Copyright C) 1985, American Society for Microbiology
Assignment
of
the Temperature-Sensitive Lesion in the Replicatio-n
Mutant
Al of Vesicular
Stomatitis
Virus
to
the
N
Gene
M. DAVIDMARKS,t JENNIFERKENNEDY-MORROW,t ANDJUDITH A. LESNAW* SchoolofBiological Sciences, University of Kentucky, Lexington, Kentucky 40506
Received 6 June1984/Accepted24September1984
The replication defect in the temperature-sensitive mutant Al of the New Jersey serotype (Hazelhurst subtype) of vesicular stomatitis viruswasconfirmedby the absence ofintracellular nucleocapsidsin infected cells incubated at the restrictivetemperature. After preamplification, the relative yield of the Al N protein accumulatedintracellularly after 1 h of incubationattherestrictivetemperaturewasdecreasedby50% that of thewild-typeorrevertantAl Nprotein. This differencewasnotasapparentinpulse-chase experiments. The functional lesion in Al was correlated with a structural alteration in the N protein on the basis of the thermolabilityof thetemplateactivityof the Al Nprotein-RNA complex in in vitro transcription reactions and thecovariance of thisphenotypewith thetemperature-sensitivephenotype inaspontaneousAl revertant.This correlation wasconsistent with adirect role of the Nprotein in replication and allowed the assignment of the Ngeneto complementation group A.
The set of 48 temperature-sensitive (ts) mutants isolated
from the New Jersey serotype (Hazelhurst subtype) of vesicular stomatitis virus (VSV) has been classified into six
nonoverlapping complementation groups (designated A
through F), each of which presumably represents a
func-tional unit on the viral genome (19). Because only five VSV geneproducts (L,G,M,NS, and N; all structural) have been
identified (23), the existence of six complementation groups
in the New Jersey collection has been of considerable
interest. We, as well as others, have been studying these
mutants with the objective of identifying viral functions
involved inRNA synthesis and correlatingthem with viral
proteins. RNA synthesis in VSV involves two distinct
pathways: transcription (the sequential synthesis of a
47-base"leader" sequenceencodedattheprecise3' endof the genome, followed by the capped, methylated, and
polyad-enylated monocistronicmRNAsfortheN,NS, M,G,and L
proteins) and replication (the synthesis ofa genome-length
positivestrand which, complexed withN protein, serves as
atemplate for the synthesis ofnew genomic RNA) (1). Of the six New Jersey complementation groups, four contain members whichdisplay an RNA-negative phenotypeat the restrictive temperature in vivo (19). Analyses of viral RNA
species synthesized in vivo at the restrictive temperature
suggested that mutants Al and El (the prototypes of
com-plementation groups A and E, respectively) were defective
inreplication,that mutant
Bi
(the prototype ofcomplemen-tationgroup B) wasdefective in primarytranscription, and
thatmutant Fl (theprototype ofcomplementation group F)
wasdefective in both transcriptionandreplication (12). The
transcription
defects
were confirmed by in vitro studies (2,10, 21).
In vitro reconstitution studies demonstrated that three of theviralproteins, N(the nucleoprotein) and L and NS(the two subunits of the viral encapsidated transcriptase), were necessary and sufficient for transcription (5). Presumably,
* Correspondingauthor.
tPresentaddress: Department of Plant Pathology, Purdue Uni-versity,West Lafayette, IN 47907.
tPresent address: Department of Biochemistry, University of Kentucky Medical Center, Lexington, KY40536.
these three proteins were also involved in the replication
pathway;therefore, thefour complementationgroups(A, B,
E, andF)displaying defects in RNAsynthesis were believed torepresentthe L, N, andNS genes. The extra complemen-tation group was postulated to represent a sixth, nonstruc-tural, virus-encoded protein involved in thereplication path-way (12).
Assignments have now been made for three of the four RNA-negative complementation groups. On the basis of in
vitro reconstitution experiments assayed in vivo, the ts
lesions in transcription mutants Bi and Fl have been as-signed to the L gene (2), and on the basis ofthe aberrant
electrophoretic mobility andpeptide map oftheNS protein
of thereplicationmutantElwith respecttothoseof the wild type(WT) and revertants, the NS gene has beenassignedto the E group (4, 11). The discovery that two of the four
RNA-negative groupsrepresented theLgeneeliminatedthe
necessity of invoking a yet-undetected sixth viral gene
product, and it seemedlikelythatcomplementationgroupA represented the N gene. Nevertheless, as a result of the initial failure to detect alterations in the Al N protein by
polyacrylamide gel electrophoresis and peptide mapping
(11), coupled with the high levels ofpolymerase activity (in excess of those in WT) in Al virions and transcribing
nucleocapsids assayedin vitroattherestrictive temperature (21), no assignment was made, and the possibility that
complementationgroupArepresentedasixth,nonstructural
protein was left open.
Inthe present communication,wepresentdatawhichnow
permit the assignment of the N gene and confirm the
assignment of a replication function to complementation
group A.
