethelevel of replication by two- to fivefold in conjunction with rep,
they alone arenot sufficient to supportreplication.
Distal placement of the BKV enhancer fails to restore replication. It was important to know whether replication could be restored by the distal insertion ofcomplete BKV enhancersinto the replication-defective plasmid pMLA56L, which contains the BKV origin with 12 bp of the enhancer
c
0)
y:D
MboIN S
-J LUI 0) (0 N
40
0O
40
S. _-ls
WI'
.1_W WF
&A
FIG. 5. Replication of BKV origin plasmids with deletions and insertions in the BKV enhancer. Constructsdepicted in Fig. 3 were transfected into COS-1cells,andHirtDNAwasanalyzedasdescribed in the legendtoFig. 2. Bands indicativeof unreplicated input DNA
forpA69LandpA26EontheDpnI blotwereseenafteroverexposureof the film. Marker bands representingthe positions of internal standard
(S)andnonreplicative (N) bandsareshown. Arrowsarethe sameasinFig. 2.
Mbo
I
2
-4 -4
N z mO z
N4 ,r a 4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.89.549.475.683.2]m
co
0
C-) m
NR Q
I j - 1-- 17
m z - - -
-CX CX C Ct CX <
--4
-K
.a
FIG. 6. Replication of BKV origin plasmids with internal alter-ations producingchanges in the stereospecific alignment of the BKV enhancer.ConstructsdepictedinFig. 3 were transfected into COS-1 cells, and HirtDNA wasanalyzedbySouthernblotting as described in the legend to Fig. 2. The DpnI blot is shown. Marker bands representing the positions ofreplicative (R) and nonreplicative (N) bands areshown. Arrows arethe same as in Fig. 2.
juxtaposedto the A-T-rich stretch (Fig. 7). In pBKen(A) the enhancer insert is positioned 2,969 bp from its natural
position. InpBKen(E) the enhancer insert is positioned on the early sideof the regulatory region, 198 bp from its natural
location, and inpBKenTag(E) the enhancer insert abuts the early boundary ofT-antigen-binding site I, 94 bp from its
1 2 3 4 5
-0
naturalposition.Theextentof plasmid replication48 hafter transfection into COS-1 cellswasdetermined bythe appear-ance of MboI bands. The control plasmid pMLBKcor-BKen+L(Gardner), which contains an intact
single-enhancerorigin, produced the expected MboI band (Fig. 7, lane 1), whereas pMLA56L (Fig. 7, lane 2) did not. Each
plasmid with a distally inserted BKV enhancer failed to
produce an MboI band, indicating that replication was not restored (Fig. 7, lanes 3 through 5). Ofnote is the fact that the distalplacement of the SV40 enhancer in pMLA56L also failed to augment replication (not shown). These results
clearly indicate that the ability of the BKV enhancer to supportreplication isposition
dependent.
Point mutagenesis of factor-binding sites in the BKV en-hancer reduced transcription but not replication. Wewanted todetermine whether therequirement of the BKV enhancer inreplicationislinkedtoitsabilitytostimulatetranscription.
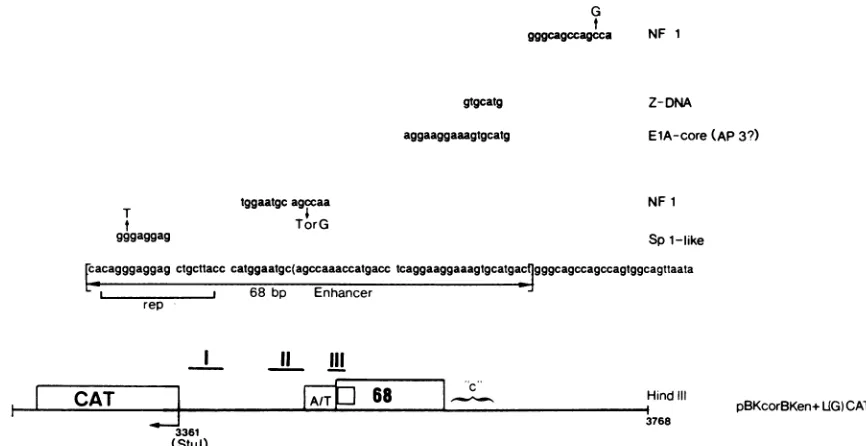
Depicted in Fig. 8 are the landmark elements of the BKV
enhancer, which eitherarebindingsitesfor cellularproteins
orhave been shown to be important in the transcription of othergenes. These include a sequencefor an Spl-like site
(14), an NFI site (11, 12, 37, 42), anadenovirus EIA core
(11,45), andastretch of DNAwhich couldpotentiallyform aZ-DNAstructure(41). Since enhancersappear tofunction through the binding of factors involved in transcription, we generated point mutations in the known NFI and the puta-tive Spl factor-binding sites to determine whether these elements play distinct roles in transcription andreplication.
