0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.15.9369–9380.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
MINIREVIEW
Stealth and Cunning: Hepatitis B and Hepatitis C Viruses
Stefan F. Wieland and Francis V. Chisari*
Department of Molecular and Experimental Medicine, The Scripps Research Institute, La Jolla, California
The hepatitis B virus (HBV) and hepatitis C virus (HCV) are noncytopathic, hepatotropic members of the hepadnavirus (HBV) and flavivirus (HCV) families that cause acute and chronic necroinflammatory liver disease and hepatocellular carcinoma (HCC) (29, 38, 79). On a worldwide basis, over 500 million people are persistently infected by these viruses and are at great risk of dying prematurely from HCC (29, 64, 79, 111). It is widely believed that the outcome of both infections and the pathogenesis of the associated liver diseases are deter-mined by host-virus interactions mediated by the immune re-sponse. It has been difficult to elucidate the viral and host factors at play in these infections, however, largely because the host range of HBV and HCV is limited to humans and chim-panzees (3, 19) and because cell culture systems and small animal models that are susceptible to HBV and HCV infection do not exist. Thus, the current state of our understanding of the biology and pathogenesis of these infections reflects what has been learned about their natural history (47, 64) and im-munobiology (29, 137) in humans and chimpanzees, by the virological and immunological analysis of related hepadnavirus (131) and flavivirus (18) infections in their natural hosts, and by biochemical, molecular, virological, and immunological analysis of cell lines (5, 15, 67, 73, 76, 88, 89, 100, 109, 114, 135, 174) and mouse models that express individual viral genes or reproduce the viral life cycles to various degrees (30, 33, 56, 63, 74, 75, 77, 83, 90, 101, 102, 107, 110, 161). Thanks to these efforts, in recent years we have gained important new insight into the viral and host factors that determine pathogenesis and outcome of HBV and HCV infection. As we will describe in this review, it now appears that HBV is a stealth virus that establishes itself very efficiently without alerting the innate immune system to its presence, although it is readily controlled when the adaptive immune response is induced. In contrast, HCV strongly induces yet cunningly evades the innate immune response and also defeats the adaptive immune response by mutation and functional inactivation.
VIRAL FACTORS
Although host factors contribute importantly to the outcome of HBV and HCV infection, viral factors are also critically involved. Perhaps the strongest evidence that viral factors play
a role in the outcome of HBV and HCV infection is that more than 95% of adult-onset HBV infections are self limited, while more than 70% of adult-onset HCV infections persist, and the outcomes for humans and chimpanzees are similar (19, 81, 145, 149, 151). Moreover, as illustrated in Fig. 1, we have recently shown that chimpanzees that have previously cleared HBV (58) (Fig. 1, upper panels) become persistently infected when subsequently inoculated with HCV (Fig. 1, lower panels) (149). These results suggest that the different outcomes of infection could not be due to host genetic differences in these animals. Figure 1 also demonstrates an interesting and underappreci-ated difference between HBV and HCV, i.e., that the logarith-mic phase of HCV amplification occurs much earlier than for HBV, even when the HBV inoculum is several orders of mag-nitude greater than that of HCV (58, 149, 152). This and other viral factors that could contribute to the usually different out-comes of HBV and HCV infection are listed in Table 1 and will now be briefly discussed.
Viral genome, products, and replication strategy.Following infection, the 3.2-kb partially double-stranded DNA HBV ge-nome is delivered to the nucleus and converted into a co-valently closed circular double-stranded HBV DNA (cccDNA) molecule that serves as a transcriptional template for the host RNA polymerase II machinery, which produces four capped and polyadenylated mRNAs that encode the viral core and envelope structural proteins and the precore, polymerase, and X nonstructural viral proteins (reviewed in reference 131). One of the major HBV transcripts is a 3.5-kb greater-than-genome-length RNA that is translated to produce the viral core and polymerase proteins and also serves as a pregenomic RNA that is encapsidated with the polymerase by the core protein in the cytoplasm of the hepatocyte (131). Viral repli-cation occurs entirely within these capsids by reverse transcrip-tion of the pregenomic RNA to produce a single-strand DNA copy, which serves as the template for second-strand DNA synthesis, producing a circular double-stranded DNA (131). Viral capsids containing double-stranded DNA traffic either to the nucleus, where they amplify the viral cccDNA genome, or to the endoplasmic reticulum, where they engage the viral envelope proteins, bud into the lumen, and exit the cell as virions that can infect other cells (131).
In contrast, the HCV life cycle is entirely cytoplasmic. The HCV genome is a 9.6-kb, uncapped, linear, single-stranded RNA (ssRNA) molecule with positive polarity that serves as template for both translation and replication. Translation of the plus-strand RNA initiates at an internal ribosomal entry site, resulting in production of a single polyprotein precursor
* Corresponding author. Mailing address: Department of Molecular and Experimental Medicine, SBR-10, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. Phone: (858) 784-8228. Fax: (858) 784-2160. E-mail: [email protected].
9369
on November 8, 2019 by guest
http://jvi.asm.org/
that is processed into structural (C, E1, E2, p7) and nonstruc-tural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) protein subunits by host and viral proteases (4, 37, 50, 51, 62, 85, 87, 98, 117, 123, 134, 146, 147, 154, 166). HCV replication occurs via a minus-strand intermediate within a membranous compart-ment in the cytoplasm of the cell (99), yielding double-stranded RNA (dsRNA) intermediates as essential compo-nents of its life cycle. Thus, while HBV RNA, RNA/DNA hybrids, and DNA replicative intermediates are sequestered entirely within capsid particles (131), the HCV ssRNA and dsRNA replicative intermediates are likely exposed to the dsRNA sensing machinery of the cell (99). It would not be surprising, therefore, if these two viruses induce very different innate cellular responses following infection. We will discuss the potential significance of this observation when we discuss
the cellular genomic responses to HBV and HCV infection later in this review.