MATERIALSAND METHODS
Cell line and virus stocks. BHK-21 C13 cellswere propa-gated as monolayers as described previously (11), except thatthegrowth mediumwassupplementedwith2mM
L-glu-tamine. Mutants Al andEl and theWT (New Jersey sero-type, Hazelhurstsubtype)fromwhichtheyhadbeenderived (19) weregenerously provided by Craig Pringle. The spon-taneous revertants of El and Al were isolated as plaques
44
on November 10, 2019 by guest
http://jvi.asm.org/
that appeared atthe restrictivetemperature duringthe
titra-tion ofmutantenrichments.
In vivo viral nucleocapsid synthesis. Duplicate monolayers
were infected and incubated at 31 or39.5°C in thepresence
of [5,6-3H]uridine (specific activity, 35 to 50 Ci/mmol;New
England NuclearCorp., Boston, Mass.) andactinomycinD
(Calbiochem-Behring, LaJolla, Calif.). After6 h of
incuba-tion, the contents of each flask (including the overlaying
medium) were frozen, thawed, and separated by low-speed
centrifugation into supernatant (unbound-virion) and pellet
(bound-nucleocapsid) fractions. Virionspresent in the
super-natant (unbound fractions) were pelleted and analyzed by
density gradient centrifugation. Fractions were collected
and analyzed for radioactivity by liquid scintillation spec-troscopy. Viral nucleocapsids wereextractedfromthe
low-speed pellet (bound fractions) with 1% Nonidet P-40-2 M
urea and analyzed by sucrose density centrifugation.
Frac-tions were collected and analyzed as described above.
Additional details of the procedure are presented in
refer-ence6.
Intracellular viral protein synthesis and stability.Replicate
monolayers of5 x 106 cellswereinfectedat amultiplicity of
infection of25, and theinfection mixtureswerepreamplified
by incubationat31°C for3 h in the presenceofactinomycin
D(5,ug/ml). For thedetermination ofyields ofviralproteins
accumulated intracellularly, the medium was replacedwith
mediumcontaining10,uCi of
[35S]methionine
(specificactiv-ity, 600 to 1,400 Ci/mmol; Amersham Corp., Arlington
Heights, Ill.) and 0.01 mM unlabeled methionine (0.1 the
normalconcentration) per monolayer. Incubation wasthen
continued for 1h at either31 or
39.8°C.
Forthedetermina-tion of the stability of pulse-labeled viral proteins, the
medium was replaced with medium containing 10 ,uCi of
[35S]methionine
per monolayer, and the monolayers wereincubatedfor10 min(pulse) at theappropriate temperature
(31 or 39.8°C). A 10-fold excess of unlabeled methionine
(final concentration, 1 mM) was then added, and the
infec-tion mixtureswereincubatedatthe
labeling
temperatureforvarious periods of time (chase). Extracellular (overlaying
medium) and intracellular (monolayer) fractions were then
disrupted with sodium dodecyl sulfate (SDS) and analyzed
by SDS-polyacrylamide gel electrophoresis in gels
contain-ing 10%polyacrylamide and 0.13 or 0.5% bisacrylamide in
thediscontinuous buffersystem(9). Aspreviously reported
(4, 11),the El NS
protein
is anelectrophoretic variant. Todisplay the WT and El NS proteins on a single gel, we
increased the
bisacrylamide
concentration to 0.5%.Addi-tional details of thelabeling protocols, electrophoresis, and
fluorography can be found in reference 6. The data were
quantitated by determiningtheamountofradioactivity
con-tainedin eachoftheviralproteinbandsexcisedfromthegel
by liquid scintillation spectroscopy asdescribedpreviously
(6).
In vitro functional analyses. WT, Al, and revertant Al
virionswerepurifiedbydifferential and rate sucrose density
gradient centrifugationsfrominfections incubated for48 h at
31°C, and transcribing cores containing the L, NS, and N
proteins and RNA were prepared as described previously
(10). The procedure for the preparation of enzyme (L and
NS) and template (N and RNA) fractions (10) has been
modified. Transcribing cores at a protein concentration (14)
of 2 mg/ml in 0.1 M column buffer (0.1 M
NaCl,
25%glycerol, 10 mM Tris-hydrochloride [pH
8.01)
were treatedwithanequal volumeof 2x disruption buffer (4 M NaCl, 2% Nonidet P-40. 10 mM
Tris-hydrochloride
[pH 8.0], 2 mMdithiothreitol) for 2 h at
0°C.
Under theseconditions, LandNS proteins were released, and the templates became
floc-culated and could be removed by low-speed centrifugation
(11,100 xg,40C,30min). Thesupernatant(enzyme) fraction
wasfreed ofresidual templates byhigh-speedcentrifugation
(176,700 x g, 4°C, 3 h) and stored at -700C. Immediately
before use, the enzyme fractionsweredialyed against0.3 M
columnbuffer. ItwasnecessarytomaintainaNaCI
concen-tration above1 M during storage at -70°C toprevent the L
protein from
aggregating.