We mutated the Spl-like site (GGGAGGAG to GTGAG GAG), which is present in the rep element required for replication. In SV40, replication and transcription are both stimulated by the G-C-rich 21-bp-repeat elements which bind Spl (14, 15, 19). Althoughourdeletionanalysisshowedthat the NF1 site in the enhancer is notabsolutely required for
E
L
E
L
[image:6.612.76.278.75.264.2]E
L
E
L
FIG. 7. Analysis of BKVorigin plasmids containing replication-defective enhancers and distallyinserted BKV enhancers. Constructs depictedwereanalyzedforreplicationby MboIdigestionasdescribed in thelegendtoFig.2.pMLBKcorBKen+L(Gardner) (lane 1)contains
aBKVoriginwithasingle complete 68-bprepeat and late-sidesequences;nonreplicative pMLA56L (lane 2)containsaBKVoriginwith 12 bpof the enhancer. Insertion of BKV enhancers intopMLA56L:372bponthe lateside of theorigin generatedpBKen(A)(lane 3);3.7kbaway
generated pBKen(E)(lane 4);abuttingT-antigensiteIgeneratedpBKenTag(E)(lane 5).Arrowsarethesame asinFig.2. E,Earlyside;L, late side.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.110.503.432.664.2]1520 DEL VECCHIO ET AL.
T
gggaggag
tggaatgcagccaa
TorG
G
gggcagccagcca NF 1
Z-DNA
ElA-core (AP 3?)
NF 1
Sp 1-like
rcacagggaggag ctgcttacccatggaatgc(agccaaaccatgacc
68bp Enhancer
rep
I
CA
IIICAT I1 rIAIT7.68 Hind IlIl
3768 3361
(Stu I)
pBKcorBKen+ UG) CAT
FIG. 8. Diagram of BKV enhancer landmarks and specific pointmutations. Pointmutationsshownweremade in theSpl-likesite andthe
NF1sites present inboth the enhancer and region c ofparental plasmid pBKcorBKen+L(G)CAT. The small box in the 68-bp enhancer
represents therep element. Nonmutated landmark sites includethe adenovirusElAcore and Z-DNA elements. ShownaretheA-T-rich
stretch andT-antigen-binding sites I, II,andIII.TheCATgenewasusedtomonitortranscriptionfrom theBKVearlypromoter.
BKV replication, we choseto mutatethis site (TGGAATG CAGCCAA to TGGAATGCAG[G]CAA) and examine how these alterations affect both replication and transcrip-tion inthecontextof thecomplete enhancer. NF1, whichis known to bind to the BKV enhancer (12, 37, 42) and is required for the replication of adenovirus DNA (9, 44), has also beenshowntobe theCAAT-binding protein involvedin thetranscription of certaingenes(26). We also mutated the
NF1 site (TGGGCAGCCAGCCAGT to TGGGCAGCCAG
GCAGT),
which ispresent inthe 30-bp regionc onthe latesideof theenhancer, eventhoughwehave shownthat these
late-sidesequences aredispensable for replication. Toassay
bothreplication andtranscription, theBKV earlypromoter of the single-enhancer origin was linked to the CAT gene.
Figure 8 depicts this parental origin plasmid, pBKcor BKen+L(G)CAT, and the specific nucleotide changes which
weremadeeither singlyorincombination in the Spl-likeand
both NF1 sites. These constructs were transfected into
COS-1cells,and halfofthecells fromeachtransfectedplate
were assayed for CAT activity and the other half were
assayedforreplication.
Table 1 summarizes the transcriptional activity obtained witheach of the site-specific point mutants. In all casesthe
level of CAT activity of the point mutants was reduced
relativetothat of the parentalplasmid, pBKcorBKen+ L(G). Thesingle-base change produced intheSpl-likesite reduced transcription by 81%. This finding supports the prediction thatthe BKVSpl-like elementmaybind transcriptionfactor
Spl, especially sinceinSV40this base has been showntobe protected fromdimethyl sulfate methylation by thebinding ofSpl (19) and since transitions atthis position have been shown to reduce SV40early transcription (54). Mutation of thebase inthe enhancerNF1 site, which isacontactpoint forNF1binding and which isaconserved base in CAAT box sequences (10, 26), caused CAT activity to be reduced by 50%. Thisresultsuggestsapotential involvementofNF1 in
the transcription of the BKV early promoter and is in agreement with therecentfindings of others(11). Consistent
with the findingthat region c augments earlytranscription,
mutation of the NF1 site present in this element caused CAT
activitytobe reducedby25%. Thisfindingsuggeststhat the stimulation of transcription by region c could involve the
binding of an NF1/CAAT-binding protein. As expected, combinations of mutations in the above sites resulted in
comparable reductions in CATactivity. These results show that the Spl-likeandNF1 sitesareinvolved intranscription of the BKV early promoter. Furthermore, the nature of these mutations suggests that these sequences potentially
function intranscription throughthebindingof thepredicted
factors.