Amplification rate, viral load, and mutation rate.As men-tioned above, another interesting difference between HBV and HCV is the rate at which they expand in the liver after inoc-ulation, and the peak viral titers that they ultimately produce. For example, experimentally infected chimpanzees inoculated
with 108genome equivalents (GE) of HBV display a prolonged
lag phase before viral DNA displays logarithmic expansion in
the liver or serum, and the peak titers are routinely in the 109
GE per ml range (58, 152). In contrast, when the same animals are subsequently inoculated with HCV, the viral RNA expands logarithmically within the first 2 weeks and reaches peak titers
that rarely exceed 106GE per ml (Fig. 1) (149). Since the onset
of the cellular immune response to both of these viruses is detectable 3 to 4 weeks after inoculation, i.e., before the log-arithmic spread of HBV but after the log phase of HCV (ref-erences 149 and 152 and see below), the two viruses could represent very different antigenic challenges to the immune response, and this could have an important impact on the outcome of the infection. Once infection is established, differ-ences in HBV and HCV antigenic burden could also have an impact on viral clearance. In particular, it has been shown by the use of highly sensitive and specific immunohistochemical reagents that HBV can infect up to 100% of the hepatocytes (58, 152). Importantly, high-level HBV antigen expression (es-pecially HBcAg) is easily detectable within infected cells (58, 152), which could contribute to their high visibility to the adap-tive immune system and, therefore, the usual outcome of viral clearance in immunologically competent adults. In contrast, there is considerable uncertainty about the number of hepato-cytes that are infected by HCV. Indeed, estimates based on HCV RNA quantification in chimp liver biopsies during HCV infection suggest that either very few hepatocytes replicate
[image:2.585.118.465.70.266.2]FIG. 1. Sequential HBV and HCV infection in chimpanzees. Upper panels, two naı¨ve chimpanzees (Ch1558 and Ch1564) were intravenously inoculated with 108GE of a monoclonal HBV isolate (genotypeayw) contained in pooled serum from HBV transgenic mice as described by Guidotti et al. (58). Lower panels, 22 months after resolution of the acute HBV infection, Ch1558 and Ch1564 were transfected with HCV RNA of genotype 1a as described previously (149). Serum HBV DNA (upper panels) and HCV RNA (lower panels) levels are expressed as percentages of the corresponding peak serum levels (% max).
TABLE 1. Viral factors influencing spread and resolution of infection
Factors influencing spread and resolution of infection with:
HBV HCV
dsDNA genome (3.2 kb) ssRNA genome (9.6 kb) Replicates via positive-stranded
RNA in capsids in cytoplasm
Replicates via negative-stranded RNA in membraneous web in cytoplasm
Spreads slowly Spreads rapidly
High titer (⬃108to 1013GE/ml) Low titers (⬃105to 107GE/ml)
Can infect all hepatocytes No. of infected hepatocytes unclear Lower mutation rate of⬃10⫺5 Higher mutation rate of⬃10⫺3
Evasion strategies Evasion strategies Mutational escape Mutational escape Precore (tolerogen) Core (inhibits Fas)
HBsAg (tolerogen [?]) E2 (inhibits NK cells [CD81]) X (inhibits proteasome) E2, NS5a (inhibit PKR)
NS3 (inhibits IRF3)
on November 8, 2019 by guest
http://jvi.asm.org/
HCV at high levels or that low-level HCV replication occurs throughout the liver (12). Both situations would be expected to result in inefficient antigen recognition by the adaptive im-mune system, which could contribute to the usual outcome of viral persistence in HCV infection. Finally, because of the lack of proofreading activity in the viral RNA-dependent RNA polymerase (NS5B), the mutation rate of HCV is very high
(10⫺3per nucleotide per year) (106). This results in the rapid
evolution of a viral quasispecies in each infected subject, pre-sumably due to immune selection pressure. Since hepadnavi-ruses replicate by reverse transcription of an RNA pregenome,
they are thought to have a high mutation rate (10⫺5per
nu-cleotide per generation) (48, 115), albeit one that is 100 times lower than that for HCV, which may account for the fact that the emergence of viral variants during HBV infection is much less frequent than for HCV infection (24, 121).
Evasion strategies.As will be described later in this review, viral clearance and disease pathogenesis with both HBV and HCV infection are mediated largely by the immune response. For these noncytopathic viruses to persist, they must either not induce a response or they must overwhelm, evade, or counter-act it. All of these scenarios have been shown to be operative in chronic HBV and HCV infection. Interestingly, as we will see later, it appears that HBV “evades” the innate response by simply not inducing it to act as a stealth virus in this regard (171). On the other hand, HBV appears to employ active evasion strategies that target the adaptive immune response (see below), which is well known to be activated by this virus (8, 10, 28, 29, 91, 97, 118–120, 150, 152). This suggests that the adaptive response plays an important role in the control of HBV infection while, interestingly, the innate response does not. Importantly, as will be discussed later in this review, both of these ideas are supported by genomic analysis of the liver in acutely HBV-infected chimpanzees (171). In contrast to HBV, HCV displays several cunning evasion strategies that target the innate and the adaptive immune responses (see below), sug-gesting that it activates and must, therefore, evade both during infection. Importantly, this concept is also supported by the gene expression profiles of the acutely HCV-infected liver (see below and references 12 and 142). The known HBV and HCV evasion strategies will now be briefly discussed.