Template fractions obtaineddur-ingthelow-speedcentrifugation described abovewerefreed
of residualpolymeraseactivity by
being
dilutedto0.2mg/ml
in 0.1 M column buffer and retreated withanequalvolumeof
2x disruption buffer for2 h at0°C.Thetemplateswerethen
collected by low-speed
centrifugation
as describedabove,
resuspended in0.1 Mcolumn buffer, adjustedto0.5 mg/ml
(on thebasis ofprotein determinations),andstoredat-70°C
until used. Theproteincompositions ofthevarious fractions
weredeterminedbySDS-polyacrylamide gel
electrophoresis
in the discontinuous Tris buffer system (9) as described
previously (10). The residual polymerase
activity
ofthesetemplate preparations was 10% or less than the
activity
obtained afterreconstitution withthe enzyme fraction.
The thermostabilities ofthese fractions were determined
byheating thematvarioustemperaturesorfor various times
at agiven temperature before reconstitution
(in
the case ofenzyme and template fractions) with the
appropriate
un-heated WTfraction. Polymerase
activity
was thenassayed
at the optimal temperature for the reaction
(310C).
Theconditions forthe
transcription
reactionand assayonDEAEfilterpaper diskswere asdescribed
previously
(10).RESULTS
In vivo viralnucleocapsid synthesis.
Replication
defects inmutantsAl and
El,
the prototype mutants ofcomplemen-tation groups A and E ofthe New Jersey serotype
(Hazel-hurst subtype) of VSV (19), were suggested by the absence
of detectable
genome-sized
viral RNA in infected cellsincubated at the restrictive temperature (12). Because the
level of
genome-sized
RNA in WT VSV infections is verylow in relationtothelevelofmessage-sized RNA, analyses
of intracellular viral
nucleocapsids
have beenused toverify
replication defects suggested by the absence of
genome-sized RNA in mutant-infected cells
(22).
Although
thisapproach was successful in thecase ofanalyses of Indiana
serotype mutants, the failure ofthe
commonly
usedfreeze-thaw
technique
(22) toreleaseviralnucleocapsids
fromWTNew Jersey infections precluded the
application
of thisapproachtothe NewJersey serotype mutants. A
technique
fortheisolation of intracellularNew
Jersey
serotypenucleo-capsids
which is based upon the extraction offreeze-thaw-disrupted
cells with Nonidet P-40 and urea wasre-cently
developed
(6); we used thistechnique
toinvestigate
mutants Al and El. The absence of detectable
nucleocap-sidsinAl-andEl-infected cells incubated attherestrictive
temperature confirmed the
replication
defects in both mu-tants (Fig. 1).Intracellular viral protein synthesisand stability.
Knipe
et al. (8) have shown that certain altered viralproteins
arerapidly degraded at the restrictive temperature in vivo.
Therefore,
in anattemptto determine thealteredprotein
inAl,
weexamined theyieldsof viralproteins synthesizedand accumulated in Al-infected cells after 1 h ofincubation at the restrictive temperature, after preamplification. Visual observation of thefluorograms ofSDS-polyacrylamide gels
ofAl and control
samples suggested
adecrease in theyield
on November 10, 2019 by guest
http://jvi.asm.org/
5
p
4
0
1,
0r-16
14
162
'10
X Q
u
6r
UNBOUND
10
20
30 40
10
20 30 40
10 20 30 40
FRACTION
NUMBER
BOUND
1-F
0
10
20 30
40
10
20
30
40
10
20
30 40
FRACTION
NUMBER
FIG. 1. Sucrose density gradient analyses of[3H]uridine-labeled virions (unbound viral particles [A]) and nucleocapsids (bound viral particles [B]) synthesized in cells infected with WT (a), mutant El (b), or mutantAl (c) virus at 31°C
(O*-
) or 39.5°C(0----
-0). Fraction1 correspondstothebottomof the gradient.ofthe N protein relative to those of theother Al proteins
andcontrolproteins(Fig. 2).
Quantitationofthe datashowninFig.2confirmedthatthe
yield oftheAl Nprotein accumulated during a 1-h labeling
period at the restrictive temperature after preamplification
wasreducedby50%with respect toAl infections incubated
atthe permissive temperature and with respect to control
infections (WT, revertant Al, El and revertant El)
incu-batedat either temperature(Table 1). Intheseexperiments,
the overlaying medium (extracellular fractions containing
mature virions and shed proteins) was removed, and the
ittracetlularand extracellularfractionswereanalyzed
sepa-rately. Thisprocedureprovidedacontrol forpossible
differ-ewesinthe stabilitiesofintracellularviralproteins and viral
proteias associated with mature virions released into the
extracellular fraction in control infections. Moreover, it ensured thatany decrease in theyield of an intracellular viral
proteinwas nottheresultof increasedshedding(13). In the
caseofthe Alinfections incubatedattherestrictive temper-ature, no Nprotein appeared in thecorresponding extrace-Ilular fraction(Fig. 2B).