Theabilityof thepointmutants toreplicateinCOS-1 cells is shown inFig.9. Surprisingly,allof thepointmutants were able to replicate as well as the parental plasmid. It is
particularly interesting that theNF1 mutationhad noeffect onreplication (Fig. 9, lanes 5through 12) sinceanidentical mutation in the NFI site of adenovirus prevented DNA
[image:7.612.91.524.67.290.2]replication(44). From this result and those obtained from the deletion analysis, we conclude that the NF1 site does not play amajorrole in BKV replication. Of key importance is
TABLE 1. Quantitationofreplicationandtranscription ofBKV site-specificmutants"
Replication Transcription
(%)
(CATactivity[%o)
None(parental) 100 100
Spl 97 19 12
NF1(T) enhancer 93 54± 23
NF1(G) regionc 88 25± 11
SpI + NF1(G)enhancer 106 17± 13
BothNF1 sites 88 32 + 27
" LevelsofreplicationandCATactivityof the mutants areexpressedas a percentage oftheparental plasmid. The intensityofreplicativebands was normalized to the internal standard. The range of CAT activity is noted. Resultsarebasedonthedetermination obtainedfrom three tofive
experi-ments. gtgcatg
aggaaggaaagtgcatg
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.324.560.600.681.2]_
1
2
-C
-C~~~~~U L
- -a - ° C
X Q z-c cn
LL o
zt
.D
3 4
5 6 7 8 9 10 11 12
6e.U ..
FIG. 9. Replication of BKV origin plasmids containing point mutations in the enhancer. Constructs depicted in Fig. 8 were
transfected into COS-1 cells, and Hirt DNA was analyzed by
Southernblottingasdescribed in thelegendtoFig. 2. Locations of thepoint mutations made in theenhancer of the BKV origin plasmid depicted in Fig. 8arenoted abovelanes. Constructsweretestedin
duplicate. The MboI blot is shown. Positions of replicative and nonreplicative bandsareshownasinFig. 2. Arrowsarethesameas
inFig. 2. WT, Wildtype.
the fact that mutations in the Spl and NFI sites which greatly reduced transcription had no effect on replication
(Fig. 9). To rule out the possibility that the point mutants
werealtered in theirrates ofreplication such thatatthe end ofa48-htransientassay they could reach thesame level of
replication as plasmids with thewild-type origin, we
exam-ined replication at 24, 36, and 48 h posttransfection. The replication rate of the mutated origins wasthe same as that
of the wild-type origin, further demonstrating that these mutations had no effect on replication (data not shown). These findings suggest that the requirements for the BKV enhancer in replication aredifferent from those involved in
transcription.
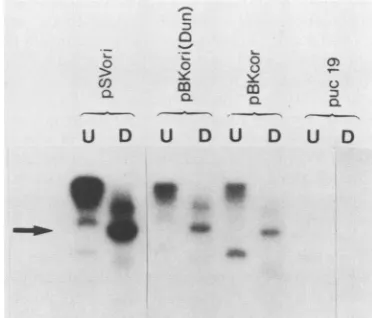
Replication ofBKV in vitro doesnot requireany enhancer sequences.It has beenshownthatreplication ofSV40invivo is dependent on the presence of either the 21-bp-repeat
elements or the 72-bp enhancer, but neither of these
ele-ments is required for replication in vitro (33, 34). We examinedwhether the BKVenhanceris alsodispensable for invitro replication. PlasmidpBKori(Dunlop), containing the complete origin of BKV with three 68-bp enhancers, and pBKcor, an enhanceless plasmid, were assayed for their
abilitytoreplicate invitro. Invitro replication of BKV does not requireany enhancer elements since pBKcor and pBK
ori(Dunlop) replicated tovirtually thesameextent(Fig. 10). It isnoted thatreplicationwasdependentonthepresenceof
largeTantigenintheextract(notshown)and that the BKV origin replicatedlessefficientlythan did theSV40 origin. The negative control, puc 19, failed to replicate. Thus, as with
SV40, replication of BKV in vitro does not require the
presenceofanyenhancerelements.