Mutational escape. Mutational inactivation of B-cell and T-cell epitopes occurs in chronic HBV infection (9, 21, 121, 165), but it is much more common in HCV (24, 36, 169; reviewed in reference 137). Indeed, mutations that abrogate recognition by antibody, CD4 T cells, and CD8 T cells in chronically HBV- and HCV-infected humans (7, 9, 20, 24, 36, 65, 82, 92, 121, 132, 153, 157) and chimpanzees (36, 169) have been described. The T-cell epitope mutations often occur in epitope residues that bind to major histocompatibility complex (MHC) molecules, thereby precluding antigen presentation (24, 36). Less frequently, inactivating mutations in residues that flank T-cell epitopes and impair the ability of those epitopes to be processed by the proteasome (132) or trans-ported by the TAP protein into the endoplasmic reticulum (133), also precluding antigen presentation, have been de-scribed. More often, mutations occur in epitope residues that are engaged by the T-cell receptor (TCR). Inactivating muta-tions of this sort preclude antigen recognition (9, 36, 49, 68, 92), which makes the cells that are infected by the mutant virus
invisible to the T cells containing the corresponding TCR. In addition, epitope mutations in which the variant residue re-mains visible to the TCR but antagonizes it have been de-scribed (7, 9, 24, 72, 157); with these mutations, the TCR no longer recognizes its cognate wild-type epitope, making cells infected by the wild-type virus and the mutant virus invisible to that TCR-bearing population of cells. All of these mechanisms have been described and probably contribute to viral persis-tence in both HBV and HCV infection, although they are much more common in HCV infection, presumably because of the higher mutation rate for HCV.
HBV proteins that inhibit the adaptive immune response.
Several mechanisms are probably responsible for viral persis-tence during HBV and HCV infection (reviewed in references 28, 64, 116, and 137). As will be discussed later in this review, chronic HBV and HCV infection are characterized by absent, weak, or narrowly focused CD4 and CD8 T-cell responses to the corresponding viral antigens (78, 97, 104, 112, 120, 145, 168). The responsible mechanisms are not entirely clear, but T-cell deletion, anergy, exhaustion, and ignorance have all been reported to occur in HBV- and HCV-infected humans and chimpanzees (reviewed in references 28, 29, and 137). Interestingly, at least two virus-specific tolerogenic mecha-nisms that can be ascribed to the HBV precore and surface proteins appear to be operative in chronic HBV infection The HBV precore protein is not required for HBV replication or infection (23, 25, 155, 156), but it is processed and secreted as HBeAg, a 17-kDa protein that is small enough to cross the placenta and induce neonatal tolerance, at least in HBV trans-genic mice (96). In addition, HBeAg has been shown to sup-press the antibody and T-cell response to HBcAg in adult T-cell receptor transgenic mice (26), functioning either to de-lete or anergize HBcAg/HBeAg cross-reactive T cells, depend-ing on their functional avidity for the tolerogenic epitope. Thus, HBeAg may suppress immune elimination of infected cells by HBcAg-specific T cells and, thereby, contribute to viral persistence in chronically infected adults. Clinical evidence supports this notion, since viral mutations that preclude the production of HBeAg are often associated with exacerbations of liver disease and, sometimes, even with viral clearance in chronically infected patients (17, 47). Thus, although the pre-core protein has no known role in the viral life cycle, it may function as an HBV-specific immunosuppressive factor that protects the virus against immune attack. The hepatitis B sur-face antigen (HBsAg) might also suppress immune elimination of infected cells by functioning as a high-dose tolerogen, since extremely high serum HBsAg titers, in the mg per ml range, are often seen with chronically infected patients (122, 167). In keeping with the high antigen load, chronically infected pa-tients display absent or subnormal levels of HBsAg-specific
CD8⫹T cells; however, these CD8⫹T-cell populations display
abnormal HLA/peptide tetramer binding properties in con-trast to the few HBcAg-positive T cells that are detectable in these patients, which are functionally normal in this regard (122). In addition, it has been reported that the HBV X pro-tein, a transcriptional transactivator that is required for initi-ation of infection (177, 180), can inhibit cellular proteasome activity when it is overexpressed (66). Thus, the HBx protein has the potential to inhibit antigen processing and presentation if it is overexpressed to a comparable degree in infected cells.
on November 8, 2019 by guest
http://jvi.asm.org/
In all instances, therefore, HBV attempts to make itself invis-ible to the various effector limbs of the immune response, functioning as a stealth virus in this regard.
HCV proteins that inhibit the innate immune response.
While HBV evasion strategies are focused primarily on the adaptive immune response, HCV has developed mechanisms that appear designed to enable it to escape both the innate and the adaptive response. As shown in Table 1, mutational escape from the adaptive response due to its high mutation rate is common in HCV-infected patients and chimpanzees (35, 36, 52, 136, 153, 162, 163, 169, 170). In addition, however, because it replicates via a dsRNA intermediate, HCV activates protein kinase R (113), interferon regulatory factor 1 (IRF-1) (42, 113), and IRF-3 (42, 143), and downstream antiviral genes that were originally shown to be activated by these factors in dif-ferent viral infections (reviewed in references 14 and 141). In keeping with this notion, as we will describe later in this review, HCV induces a large number of interferon (IFN)-inducible genes in the liver during acute and chronic infection (12, 13, 142). Nonetheless, HCV appears to be resistant to these anti-viral pathways (128, 141). Indeed, several HCV structural and nonstructural proteins (E2 and NS3 and NS5, respectively) have been shown to inhibit nonoverlapping functions of the innate immune response. E2 and NS5A have been shown to bind to the kinase domain of PKR and inhibit IRF-1 activation (46, 113, 148). The NS3/4A protease has been shown to cleave the toll-like receptor 3 adaptor protein (TRIF) (84) and to disrupt RIG-I signaling (39), thus blocking the phosphoryla-tion and effector acphosphoryla-tion of IRF-3 (40). In addiphosphoryla-tion, some in-vestigators have demonstrated that overexpression of the HCV core protein can inhibit Fas-mediated apoptosis (93, 144), al-though this is controversial since it has also been shown to stimulate apoptosis by others (178, 179), and overexpression of the HCV nonstructural protein E2 inhibits natural killer cell function by binding to CD81 (32, 158). It is important to point out, however, that all of these effects have been demonstrated in transfection and replicon systems where the HCV proteins are overexpressed. Therefore, these functions should be con-sidered speculative until they can be tested with a cell culture model of natural HCV infection.