To ruleout thepossibility that the decrease in the relative accumulation of the Al Nproteinwas asecondaryeffectof theblock inreplication, mutant El, whichis also defective
in replication, was included in the preceding experiment.
Therationale was thatablock in 40S RNAsynthesis would preventnucleocapsid assembly,which inturncould resultin theaccumulation of WT N protein inan abnormal cellular compartment, where it could be degraded. Because El,
which codesfor analteredNS proteinbut a WT Nprotein,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.114.493.74.532.2]failsto synthesize40S RNA and therefore cannotassemble nucleocapsids at the restrictive temperature in vivo, it constituted the appropriate control. Indeed, the relative accumulation of the El N protein did not decrease at the restrictivetemperature(Table 1). It is interestingto notethat the relative accumulation of the altered El NS protein did also not decrease at the restrictive temperature (Table 1). This finding is inagreement with the earlierobservation by Knipeetal. (8) that notall alteredproteins aredegraded at the restrictive temperature.
The decrease in the relative accumulation of the Al N protein could have resulted from either its degradation orits reduced synthesis atthe restrictive temperature. We there-fore determined the stabilities ofWT, Al, andrevertantAl intracellular proteins pulse-labeled and chased for various time intervals at the restrictive and permissive (control) temperatures. Fluorograms of the gels are shown in Fig. 3. The data were quantitated and expressed as relative ther-mostabilitiestodetermine whetheradecrease in the relative yield ofa proteinat 39.8°C reflected thermolability of that protein. The relative thermostability of the Al N protein but notthose of the WT andrevertantAl Nproteins decreased slightly asthelength of the chase increased (Table 2).
0IE
L
G N NS
WT Al RAI El REI
V P R P R P R P R P R
-.- eO
V.- V
-MEW
M
0FI
WT Al RAI REI El RElV PR P R P R P R P R
L _
G _
N w
NS
M _
FIG. 2. SDS-polyacrylamide gel electrophoresisof WT and
mu-tant viral proteinsaccumulated intracellularlyat thepermissive or
restrictivetemperature.After 3 h ofpreamplificationatthe
permis-sive temperature, duplicate monolayers infected with WT, Al,
revertantAl(RAl), El,orrevertantEl(REl)viruswereincubated
for 1 h at31°C (P)or39.8°C (R)inthepresenceof[35S]methionine
and actinomycin D. The proteins in the extracellular fractions
(overlayingmedium[B])and intracellularfractions(disrupted
mono-layers [A])wereelectrophoresed throughSDSgels containing10%
acrylamideand0.5%bisacrylamide. Shownarefluorogramsofthe
driedgels. 14C-labeled marker virion proteins (V) are identifiedto the left of eachgel. Detailsof the infections andelectrophoresisare
[image:4.612.320.565.95.171.2]givenin the text.
TABLE 1. Temperature dependenceof relative yields of viral proteins accumulated intracellularlya
Relativeaccumulationb of indicated protein
Virus
L G N NS M
WT 86 119 107 84 106
Al 110 130 58 100 126
Revertant Al 86 126 105 85 102
El 89 111 96 96 117
Revertant El 84 117 106 95 100
aBandscorrespondingtothefive viral proteinswere cutfromeachlaneof
the gel containing the intracellular fractions (Fig. 2A) byusingthefluorogram
as atemplate. The.radioactivity in each bandwasdeterminedasdescribedin the text.
bRelative accumulation=(relative yieldat39.8°C/relative yieldat31°C) x
100, where relative yield at31°C =(cpminagiven protein/sumofcpm in all
proteins inthe sample) x 100.
In vitro functional analyses. Because the data suggested
that the N protein was a likely candidate for the gene
assignmentin complementationgroup A,we examined
mu-tant Al with an in vitro functional analysis designed to detect lesions in the three viral proteins (L, NS, and N)
required fortranscription(10). The rationale is based on the
role ofthe N protein-RNA complex as atemplate for both
replication andtranscription.Wereasoned thatalthough the
Al mutant wascapable ofdirecting primarytranscriptionat therestrictive temperature(39.5°C)invivo (12),analteration inthe N protein could affect the relative thermostability of
the N protein-RNA complex at elevated temperatures in vitro. The thermal inactivation could be detected by the
failure of the heat-treated complexes to direct in vitro
polymerization.
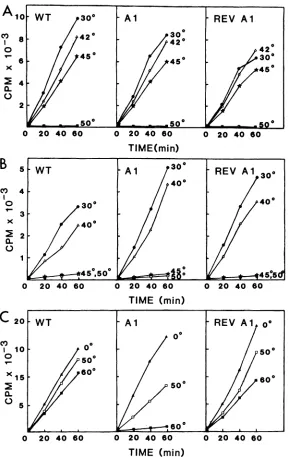
In thefirstsetofexperiments, thethermolabilitiesof WT,
Al, and revertant Al transcribing cores (L, NS, N, and RNA), enzymefractions(L andNS), andtemplatefractions
(Nand RNA)werecomparedas afunction oftemperatureof
inactivation. The thermostabilities ofall three transcribing
cores were comparable (Fig. 4A). Similarly, the three
en-zyme preparations were indistinguishable in this assay, in
agreementwith a previousreport (2). Whereas transcribing cores withstood heating for 15 min at 45°C before being
assayed for polymerase activity (Fig. 4A), the enzyme
fractions, when treated for 15 min at 45°C before being
reconstitutedwith the WTtemplate, wereinactive(Fig.4B).