DISCUSSION
The datapresented here show that the 68-bp enhancer of BKVisdirectlyinvolved in DNAreplication in COS-1 cells. Althoughanorigin containingonecopyofthe68-bprepeatis able to replicate equally well as one that contains three
repeats, the elimination of all enhancer sequences
com-
1-L a
U D - DU
C/) co ma
US D
UJ
D UDX
0)
U D
FIG. 10. Invitroreplication ofBKVorigins.Anin vitro replica-tionsystemcontainingaHeLacellextract,immunoaffinity-purified SV40Tantigen, and BKVoriginconstructs wasincubatedat37°C for4h.Halfofthe32P-labeledproductswerecodigestedwithEcoRI andDpnI. Both the digested (D) and undigested (U) samples were electrophoresed through 1.5%agarose gels, dried, and autoradio-graphed. Plasmids representedare pBKori(Dunlop),a BKVorigin with three enhancers; pBKori, an enhancerless BKVorigin; pSVori, an SV40 origin-positive control; and puc 19, a negative control. Replicative bandsareindicatedby the arrow.
pletely abolishes replication. Replication from BKV origins occurs when only the first 21 bp of the enhancer are juxtaposed to the A-T-rich stretch. We refer to these 21 bpof the BKV enhancerasthe rep element.Deletion of 9bp ofthe rep elementfrom the late side reduced replication to unde-tectable levels, but origins containing only the rep element could replicate at 20 to40% of the level oforigins with a complete enhancer. Juxtaposing the remaining 47 bp ofthe enhancertotheA-T-rich stretch in the absence of rep would not support replication. The distal placement of a complete enhancer to an origin with a partially deleted rep element could not restore replication, demonstrating a positional
requirementof the rep element. Additionally, internal dele-tionsorinsertions in sequences outside the rep element that
changedthe helical register in the enhancer had little or no effectonthe levelofreplication,furtherdemonstrating that they are nonessential for replication. It has been similarly shown that stereospecific alignment within the BKV en-hancer is notcritical fortranscriptionof theearly promoter (12). Furtheranalysis is needed to delineate the boundaries of the rep element and critical nucleotides within this ele-ment which are essential for replication. Additionally, al-though the known variants of BKV differ with respect to their enhancer sequence arrangements (18, 49, 57), the element thatwedefinedasrepis conserved inall sequenced variants.
In contrast to the results obtained in vivo, enhancerless BKV origins were able to replicate in vitro. Using immu-noaffinity-purified SV40 T antigen and a HeLa cell extract capable ofsupporting replicationinvitro,wefound thatthe enhancerless origin could replicate tothe samelevel as an origin containing three enhancer repeats. Thus, whatever function theBKVenhancerservesin stimulatingreplication in COS-1 cells is notrequiredforreplicationin vitro.
Itis important tocompare these results on BKV
replica-tion with those obtained withSV40 and Py.ReplicationofPy
is dependent on the presence of its enhancer element up-stream of the site of initiation of replication. The Py
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.57.293.79.245.2] [image:8.612.337.523.81.240.2]hancer contains genetically defined subdomains a and 3, either of which must be juxtaposed next to the origin for replication to occur. These elements function in a position-dependent, but orientation-inposition-dependent, manner (40). Addi-tionally, heterologous enhancer elements such as the 72-bp repeats of SV40 can substitute for the Py enhancer in replication of Py (4). Analogous to the dependence of Py replication on the positions of the a and a elements, repli-cation of BKV in COS-1 cells absolutely requires the pres-enceof the rep element juxtaposed to the A-T-rich stretch; it remains to bedetermined whether the rep element exhibits a similar orientation independence. Replication of SV40
re-quires the presence of either the G-C-rich 21-bp repeats or the 72-bp enhancers. Deletion of both of these elements prevents replication in vivo; however, replication in vitro canoccur in their absence (34). Similarly, as noted above, replication of BKV origins in vitro does not require any enhancer sequences. In SV40,juxtaposing either the 21-bp repeats or the 72-bp enhancer elements to the A-T-rich stretch supports replication equally well. Incontrast, repli-cation ofBKV in vivo absolutely requires the presence of
the rep element juxtaposed to the A-T-rich stretch.
Juxta-posing the remaining 47 bp of the BKV enhancer to the A-T-rich stretch does not restore replication. This demon-strates aspecific requirementfor theseparticular21bp ofthe BKVenhancer,which is in sharp contrast to thereplication
requirements of SV40.