Thus, because it is so visible to the innate immune system, HCV appears to employ several evasion strategies that con-tribute to its high propensity to persist. There is a problem with this hypothesis, however, at least in terms of evasion of the interferon response. Since activated IRF-1 and IRF-3 are both
known to induce beta interferon (IFN-) gene expression (45,
164), one would not expect interferon-regulated genes to be strongly induced in the liver during HCV infection if E2, NS3, and NS5A inhibit their activation. Nonetheless, genomic anal-ysis of the livers of several HCV-infected chimpanzees (see below) indicates that a strong correlation exists between the level of HCV viremia and the intrahepatic expression of a large number of interferon-regulated genes (12, 13, 142), which shouldn’t occur if HCV proteins inhibit interferon induction. Clearly, more studies are needed to understand the basis for these apparently contradictory observations.
Until then, we favor the notion that HCV circumvents the interferon signaling cascade not by blocking its induction but by inhibiting the antiviral effector functions of interferon-in-duced target genes. Alternatively, if IRF-1 and IRF-3
activa-tion are partially blunted by HCV, instead of being totally blocked, the interferon induction might be at a sufficient level to keep viral titers relatively low without completely preventing HCV infection. Thus, despite the interesting and provocative reports about HCV-induced IRF-1 and IRF-3 blockade, the potential impact of these processes on HCV infection will not be testable until the development of cell culture and small animal models of HCV infection.
HOST FACTORS
The fact that most adult-onset HBV infections are self-limited while nearly all neonatal HBV infections persist serves as strong evidence that host factors play a critical role in the outcome of HBV infection (28, 29). This relationship is rein-forced by many studies showing a close association between the vigor, diversity, and effector functions of the cellular immune response to HBV and HCV and the outcomes of these infec-tions, not only in naturally infected humans (81, 104, 112, 151) but also in transgenic mice (56, 175) and experimentally in-fected chimpanzees (6, 31, 149, 152). These observations will now be considered, using the host pathways that contribute to these differential outcomes as the starting point for discussion.
Pathways to viral persistence.Neonatal tolerance to HBV is probably responsible for viral persistence following mother-infant transmission (29). On the other hand, the basis for the inadequate immune response that is characteristic of adult-onset chronic HBV and HCV infections is not well understood and may, in fact, be multifactorial in origin. For example, while viral clearance during self-limited HBV and HCV infection is characterized by a vigorous, polyclonal CD4 and CD8 T-cell response to these viruses (31, 91, 120, 138, 149, 151, 152), primary failures to establish a CD4 and CD8 T-cell response in a patient who was studied prospectively after accidental needle stick exposure to HCV (151) and in HCV-inoculated chimpan-zees (149) have been clearly described, suggesting that primary immunological nonresponsiveness to HCV probably led to persistent infection in those cases. Whether, as has been shown with other viral systems, the failure to produce a vigorous T-cell response to HCV was due to the possible negative im-pact of antigen overload during immunological priming (re-viewed in references 34 and 130), to virus-induced defects in antigen presentation (reviewed in reference 176), to the hy-perinduction of regulatory T cells (reviewed in reference 127), to a genetically determined restriction of the virus-specific T-cell repertoire (reviewed in reference 105), or occurs for other reasons remains to be determined. It is also thought that the diminished T-cell responses in chronically infected patients could be due to the induction of anergy and/or the exhaustion of an initially vigorous T-cell response by high viral load, pre-sumably reflecting excessive antigen stimulation of virus-spe-cific T cells. If this occurs, it would contribute to the mainte-nance of persistent infection, although it would obviously be a secondary event. There is a precedent for this notion, since Lechner et al. (80) and Ulsenheimer et al. (160) have shown
that CD8⫹ and CD4⫹ T-lymphocyte responses are induced
during acute HCV infection but are not sustained during pro-gression to chronicity. There is also evidence that the ability of
HCV-specific CD8⫹T cells to produce gamma interferon
fol-lowing in vitro exposure to antigen is compromised
on November 8, 2019 by guest
http://jvi.asm.org/
(“stunned”) as a consequence of antigen overstimulation in vivo during persistent infection (53, 81, 151, 168). Further-more, Boni et al. (16) have shown that antiviral treatment can
overcome CD8⫹ T-cell hyporesponsiveness in subjects with
chronic HBV infection, suggesting that the T cells are present in these subjects but are suppressed. Thus, primary and sec-ondary immunological hyporesponsiveness to both viruses can occur in persistent HBV and HCV infection.