As has been pointed out previously (10), the enzyme is
apparentlystabilized by its interactionwith the template in
transcribingcores. Incontrasttothetranscribing cores, the
ability of the WT template to direct polymerization was
retainedafter 15 minof heatingat60°C(Fig.4C). The greater
thermolabilityof thetranscribingcoresreflectedthatoftheir
least stable component, the enzyme (10). However, in contrast toboth WT andrevertantAltemplatefractions,the
Al template fraction lost50% ofits activity after 15 minof
incubationat50°Candwascompletelyinactivatedby 15 min
of incubationat 60°C (Fig. 4C).
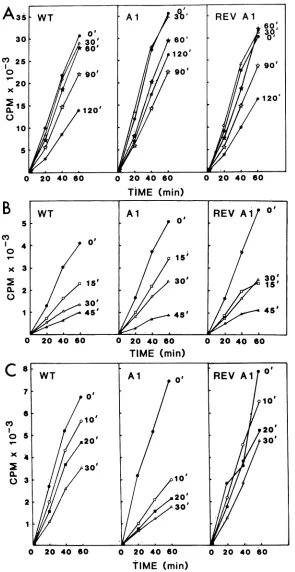
The thermostabilities of these fractions were more
pre-cisely determined in a related set ofexperiments in which
theviralfractions were preheated forincreasingtime inter-vals at a temperature determined from the data in Fig. 4.Al
transcribing cores preheated at 42°C for 0 to 120 min
appeared to be more stable than WT or revertant Al
transcribingcores(Fig.SA).Nodifferences in the stabilities
of the three enzyme fractionsweredetected after0 to45min of incubationat41°C (Fig. SB). In contrast,theAl template
fraction was distinguished from both WT and revertant
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.68.302.308.602.2]WT Al RAl (E) V WT Al RAI
NS N :'W
o 30 60 0 30 60 0 30 60
MINUTES OF CHASE
0 30 60 0 30 60 0 30 60 MINUTES OF CHASE
Al RAl @ V
~
- L-fr) V W T L
G
NS
N
M -
_-0 3_-0 60 0 30 60 0 30 60
MINUTES OF CHASE
WT Al RAI
Gso NS
N _
M _
0 30 60 0 30 60 0 30 60 MINUTES OF CHASE
FIG. 3. SDS-polyacrylamidegelelectrophoresis of pulse-labeled intracellularviral proteins chased for various times. Duplicate WT,Al, andrevertantAl(RAl) infections preamplifiedfor 3 hat31°Cin thepresenceof actinomycinDwerepulse-labeledwith [35S]methionine for 10minat31 (A andB)or39.8°C (Cand D). For the chase, excessunlabeled methionine (1 mM)wasadded, and incubationwascontinued
atthelabelingtemperature for 0, 30,or60min. Extracellular fractions (overlaying medium[BandD])andintracellular fractions (disrupted monolayers [A and C])weredissociated with SDSand electrophoresedonpolyacrylamidegels (10%acrylamide-0.13%bisacrylamide) in the
discontinuous Tris buffersystem. Shownarefluorograms ofthedriedgels.'4C-labeledmarkervirion proteins(V)areidentifiedtothe leftof eachgel. Details of thelabeling protocols and electrophoresis aregiveninthetext.
Altemplate fractions by the loss of60% of its activity after 10 minof incubation at52°C (Fig. 5C).
DISCUSSION
Attempts to assign the lesion in the Al mutant to a structuralprotein by in vitro transcription analyses of
tran-TABLE 2. Relativethermostabilities of pulse-labeled intracellular viral proteins as a function of the length of the chasea
Length of Relativethermostabilitybof indicated
Virus chase protein
(min) L G NS N M
WT 0 119 104 100 94 95
30 104 109 99 98 94
60 89 112 97 100 103
Al 0 121 109 99 92 95
30 84 120 113 87 100
60 92 133 102 80 111
RevertantAl 0 131 100 98 93 95
30 98 102 107 100 91
60 92 96 103 108 94
aTheradioactivitycontained in the bands corresponding to thefiveviral
proteins ineach lane of thegelscontainingthe intracellular fractions shown in
Fig.3A andCwasdetermined asdescribedin the text.
b Relativethermostability=(relativeyield at39.8°C/relative yieldat31°C)
x 100, where relative yield = (cpm in a given protein/sum of cpm in all
proteinsinthesample) x 100.
scribingcoreshadpreviously beenunsuccessful(21).