Through site-directed mutagenesis of the Spl-like and NF1 sites of the 68-bp repeat, as well as the NF1 site in
region c, we were able to reduce the level oftranscription
without affecting replication. Mutation of the Spl-like and
both NF1sites reduced the level oftranscription bytwo- to fivefold, but had no effect on replication. Biochemical and
geneticdata suggest that both the enhancer andregioncbind NF1 since purified NF1 willfootprint in theseregions (37).
Our mutation in the NF1 site inregion c,previously shown toreduce NFIbindingtotheadenoviraloriginofreplication
by 10-fold (44), reduced transcription from the BKV early
promoterby 4-fold. Itisinterestingthat the mutationin the NF1 site in region c appeared to exert a greater effect on transcription that did the mutation which affected the NF1 site in the enhancer, eventhoughthemutation in this latter sitewasestimated to reduce NF1bindingto asyntheticNF1 siteby 250-to400-fold(48). Ourresultsonthereplication of
the point mutants indicate that the binding of NFI to the BKV enhancer is not involved in replication. Indeed, the genetic analysis clearly shows that replication can occur despite deletionormutation of both the NFI site in the68-bp
enhancerand the NF1 site ofregion c. Additionally,
plas-mids with the 21-bp rep element deleted which have the
remaining 47 bp of the enhancer containing the NF1 site
juxtaposed to the A-T-rich stretch did not replicate.
Simi-larly,webelieve thatfactor bindingtotheSpl-like sitein the rep element is not critical for replication. In fact, it is important to note that there are somewhat conflicting
find-ings as to whether a factor(s) actually does bind to the
Spl-likesite (12, 37). Markowitz and Dynan (37) showed that neither HeLa cell proteins from heparin-agarose column fractions nor affinity-purified Spl from HeLa cells binds to the Spl-like site in the BKV enhancer. However, Deyerle andSubramani(12)found weak protection of this site, using heparin-agarose-purified HeLa cell protein fractions. It is
possible that afactor other than Spl bindshere or that the
binding of Spl requires interaction with other factors. In agreementwith our study, linker-scanning mutation analysis
by DeyerleandSubramani(12) showed that mutations in the
Spl-like site caused a fivefold decrease in
transcription.
Thus, although the element that we defined as rep doescontain an
Spl-like site,
oursite-directedmutagenesis
sug-geststhat thebinding
ofSpl
tothe enhancer isnotinvolved in BKVreplicationinCOS-1 cells. Finally,purified
AP-1hasrecently been shown to bind to the BKV enhancers atthe
junctions
ofthe68-bp
repeats(37).
Since anorigin
with asingle
68-bprepeatwhichcontainsonly
halfofthe AP-1 site is abletoreplicate
asefficiently
as anorigin
with three68-bp
repeats, we
postulate
that thebinding
of AP-1to the BKV enhanceris notrequired
forreplication.
In the natural
situation,
BKV uses its own Tantigen
toreplicate
its DNA inahuman host. It shouldbe noted that thesestudiesweredoneusing
asimiancelllinethatproduces
SV40 Tantigen. Although
thefirstBKVisolatewaspropa-gated in
monkey (Vero)
cells(18),
cis elementsrequired
forreplication of BKV could
differ,
depending
on thespecies
from which the cell line was derived or the available T
antigen.
Wefavor the
hypothesis
that themechanismby
whichthe BKVenhancerstimulatestranscription
is different fromthe mechanism by which it augmentsreplication.
Ouranalysis
shows that elementsofthe BKV enhancer definedas
impor-tant fortranscription
were not essential forreplication.
These results are critical not
only
inunderstanding
how elementsclassically
definedastranscriptional
enhancerscan augmentreplication,
but also indistinguishing
between elements of the BKV enhancer involved inreplication
ortranscription.
The repelement thatwedefinedascritical forthe
replication
from BKVorigins
could function eitherby
binding
acellular factor involved in DNAreplication
orby
assuming
acertain DNA conformationrequired
forreplica-tion. Since rep appears to be
dispensable
forreplication
invitro,
it may beinvolved indetermining
chromatinstructure. Forexample,
repcouldpreventhistonesfrombinding
totheorigin
and allow thereplication
machinery
better accessto theorigin.
Since chromatin structure doesnot appeartobeimportant
invitro,
repwouldnotbe needed.Alternately,
repcould binda
specific
factorrequired
fortheinitiationofDNAreplication (56).
Replication
in vivo wouldrequire
thebind-ing
ofacellular factortorepfor theefficient formationofaninitiation
complex; however,
in vitroproteins
such as Tantigen
andpolymerase
ao-primase
could form an initiationcomplex
on naked DNA without the aid of aputative
rep-binding
protein.