Pathways to viral clearance.It is widely believed that the cytotoxic T-lymphocyte (CTL) response clears viral infections by killing infected cells. CTL killing is an inefficient process, however, requiring direct physical contact between the CTLs and the infected cells. Thus, it may not be possible for CTLs to kill all infected cells if the CTLs are greatly outnumbered, as
occurs during these infections, in which as many as 1011
hepa-tocytes can be infected. Thus, although the liver disease in these infections is clearly due to the cytopathic activity of the CTL response, viral clearance may require more efficient CTL functions than killing. Important insights into the pathogenetic and noncytopathic antiviral functions of the CTL response have come from studies in HBV transgenic mice that develop an acute necroinflammatory liver disease after adoptive trans-fer of HBsAg-specific CTL clones (1, 56, 103). In that model, the CTLs rapidly enter the liver and recognize viral antigen, triggering two events, (i) apoptosis of the hepatocytes that are
physically engaged by the CTLs and (ii) secretion of IFN-␥,
which noncytopathically inhibits HBV gene expression and replication in the rest of the hepatocytes (54, 56) by preventing the assembly of the RNA-containing capsids in the cytoplasm (172) in a proteasome- (126) and kinase-dependent (125) pro-cess. During this remarkable process, the viral nucleocapsids disappear from the cytoplasm of the hepatocytes (56, 172) and the viral RNAs are destabilized by a SSB/La-dependent mech-anism in the nucleus (59–61, 159), yet the hepatocytes remain perfectly healthy (54, 159). As a result, all of the viral gene products and virions decrease in the liver and the serum (56), inhibiting further viral spread. This antiviral process is
com-pletely blocked by the administration of antibodies to IFN-␥
before the CTLs are injected, and it is not induced by
HBsAg-specific CTLs derived from IFN-␥knockout mice whose
cyto-pathic functions are perfectly normal (56), indicating that
IFN-␥production by the CTLs is responsible for the
noncyto-pathic antiviral effect. Furthermore, HBsAg-specific, Fas li-gand-deficient, and perforin-deficient CTL clones that do not cause hepatitis in these animals do inhibit viral replication (56), proving genetically that the cytopathic and antiviral func-tions of CTLs are completely independent of each other. These results suggest that a strong intrahepatic CTL response to HBV can suppress viral gene expression and replication non-cytopathically. In addition, it may even “cure” infected hepa-tocytes of the virus, provided that the HBV transcriptional template (cccDNA) is also eliminated from infected cells. This could not be tested with HBV transgenic mice, because they do not produce cccDNA (57). However, as will be discussed be-low, the kinetics of cccDNA elimination during experimental HBV infection in chimpanzees suggests that cccDNA could, at least partially, be eliminated from hepatocytes by a noncyto-lytic mechanism (58, 173).
Interestingly, it has been shown that HBV replication is also suppressed by the antiviral effects of alpha/beta interferon (55,
95, 172). Indeed, in the transgenic mouse model, at least, HBV
replication is inhibited by any stimulus that induces IFN-␥or
IFN-␣/ in the liver, including CD4⫹ T cells (41), NK and
NKT cells (70), and other hepatotropic viral (22) and parasitic infections (94, 108). This raises the possibility that HBV infec-tion can be controlled by many arms of the immune response and perhaps explains why HBV infection is almost always self-limited in immunologically normal adults.
To investigate whether these principles apply to the clear-ance of HBV and HCV infection, we extended these studies to HBV- and HCV-infected chimpanzees (58, 149, 152). In these studies, we showed that the early phase of clearance of HBV was temporally associated with the appearance of CD3, CD8,
and IFN-␥mRNA in the liver (152), which reflected the influx
of virus-specific CD8⫹ T cells into the liver (152). But,
al-though HBV replicative intermediates (152, 173) and cccDNA templates (152, 173) decreased as much as 50-fold and 8-fold from peak levels during this time, respectively, there was little or no attendant liver disease, despite the fact that virtually 100% of the hepatocytes were infected (58, 152), suggesting that noncytopathic mechanisms were active during this early phase of viral clearance. Furthermore, we also showed that
monoclonal antibody-mediated depletion of CD8⫹T cells (but
not CD4⫹T cells) at the peak of infection delayed the onset of
viral clearance and liver disease for several weeks, until the
antibody titers waned and virus-specific CD8⫹T cells became
detectable in the liver (152). Thus, we conclude that the prin-ciple of CD8-dependent noncytopathic clearance of HBV, which was discovered by using the HBV transgenic mouse model, is operative in the context of the full-fledged viral infection. Interestingly, several lines of evidence suggest that HCV may also be susceptible to CD8-dependent noncytolytic clearance mechanisms. For example, in a study of the immune response to HCV with five individuals who became infected after accidental needle stick exposure to HCV-positive blood (151), the only subject to clear the virus did so in the context of
an IFN-␥-producing HCV-specific CD8⫹T-cell response, and
clearance occurred without a corresponding surge of liver dis-ease activity (151). Furthermore, viral clearance has been
re-ported to occur in the context of an intrahepatic CD8⫹T-cell
response in experimentally HCV-infected chimpanzees in the absence of elevated serum alanine aminotransferase activity (a marker of liver cell destruction) (149), and viral clearance has
been shown to be delayed by CD8⫹T-cell depletion in such
animals (138). Lastly, both IFN-␥ and IFN-␣/ have been
shown to inhibit the replication of an HCV replicon without evidence of toxicity in Huh-7 cells in vitro (15, 27, 43, 44), demonstrating that HCV is susceptible to interferon-induced noncytolytic control mechanisms in the replicon system.