How-ever,wereasoned that because thestability of the transcrib-ing cores reflects that of their least stable component (the enzyme),alesioninthe N
protein
which lowersthestability of the template fraction could be masked by the inherent lability of the enzyme fraction. Indeed, the transcribingcores as well as the enzyme fractions were completely inactivated by being heated for 15 min at 50°C (Fig. 4). Under theseconditions, the WTand revertantAl template fractions were relatively unaffected, whereas the Al tem-plate fraction lost 50% ofitsactivity (Fig. 4). Onthe basis of theseobservations, werejectedthepossibility that comple-mentationgroup Arepresenteda nonstructuralprotein.
The thermolability of the Al virion template containing the N protein and the genomic RNA coupled with the covariance of this phenotype with the ts phenotype in a
spontaneous revertantof Alallowedustoassignthe Ngene
tocomplementationgroupA.Thelowrelative accumulation of the Al Nproteinatthe restrictivetemperatureinvivowas consistent with this assignment. The small degree of Al N protein degradationdetected in thepulse-chase experiment (Table 2)doesnot accountfor thelowrelative accumulation of the Al N protein observed duringthe 1-h continuous-la-beling experiment (Table 1). We have not resolved this discrepancy. The low relative accumulation may reflect differential synthesis ofWT, revertant Al, and Al N
pro-teins at the restrictive temperature. However, this
discrep-ancy may alternativelyreflect differences in the
experimen-tal conditions used in the twosetsoflabeling experiments.
V
G NS
N
M
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.142.477.69.360.2] [image:5.612.56.296.540.674.2]A10 WT 300 A1 REV Al
s ~~~420 300
o 20 40 so o 20 40 eo40420
n
42X
.
2
500 .500 0
0 20 40 60 0 20 40 60 0 20 40 60
TIME(min)
B s WT - A1 - REV A1 30
400
0~ 0 40603000 020 40 o20 40
X 400
450,500 04 ,50
0 20 40 60 0 20 40 60 0 20 4060
TIME
(min)
C20 WT Al REV Al o
00
Cf) 0 , o
I 10 500
~~~500
600
x ~
~~~~5060
0.~~~~~~~~~~~~~~s
05
0 20 40 60 0 20 40 60 0 2040680
TIME (min)
FIG. 4. Thermal inactivation ofWT, Al, andrevertant Al(REV Al)transcribingcores, enzyme fractions, and templatefractionsas a
function oftemperature. (A)Transcribingcores(5jig)in 50 of 0. 1 M column bufferwereheated for 15minat30,42, 45,or50'Candassayed
forpolymeraseactivityat30°C ina100-,ul reaction mixture.(B) Enzymefractions(25 p.l)in 0.3 Mcolumn bufferwereheated for 15minat
30,40, 45,or50°C,reconstituted with 5 p.gof unheated WT template, andassayedforpolymerase activityat30°Cina100-p.lreaction mixture.
(C)Template fractions (10 p.g)in0.1 Mcolumn bufferwereheated for 15 minat0, 50, or60°C, reconstituted with 25 p.1 ofunheated WT
enzyme,andassayed for polymerase activity at30'Cina200-p.l reaction mixture. Details of thepreparationoftranscribingcores, enzyme
fractions, and template fractions, the polymerase reaction conditions, and the assaying of 20-,ul samples (inthecaseof the 100-,ulreaction
mixtures) and 40-,ul samples (in thecaseof the200-,ul reactionmixtures)aregiveninthetext.
Similarresults were obtained during an analysis of the Dl mutant(6). This mutant also encodes an altered N protein which exhibits low relative accumulation but minimal deg-radation during pulse-chase analysis. This discrepancy ap-pears to be restricted to altered N proteins, as the data obtained from continuous-labeling and pulse-chase experi-mentsareconsistentfor themutantCl Mprotein(6)and the WT Lprotein (6;Tables 1 and2).
The assignment of the N protein to complementation
groupAcompletesthegeneassignmentsof theexistingNew Jersey serotype (Hazelhurst subtype) complementation
groups: Lgene, complementationgroups B and F(2, 10);G
gene, none;Mgene,complementationgroupC(6);NSgene,
complementationgroup E(4, 11); andNgene, complemen-tationgroupA(this paper). Asetoftsmutantsof the New Jerseyserotype(Concansubtype)hasrecentlybeen isolated andclassified intotwo complementation groups: CC/A and CC/B.On the basis ofintersubtypic complementation,these twogroupscorrelated with the HazelhurstsubtypegroupsA and B, respectively. (A. D. Byrd, J. Kennedy-Morrow, M. D. Marks, and J. A. Lesnaw, J. Gen. Virol., inpress). Although nointerserotypic complementation at the level of
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.168.457.71.528.2]A35
WT Al3%'
REVAl
60:
30 0'
301
60' 30O 25 1' 90
20 ' 1
9R'
90V'C
20-
0o
04
o
o2
0e
2
~~~~~~~~~~~~120"0.15
120'
10
5
0 20 4060 0 204060 0 204060
TIME
(min)
BIG.