The greatest
divergence
among thepapovaviruses BKV,
SV40, and JC virus occurs in their enhancerregions
(16).
This may in part
explain
their hostspecificities
and tissuetropisms.
Since theseviruses have evolvedcomplex
controlregions
with theorigin
ofreplication
overlapping
theearly
and late promoters, it is often difficult to examine one
function ofthis
region
withoutperturbing
another. We havegenetically
defined elements of theBKV enhancerinvolved in DNAreplication
and suggest that the role of the enhancer inreplication
canbeuncoupled
from its role intranscription.
Further
analysis
of the ciselementsandtrans-acting
factors involved in theregulation
of thisregion
maynotonly
provide
a model for the mechanism of enhancer action in bothreplication
andtranscription,
but alsohelp
inunderstanding
howBKV
regulates
its lifecycle.
ACKNOWLEDGMENTS
Wethank PeterHowley(NationalInstitutesof Health
[NIH])
for the generous gift ofplasmids p33-1 and p34-2; Ed Harlow (ColdSpringHarborLaboratory)forpAb419hybridoma cells;JimAlwine (UniversityofPennsylvania)andthemembersof hislaboratoryfor J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
COS-1 cells and helpful discussions; Mark Wold and David Wein-berg for reagents, protocols, and helpful advice about in vitro replication; and Rhea-Beth Markowitz for communicating results
priortopublication.
A.M.D.wassupported byPublic Health Service virology training
grantAI-07325 from the National Institutes of Health; R.A.S. was
supported by the Medical Scientist Training Program and Public
Health Service traininggrantCA-09171 fromtheNational Institutes
ofHealth. This workwassupported by Public HealthServicegrant
CA-44960toR.P.R. from the National Institutesof Health.
LITERATURE CITED
1. Arthur, R. R., K. V. Keerti, S. J. Baust, G.W.Santos,and R.S.
Saral. 1986.Association of BK viruria with hemorrhagic cystitis in recipients of bone marrow transplants. N. Engl. J. Med.
315:230-234.
2. Baty, D., H. A. Barrera-Daldana, R. D. Everett, M. Vigneron,
and P. Chambon.1984. Mutational dissection ofthe 21 bprepeat
region of the SV40 early promoter reveals that it contains
overlapping elements oftheearly-early and late-early promot-ers. Nucleic AcidsRes. 12:915-932.
3. Bullock, W.O., J. M. Fernandez, and J. M. Short. 1987. XL
1-Blue:ahigh efficiency plasmid transformingrecAEscherichia coli strain with beta-galactosidase selection. Biotechniques 5:
376-379.
4. Campbell, B. A., and L. P. Villarreal. 1985. Host species specificity of polyomavirus DNA replication is not altered by
simian virus 40 72-base-pair repeats. Mol. Cell. Biol.
5:1534-1537.
5. Caputo, A., G. Barbanti-Brodano,E. Wang, and R. P. Ricciardi.
1986. Transactivation of BKV and SV40 early promoters by
BKV andSV40 T-antigens. Virology152:459-465.
6. Chesters, P. M., J. Heritage, and D. J. McCance. 1983.
Persis-tence of DNA sequences ofBK virus and JC virus in normal
human tissues and in diseased tissues. J. Infect. Dis. 147:
676-684.
7. DeLucia, A. L., S. Deb, K. Partin, and P. Tegtmeyer. 1986.
Functional interactions of the simian virus 40 core origin of
replication with flanking regulatory sequences. J. Virol. 57:
138-144.
8. de Villiers,J., W. Schaffner, S. Lupton, and C. Tyndall. 1985.
Enhancerfunction is essential for polyomavirus DNA replica-tion. Nature (London) 312:242-246.
9. de Vries, E.,W.vanDriel, M. Tromp, J. vanBoom, andP. C.
van der Vliet. 1985. Adenovirus DNA replication in vitro:
site-directedmutagenesis of the nuclear factorIbinding site of the Ad2origin. Nucleic AcidsRes. 13:4935-4952.
10. de Vries, E., W.vanDriel,S.J. L.vanden Heuvel, and P.C.van
der Vliet. 1987. Contactpoint analysis of the HeLa nuclear
factor Irecognition site reveals symmetrical bindingatoneside
ofthe DNAhelix. EMBO J. 6:161-168.
11. Deyerle, K. L., J.A.Cassill,andS. Subramani. 1987. Analysis ofthe early regulatory region of the human papovavirus BK.
Virology 158:181-193.
12. Deyerle, K. L., and S. Subramani. 1988. Linkerscananalysis of
the early regulatory region of the human papovavirus BK. J.