Gene expression profiling. The foregoing results suggest
that one or more cellular genes that are induced by IFN-␥
and/or IFN-␣/are likely to inhibit both HBV and HCV
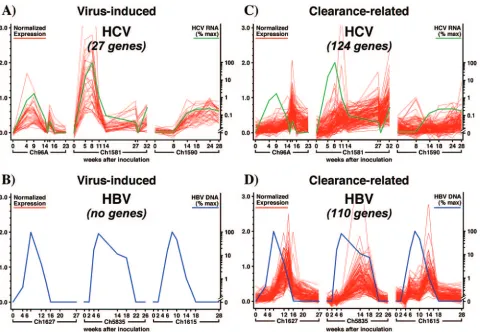
rep-lication. In order to begin to identify these genes, as well as genes that might be transcriptionally regulated by the viruses, global gene expression profiling (Affymetrix gene chip analy-sis) was performed using liver RNA obtained at multiple time points after both infections (142, 171). As shown in Fig. 2, three HCV-infected chimpanzees and three HBV-infected an-imals were included in these studies (142, 171). As illustrated in Fig. 2, three different courses and outcomes of infection
on November 8, 2019 by guest
http://jvi.asm.org/
were seen with the HCV-infected animals (shown in green). Chimpanzee 96A developed self-limited infection that reached
a viral titer of 105GE per ml and cleared the virus, similar to
the outcomes seen with all three of the HBV-infected animals (shown in blue). In contrast, chimp 1581 (which developed the
highest HCV titer, 2.5⫻106GE per ml) initially controlled the
infection by 3 to 4 logs before becoming persistently infected,
and chimp 1590 (which displayed the lowest viral titer, 104GE
per ml) became persistently infected without a period of initial control. In contrast, all three HBV-infected chimpanzees (1627, 5835, and 1615; shown in blue) developed a self-limited
infection, reaching very similar maximal viral titers of 109GE
per ml, and they cleared the virus with remarkably similar kinetics. When the gene expression profiles were established for all of the animals, we searched for virus-induced cellular genes, i.e., genes whose expression levels correlated with the viral titers in all three of the HBV-infected animals or in all three of the HCV-infected animals, and for clearance-related cellular genes, i.e., genes whose expression was correlated with the clearance of HBV in all three of those animals or whose
expression correlated with at least a 3 log reduction of viral titer in the corresponding HCV-infected animals.
HCV-induced genes.Initially, we searched for genes whose expression patterns correlated (directly or inversely) with the amount of HCV RNA in the liver over the entire time course profiled, irrespective of the outcome of infection. We reasoned that these genes would be regulated by HCV and that they might reflect activation of the dsRNA sensing machinery of the cell, might be required for HCV to establish and/or maintain itself in the liver, or both. Using Pearson correlation analysis to reduce the chance of misinterpreting random fluctuations in gene expression in individual animals, we searched for tran-scripts that correlated with the changing HCV RNA titer with
a correlation coefficient of at least 0.7 for all three animals (P
⬍0.05) (shown in red in Fig. 2A). We found 27 unique
tran-scripts that fulfilled these criteria, and we noted that they displayed an average peak fold change (FC) value of 8.8 (142). A complete list of gene names is provided in Table 2.
Impor-tantly, consistent with the time course of the IFN-␣/
[image:6.585.54.534.68.400.2]induc-tion in the liver of these chimpanzees as reported by us (149)
FIG. 2. Liver gene expression profile during acute HBV and acute and chronic HCV infection in chimpanzees. The HCV and HBV infection protocols and the gene selection criteria are described by Su et al. (142) and Wieland et al. (171), respectively. Briefly, gene expression profiles of virus-induced genes from all three HCV- or HBV-infected animals were required to positively or negatively correlate with viremia with a Pearson’s correlation of better than 0.7. Gene expression profiles related to clearance in HCV-infected chimpanzees were identified as described by Su et al. (142), and that list was extended by including genes whose expression correlated with the profiles of the prototype genes RANTES, MIG, TcR and MHC by a Pearson’s correlation of better then 0.7 for all three chimpanzees. HBV clearance-related genes were identified as described by Wieland et al. (171). HBV liver DNA levels (blue) and HCV serum RNA levels (green) are presented as percentages of the peak level (% max) for the chimpanzee with the highest peak level of HBV and HCV, respectively.
on November 8, 2019 by guest
http://jvi.asm.org/
and in independent studies by Bigger et al. (12, 13), expression
of many of these genes is known to be stimulated by IFN-␣/.
This suggests that HCV-infected cells probably detect the virus by sensing the presence of dsRNA replication intermediates,
thereby inducing the transcription of many IFN-␣/-stimulated
genes. Nonetheless, this response apparently fails to eradicate HCV from the infected cells, either for quantitative reasons or because of HCV-induced evasion mechanisms downstream of
IFN-␣/-stimulated transcription, as we discussed earlier in
this review. These unexpected findings might also suggest that, in contrast to other viruses that are inhibited by these genes, HCV might actually use the product(s) of one or more of these early response genes to facilitate infection. While these ques-tions are not approachable for the chimpanzee, they are readily addressable in vitro with HCV replicon-containing Huh-7 cells, and experiments designed to examine these issues are in progress in many HCV laboratories, including our own, at this time.
HBV-induced genes. The same analysis was performed to identify cellular genes that correlate with HBV DNA titers and might, thus, be induced by HBV infection (171). Accordingly, as we did for the HCV-infected animals, we searched for genes with expression patterns that correlated (directly or inversely) with the amount of HBV DNA in the livers of all three animals over the entire time course profiled, and we restricted our focus to genes whose changing expression levels correlated with the changing viral DNA content in the liver with a corre-lation coefficient of at least 0.7. As shown in Fig. 2B and Table 2, no genes fulfilled these criteria (171). Since virtually 100% of the hepatocytes in all three animals were infected (152), the failure of the virus to induce cellular gene expression as it spread throughout the liver suggests that HBV behaves as a “stealth” virus in that it does not induce an innate response in the cells it infects. This surprising and unprecedented obser-vation may explain why HBV replication is so highly sensitive to the antiviral effects of alpha/beta interferon, since it has evolved without the need to establish any defense mechanisms against this cytokine and, by extension, against any other cy-tokines that display similar gene expression profiles (e.g.,
IFN-␥). This is strikingly illustrated by the fact that HBV replication
is readily suppressed by antiviral mechanisms induced by toll-like receptor ligands that activate the innate immune response in HBV transgenic mice (69).