5TiWT AlREV Al
0
Irl 15
X3 ~ ~ 5'3' 0
0 30'~~~~~~~~~~0 0
1 45'445' 45'
0 204060 0 2040c60 0 20 4060
TIME
(min)
WT
Al ofREV
A 00' 10'
6
Cv) ~~10
20'
05
20 30'
30'
3
~~~~~~~~10'
2-20 30'
I
0 20 40 60 0 20 40 60 0 20 4060
tIME
(min)
FIG. 5. Thermal inactivation ofWT, Al, andrevertantAl(REV Al) transcribingcores, enzy'mefractions, andtemplatefractions as a
function of time.(A)Transcribingcores(20p.g)i'n100p.1of 0.1 M columnbufferwereheatedat420Cfor0, 30, 60, 90,or120,min andassayed forpolymerase activityat30°Cin a200-p.lreactionmixture.(B) Enzymefractions (25,ul) in 0.3Mcolumnbufferwere heated at41°C for0, 15, 30,or45min, reconstitutedwith 5p.gof unheated WTtemplate,andassayedforpolymeraseactivityat30°Cin a 100-,ulreactionmixture. (C)Template fractions (5p.g)wereheated at52.5°C for 0, 10, 20,or30min, reconstituted with15 ,ulof unheatedWT enzyme, andassayed forpolymerase activityat30°C in a100-pLIreactionmixture. Additionaldetails are given in the text.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.150.445.75.647.2]PFU productionhas been detected between NewJerseyand Indianamutants, thesetwo setsof NewJersey complemen-tationgroups can now be correlated with Indiana comple-mentationgroupsIthroughV(17)onthebasisof theirgene
assignments: complementationgroups B and Fcorrespond to complementation group I (L gene); complementation group C corresponds to complementation group III (M gene); complementationgroupE correspondsto complemen-tation group II (NS gene); and complementation group A
corresponds to complementation group IV (N gene). It is interestingto note that although theextracomplementation
group in the New Jersey mutant collection was originally
considered to represent a sixth viral protein, in factnot all the viral structural proteins are represented; nogroup
cor-respondsto the G protein. Nounique gene assignmentcan
be made formutant Dl, the sole representative of comple-mentation group D (6, 18), and the complementation
ob-served between mutants belonging to complementation
groupsB and F wasapparentlyihtracistronic (2, 19). On the basis of the absence of intracellularnucleocapsids (Fig. 1) andgenome-sized viral RNA(12) and the presence
ofprimary transcription (12) in Al-infected cells incubated at the restrictive temperature, a replication defect can be
assignedtothe Al mutant.Thestructural lesion in the Al N protein, togetherwith thetslesion in Alreplication, implies areplicativerole for the Nprotein.A similarcorrelation has been made in thecaseof the Indianaserotype mutanttsG41 (complementation group IV) (20). These genetic data are consistent with thereplicative role for the N protein postu-lated by Kingsbury (7) andBlumbergetal. (3). Accordingto this model, genome-sized RNA synthesis is coupled to nucleocapsid assembly, and the dependence ofreplication
upon continuous protein synthesis (16, 24) reflects the re-quirement for available N protein. Direct evidence for this model hasrecentlybeenreported byPattonet al. (15), who demonstrated that N protein alone satisfies the requirement forprotein synthesis in acoupled in vitro translation-repli-cation system.
The Nproteinappearstoplayadual role inreplication: (i) complexed with genomic RNA, the N protein serves as a template for replication; and (ii) newly synthesized N
pro-tein is required fornucleocapsid assembly. We have not at present resolved which of these functions isimpairedin the Al mutant. The ability of the Al N protein to function in primary transcription atthe restrictive temperature in vivo (12)suggeststhatthetemplateisnotaffected.However, the templateroles of the N proteinin transcription and replica-tionmaynotbe identical. Moreover, the increased
polymer-ase activity observed in in vitro reactions containing
heat-treated Al transcribingcores (21; Fig. 5), togetherwith the decreased polymerase activity observed in reactions di-rectedby heat-treated Al template fractions(Fig. 4and 5), suggestedapossible alterationintemplate function. Weare currently analyzing the products synthesized in these in vitro reactionsin an attempt toshed lighton thismatter.
ACKNOWLEDGMENTS
We thank M. S.Coleman, S. G. Zimmer, and A. Byrd for valuable
discussions.
Thisworkwassupported by Public Health Servicegrant
R01-AI-13574 from the NationalInstitute ofAllergy and Infectious Diseases
andby Biomedical Researchgrant5-505-07-115-07.
LITERATURE CITED
1. Ball, L.A., and G. W. Wertz. 1981. VSV RNAsynthesis: how
canyoubepositive?Cell 26:143-144.
2. Belle Isle, H. D., and S. U. Emerson. 1982. Use ofa hybrid
infectivityassay to analyze primarytranscription of tempera-ture-sensitivemutantsof theNewJerseyserotype of vesicular stomatitis virus.J. Virol. 43:37-40.