Virol. 62:3378-3387.
13. Dorries, K., G. Loeber, and J.Meixensberger.1987.Association ofpolyomavirus JC, SV40, and BK with humantumors.
Virol-ogy160:268-270.
14. Dynan, W. S., and R. Tjian. 1983. The promoter-specific
tran-scription factor Spl bindsto upstream sequences in the SV40 earlypromoter. Cell 35:79-87.
15. Everett,R. D., D. Baty, and P. Chambon. The repeated GC-rich
motifsupstreamfrom the TATA boxareimportant elements of
theSV40 earlypromoter. Nucleic Acids Res. 11:2447-2464.
16. Frisque, R. J., G.L. Bream, and M. T. Cannella. 1984. Human
polyomavirus JC virusgenome. J. Virol. 51:458-469.
17. Gardner, S. D. 1973. Prevalence in England of antibody to
humanpolyomavirus (B.K.). Br. Med. J. 1:77-78.
18. Gardner, S. D., A. M. Field, D. V. Coleman, and B. Hulme. 1971. New human papovavirus [BK] isolated from urine after renaltransplantation. Lancet i:1253-1257.
19. Gidoni, D., W. S. Dynan, and R. Tjian. 1984. Multiple specific contacts between a mammalian transcription factor and its cognate promoters. Nature (London) 312:409-413.
20. Gluzman, Y. 1981. SV40-transformed simian cells support the replication of earlySV40 mutants. Cell 23:175-182.
21. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant genomes which express chloramphenicol acetyl-transferase in mammalian cells. Mol. Cell. Biol. 2:1044-1051. 22. Hertz, G. Z., and J. E. Mertz. 1986. Bidirectional promoter
elements of simian virus 40 are required for efficient replication of the viral DNA. Mol. Cell. Biol. 6:3513-3522.
23. Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. 24. Howley, P. M. 1980. Molecular biology ofSV40and thehuman
polyomavirusesBK and JC, p. 489-514. In G. Klein (ed.), Viral oncology. Raven Press, New York.
25. Innis, J. W., and W. A. Scott. 1984. DNA replication and chromatin structure of simian virus 40 insertion mutants. Mol. Cell. Biol. 4:1499-1507.
26. Jones, K. A., J. T. Kadonaga, P. J.Rosenfeld,T. J. Kelly, and R. Tjian. 1987. A cellular DNA-binding protein that activates eukaryotic transcription and DNA replication. Cell 48:79-89. 27. Jones, N. C., P. W. J. Rigby, and E. B. Ziff. 1988. trans-acting
protein factors and the regulation ofeukaryotic transcription: lessons from studies on DNA tumor viruses. Genes Dev. 2:267-281.
28. Keller, J. M., and J. C. Alwine. 1985. Activation of the SV40 latepromoter: direct effects of T antigen in the absence of viral DNAreplication. Cell 36:381-389.
29. Kenney, S., V. Natarajan, D. Strike, G. Khoury, and N. P. Salzman. 1984. JC virus enhancer-promoter active in human brain cells. Science 226:1337-1339.
30. Kraft, R., J. Tardiff, K. S. Krauter, and L. A. Leinwand. 1988. Using miniprep plasmid DNA forsequencing double stranded templates withSequenaseM. Biotechniques 6:544-547. 31. Kunkel, T. A. 1985. Rapid and efficientsite-specificmutagenesis
without phenotypic selection. Proc. Natl. Acad. Sci. USA 82:488-492.
32. Lee-Chen,G.-J., and M.Woodworth-Gutai. 1986. Simian virus 40 DNA replication: functional organization of regulatory ele-ments. Mol. Cell. Biol. 6:3086-3093.
33. Li, J. J., and T. J. Kelly. 1984. Simian virus 40 DNA replication in vitro. Proc. Natl. Acad. Sci. USA81:6973-6977.
34. Li, J. J., K. W. C. Peden, R. A. F. Dixon, and T. Kelly. 1986. Functional organizationof the simian virus 40 origin of replica-tion. Mol. Cell. Biol. 6:1117-1128.
35. Lusky, M., and M.Botchan. 1981.InhibitionofSV40replication in simian cells by specific pBR322 DNA sequences. Nature (London) 293:79-81.
36. Major, E. O., and P. Matsumura. 1984. Human embryonic kidney cells: stable transformation with an origin-defective simian virus 40 DNA and use as hosts for humanpapovavirus replication. Mol. Cell. Biol.4:379-382.
37. Markowitz, R., and W. Dynan. 1988. Binding of cellular proteins to the regulatory region of BK virus DNA. J. Virol. 62: 3388-3398.