HBV and HCV clearance-related genes. To identify genes associated with HBV clearance, we searched for genes whose expression was induced or suppressed only during the phase of viral clearance in the three HBV-infected chimpanzees. To identify similar genes in the HCV-infected animals, we searched for genes whose expression was induced or sup-pressed during the phase of viral clearance in chimpanzees 96A and 1581 but that were not related to viremia in chim-panzee 1590, since the viral titer never decreased in that ani-mal. We identified 124 genes that were induced during viral clearance in HCV-infected chimpanzees 96A and 1581, but not in 1590, and correlated with HCV-specific T-cell infiltration
and IFN-␥expression (149) or IFN-␥-inducible genes, such as
RANTES, MIG, and MHC (Fig. 2C) (142). Similarly, as shown in Fig. 2D, we identified 110 genes that were induced during viral clearance and correlated with the same marker genes in all three HBV animals (152, 171). Table 3 lists the genes that were induced during clearance of both infections. Not surpris-ingly, it is heavily weighted towards genes expressed by alpha/ beta T cells (e.g., T-cell receptor beta), gamma/delta T cells (e.g., T-cell receptor gamma), CD3, cytolytic effector mole-cules (granzyme A), and T-cell growth regulatory genes, in-cluding the receptor for interleukin 10 (IL-10). In addition,
many genes that are known to be induced by IFN-␥are
rep-resented on the list, including those involved in antigen pre-sentation (MHC class I and class II, ubiquitin D, and the immunoproteasome subunits LMP2, MECL1, and PA28),
che-mokines (RANTES, MIG, and MIP-1) that can recruit
anti-gen-nonspecific inflammatory cells into the liver, the GTPase guanylate binding protein 1 (GBP1) and GBP2, and other
IFN-␥-induced genes, e.g., tryptophanyl-tRNA synthetase
(TrpRS) (86), solute carrier 7A, and ubiquitin D, whose rele-vance to the T-cell response and viral clearance are not imme-diately apparent. Interestingly, some of the proteins in the miscellaneous group are involved in host-pathogen interac-tions. For example, tyrosine kinase binding protein is involved in NK cell signaling and has been shown to be important in the host defense against murine cytomegalovirus (140). The un-coupling protein 2 limits oxygen radical production and mac-rophage-mediated immunity (2).
[image:7.585.301.542.88.243.2]It is important to point out that, because some of these genes
TABLE 2. Cellular genes that correlate with viremia in HCV and HBV infection
Genes correlating with viremia in infection with:
HCV HBV
Agrin MX1 None
GalS3BP OAS
Histone H2B Sp100
IFI44 STAT1
IFI-6-16 TRIM 14
IFIT1 TRIM 22
IFIT4 Trp-oxy 2
IFITM1 UBE2L6
ISG15 Vipirin
[image:7.585.42.283.90.216.2]Ly6E XIAPAF1
TABLE 3. Cellular genes induced during clearance of both HBV and HCV from the liver
Cellular gene induced
T cell IFN-␥inducible Miscellaneous
CD3 GBP1 Adenylate cyclase
CD53 GBP2 Butyrophilin
Granzyme A LMP-2 C1q
IL 10-R Mec11 CD5 antigen-like
Leupaxin MHC class I CD48
TcR MHC class II CD74
TcR␥ MIG IG lambda C2
MIP-1 IgG Fc receptor
PA28␣ Tyrosine kinase binding protein RANTES Uncoupling protein 2
Solute carrier 7A TrpRS
Ubiquitin D
on November 8, 2019 by guest
http://jvi.asm.org/
are likely to be expressed in the inflammatory cells that infil-trate the liver during viral hepatitis, their role in viral clearance is probably indirect. However, the expression of many genes on this list is not limited to inflammatory cells, and these genes have the potential to exert a more direct effect on these infec-tions by modulating viral replication within the hepatocytes. The unique subunits of the immunoproteasome (LMP2 and LMP7) are in this category, since the proteasome is known to influence various aspects of the life cycle of a number of vi-ruses, including human immunodeficiency virus (129) and HBV (139). The chemokines RANTES and MIG are also in this category, since they are known to be induced in
hepato-cytes by IFN-␥(71). Furthermore, because of the known ability
of IFN-␥to inhibit HBV in vivo (56, 109) and its demonstrated
ability to inhibit HCV replication in vitro (27, 44, 56, 76, 109), it is possible that one or more of the genes in this set may contribute to the noncytopathic clearance of these viruses, as with the previously observed antiviral effect on HBV replica-tion in the livers of HBV transgenic mice (54).