3. Blumberg, B. M., M. Leppert, and D. Kolakofsky. 1981. Inter-action of VSV leader RNA and nucleocapsid protein may control VSVgenomereplication. Cell 23:837-845.
4. Evans, D., C. R. Pringle, and J. F.Sziaigyi.1979. Temperature-sensitive mutants of complementation group E of vesicular stomatitis virusNewJerseyserotype possess alteredNS poly-peptides. J. Virol. 31:325-333.
5. Hunt, D. M., M. G. Mellon, and S. U. Emerson. 1979. Viral transcriptase, p. 169-183. In D. H. L. Bishop (ed.), Rhabdo-viruses, vol. 1.CRC Press, BocaRaton, Fla.
6. Kennedy-Morrow, J., and J. A. Lesnaw. 1984. Structural and functionalcharacterization of theRNA-positive complementa-tiongroups, C andD, ofthe NewJerseyserotype of vesicular stomatitis virus:assignment ofthe M genetotheC complemen-tationgroup. Virology 132:38-52.
7. Kingsbury, D. W. 1974. The molecularbiology of paramyxo-viruses. Med. Microbiol. Immunol. 16:73-83.
8. Knipe, D., H. F.Lodish,and D. Baltimore.1977.Analysis of the defects oftemperature-sensitive mutantsof vesicular stomatitis virus: intracellular degradation of specific viral proteins. J. Virol.21:1140-1148.
9. Laemmli,U. K.1970.Cleavage of structuralproteins duringthe assembly ofthe head of bacteriophage T4. Nature (London) 227:680-685.
10. Lesnaw, J. A., and L. R. Dickson. 1978. In vitro functional analysis ofatemperature-sensitive mutantof vesicular stoma-titis virus, New Jersey serotype, defective in transcription. Virology91:51-59.
11. Lesnaw, J. A., L. R.Dickson,andR. H.Curry. 1979.Proposed replicative role of the NS polypeptide of vesicular stomatitis virus: structural analysis ofanelectrophoretic variant. J. Virol. 31:8-16.
12. Lesnaw, J. A., andM. E. Reichmann. 1975. RNAsynthesis by temperature-sensitive mutants of vesicular stomatitis virus, NewJersey serotype. Virology63:492-504.
13. Little,S. P.,and A. S.Huang. 1978.Shedding of the glycopro-tein from vesicular stomatitis virus-infected cells. J. Virol. 27:330-339.
14. Lowry,0.H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275.
15. Patton, J. T., N. L. Davis, and G. W. Wertz. 1984. Nprotein alonesatisfiestherequirementforprotein synthesis duringRNA replication of vesicular stomatitis virus. J. Virol. 49:303-309. 16. Perlman, S. M., and A. S. Huang. 1973. RNA synthesis of
vesicular stomatitis virus.V.Interactions betweentranscription andreplication.J. Virol. 12:1395-1400.
17. Pringle, C. R. 1982. The genetics of vesiculoviruses. Arch. Virol.72:1-34.
18. Pringle,C. R., V. Devine, M. Wilkie, C. M. Preston, A. Dolan, and D.J.McGeoch. 1981. Enhancedmutability associated with
atemperature-sensitivemutantof vesicular stomatitis virus.J. Virol. 39:377-389.
19. Pringle,C.R.,1. B.Duncan,and M.Stevenson. 1971.Isolation andcharacterization oftemperature-sensitivemutantsof vesic-ularstomatitisvirus,NewJerseyserotype.J.Virol.8:836-841. 20.
Pringle,
C. R., andJ. F. Szilagyi. 1980. Gene assignment and complementationgroup, p. 141-161. In D. H.L. Bishop (ed.), Rhabdoviruses,vol. 2. CRCPress,BocaRaton, Fla.21. Szilagyi, J. F.,andC. R.Pringle. 1979. Effect of temperature-sensitive mutation on activity of the RNA transcriptase of vesicular stomatitis virusNewJersey. J. Virol. 30:692-700. 22. Unger,J. T., and M. E. Reichmann. 1973. RNA synthesis in
temperature sensitive mutantsof vesicular stomatitis virus. J. Virol.12:570-578.
23. Wagner,R. R.1975.Reproductionofrhabdoviruses,p. 1-93.In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology,vol. 4. Plenum
Publishing
Corp.,NewYork. 24. Wertz,G.W.,and M. Levine.1973. RNAsynthesis byvesicularstomatitis virus and a small plaque mutant: effects of cyclo-heximide.J.Virol. 12:253-264.
![FIG. 1.particlesFraction Sucrose density gradient analyses of [3H]uridine-labeled virions (unbound viral particles [A]) and nucleocapsids [B]) synthesized in cells infected with WT (a), mutant El (b), or mutant Al (c) virus at 31°C (O*- 1 corresponds to the bottom of the gradient.](https://thumb-us.123doks.com/thumbv2/123dok_us/1413210.94174/3.612.114.493.74.532/particlesfraction-sucrose-gradient-particles-nucleocapsids-synthesized-infected-corresponds.webp)