38. Mason, D. H., Jr., and K. K.Takemoto.1976.Complementation between BK human papovavirus and a simian virus 40 tsA mutant. J. Virol. 17:1060-1062.
39. Milanesi, G., G. Barbanti-Brodano, M. Negrini, D. Lee, A. Corallini, A. Caputo, M. P. Grossi, and R. P. Ricciardi. 1984. BKvirus-plasmid expression vector thatpersistsepisomallyin human cells andshuttles into Escherichiacoli.Mol. Cell. Biol. 4:1551-1560.
40. Muller, W. J., C. R. Mueller, A.-M. Mes, and J. A. Hassell. 1983. Polyomavirusorigin for DNA replicationcomprises mul-tiplegenetic elements. J. Virol. 47:586-599.
41. Nordheim, A., and A. Rich. 1983. Negativelysupercoiledsimian virus 40 DNA contains Z-DNA segments withintranscriptional enhancersequences. Nature(London) 303:674-678.
42. Nowock, J., U.Borgmeyer, A. W. Pukschel,R.A. W.Rupp, and A.E. Sippel. 1985. The TGGCA protein binds to the MMTV-LTR, the adenovirus origin of replication, and the BK virus
on November 10, 2019 by guest
http://jvi.asm.org/
enhancer. Nucleic Acids Res. 13:2045-2060.
43. Ondek, B., A. Shepard, and W. Herr. 1987. Discrete elements within the SV40 enhancer region display different cell-specific enhancer activities. EMBO J. 6:1017-1025.
44. Rosenfeld, P. J., E. A. O'Neill, R. J. Wides, and T. J. Kelly. 1987. Sequence-specific interactions between cellular DNA-binding proteins and the adenovirus origin ofDNAreplication. Mol. Cell. Biol. 7:875-886.
45. Rosenthal, N., M. Kress, P. Gruss, and G. Khoury. 1983. BK viral enhancer element and a human cellular homolog. Science 222:749-765.
46. Ryder, K., A. L. DeLucia, and P. Tegtmeyer. 1983. Binding of SV40 A protein of the BK virus origin of DNA replication. Virology 129:239-245.
47. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
48. Schneider, R., I. Gander, U. Muller, R. Mertz, and E. L. Winnacker. 1986. Asensitive andrapid gel retention assayfor nuclear factor I and other DNA-binding proteins in crude nuclear extracts. Nucleic Acids Res. 14:1303-1317.
49. Seif, I.,G.Khoury, and R. Dhar. 1979. The genomeofhuman papovavirusBKV. Cell 18:963-977.
50. Shah, K. V., R. W. Daniel, and R. M. Warszawaski.1973. High prevalence of antibodiestoBKvirus, an SV40-related papova-virus, in residents ofMaryland. J. Infect. Dis. 128:784-787. 51. Southern, E. 1975.Detectionofspecificsequences among DNA
fragments separated by gel electrophoresis. J. Mol. Biol. 98: 503-517.
52. Takahashi, K., M. Vigneron, H. Matthes, A. Wildeman, M. Zenke, and P. Chambon. 1986. Requirement of sterospecific alignments for initiation from the simian virus 40 early pro-moter. Nature(London) 319:121-126.
53. Veldman, G. M.,S. Lupton, and R. Kamen. 1985.Polyomavirus enhancercontains multiple redundant sequence elements that activate bothDNAreplicationand geneexpression.Mol. Cell. Biol.5:649-658.
54. Weiher, H., M. Konig, and P. Gruss. 1983. Multiple point mutationsaffecting the simian virus 40 enhancer. Science 219: 626-631.
55. Wildeman, A. G., M. Zenke, C. Schatz, M. Wintzerith, T. Grundstrom, H. Matthes, K. Takahashi, and P. Chambon. 1986. Specific protein bindingtothe simian virus 40 enhancer in vitro. Mol. Cell. Biol. 6:2098-2105.
56. Yamaguchi, M., and M. L. DePamphilis. 1986. DNAbindingsite forafactor(s) requiredtoinitiate simianvirus 40 DNA replica-tion. Proc. Natl. Acad. Sci. USA83:1646-1650.
57. Yang, R. C. A., and R. Wu. 1979. Comparative study of papovavirusDNA: BKV[MM],BKV[WT]andSV40. Nucleic AcidsRes.7:651-658.
58. Zenke, M., T. Grundstrom, H. Matthes, M. Wintzerith, C. Schatz,A.Wildeman,and P. Chambon.1986.Multiplesequence motifs are involved in SV40 enhancer function. EMBO J. 5:387-397.