SUMMARY
Collectively, the results summarized in this review suggest that HBV acts like a stealth virus early in infection, remaining undetected and spreading until the onset of the adaptive im-mune response several weeks later. We suspect that the rela-tive invisibility of HBV to the innate sensing machinery of the cells reflects its replication strategy, which sequesters the tran-scriptional template in the nucleus, entails the production of capped and polyadenylated viral mRNAs that resemble the structure of normal cellular transcripts, and the replicating viral genome is sheltered within viral capsid particles in the cytoplasm and, therefore, does not elicit a response. In con-trast, HCV activates a strong intracellular antiviral response in the liver, presumably because it replicates via a dsRNA inter-mediate in the cytoplasm, where it can readily induce the cellular dsRNA sensing apparatus and initiate the signaling cascade. In turn, this results in the induction of many genes,
such as the 2⬘,5⬘-oligoadenylate synthetase Mx1, that have
known antiviral activity (reviewed in references 128 and 141); TRIM14 and 22, members of the same gene family as TRIM5, which has been shown to be part of the host defense against retroviruses (reviewed in reference 11); and ISG15, which has recently been proposed to have a role in innate immunity (124). Impressively, however, HCV cunningly manages to spread through the liver despite induction of these genes, pre-sumably because its E2, NS3, and NS5A proteins can defeat them. On the other hand, both viruses can be controlled when
CD8⫹T cells enter the liver, recognize antigen, kill whatever
infected cells they encounter, and secrete IFN-␥, which triggers
a broad-based cascade that amplifies the inflammatory process and has antiviral activity, at least in HBV-transgenic mice and the HBV and HCV cell culture systems. The unexpected find-ing that HBV does not modulate host cellular gene transcrip-tion and apparently does not induce an innate immune re-sponse when spreading through the liver raises the possibility that HBV, unlike HCV and other viruses, has evolved to evade innate immunity by not inducing it rather then actively coun-teracting it; this, in turn, might leave HBV very sensitive to intracellular antiviral mechanisms when they are induced by
the adaptive immune system or an unrelated viral infection. Although we suggest that HBV and HCV infections can be inhibited noncytopathically by the cellular genes that are in-duced during this process, the identities of those genes, the antiviral mechanisms they elicit, and the unique evasion strat-egies of each virus remain to be determined, marking a new starting point in the quest to understand the cellular and mo-lecular immunobiology of these viral infections.
ACKNOWLEDGMENTS
We thank all of our colleagues who contributed importantly to the work cited in this review, especially Robert Purcell and Jens Bukh (NIAID, National Institutes of Health, Bethesda, MD), Andrew Su and Peter Schultz (Genomics Institute of the Novartis Foundation, La Jolla, CA), and Luca Guidotti (The Scripps Research Institute, La Jolla, CA). We are particularly grateful to B. Rehermann (Liver Dis-eases Section, NIDDK, National Institutes of Health, Bethesda, MD) and R. Lanford (Department of Virology and Immunology, Southwest Foundation for Biomedical Research, San Antonio, TX) for critical reading of the manuscript.
This work was supported by NIH grants AI20001, CA76403, and CA40489 to F.V.C.
This is manuscript number MEM-17106 from the Scripps Research Institute.
REFERENCES
1.Ando, K., L. G. Guidotti, S. Wirth, T. Ishikawa, G. Missale, T. Moriyama, R. D. Schreiber, H. J. Schlicht, S. N. Huang, and F. V. Chisari.1994. Class I-restricted cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. J. Immunol.152:3245–3253.
2.Arsenijevic, D., H. Onuma, C. Pecqueur, S. Raimbault, B. S. Manning, B. Miroux, E. Couplan, M. C. Alves-Guerra, M. Goubern, R. Surwit, F. Bouil-laud, D. Richard, S. Collins, and D. Ricquier. 2000. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet.26:435–439.
3.Barker, L. F., F. V. Chisari, P. P. McGrath, D. W. Dalgard, R. L. Kir-schstein, J. D. Almeida, T. S. Edington, D. G. Sharp, and M. R. Peterson. 1973. Transmission of type B viral hepatitis to chimpanzees. J. Infect. Dis. 127:648–662.
4.Bartenschlager, R., L. Ahlborn-Laake, J. Mous, and H. Jacobsen.1993. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type proteinase required for cleavage at the NS3/4 and NS4/5 junctions. J. Virol. 67:3835–3844.
5.Bartenschlager, R., A. Kaul, and S. Sparacio.2003. Replication of the hepatitis C virus in cell culture. Antivir. Res.60:91–102.
6.Bassett, S. E., B. Guerra, K. Brasky, E. Miskovsky, M. Houghton, G. R. Klimpel, and R. E. Lanford.2001. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infec-tion. Hepatology33:1479–1487.
7.Bertoletti, A., A. Costanzo, F. V. Chisari, M. Levrero, M. Artini, A. Sette, A. Penna, T. Giuberti, F. Fiaccadori, and C. Ferrari.1994. Cytotoxic T lym-phocyte response to a wild type hepatitis B virus epitope in patients chron-ically infected by variant viruses carrying substitutions within the epitope. J. Exp. Med.180:933–943.
8.Bertoletti, A., C. Ferrari, F. Fiaccadori, A. Penna, R. Margolskee, H. J. Schlicht, P. Fowler, S. Guilhot, and F. V. Chisari.1991. HLA class I-re-stricted human cytotoxic T cells recognize endogenously synthesized hep-atitis B virus nucleocapsid antigen. Proc. Natl. Acad. Sci. USA88:10445– 10449.
9.Bertoletti, A., A. Sette, F. V. Chisari, A. Penna, M. Levrero, M. De Carli, F. Fiaccadori, and C. Ferrari.1994. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature369:407– 410.
10.Bertoni, R., A. Sette, J. Sidney, L. G. Guidotti, M. Shapiro, R. Purcell, and F. V. Chisari.1998. Human class I supertypes and CTL repertoires extend to chimpanzees. J. Immunol.161:4447–4455.
11.Bieniasz, P. D.2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol.5:1109–1115.
12.Bigger, C. B., K. M. Brasky, and R. E. Lanford.2001. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infec-tion. J. Virol.75:7059–7066.
13.Bigger, C. B., B. Guerra, K. M. Brasky, G. Hubbard, M. R. Beard, B. A. Luxon, S. M. Lemon, and R. E. Lanford.2004. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 78: 13779–13792.
on November 8, 2019 by guest
http://jvi.asm.org/
14.Biron, C. A.1999. Initial and innate responses to viral infections—pattern setting in immunity or disease. Curr. Opin. Microbiol.2:374–381. 15.Blight, K. J., A. A. Kolykhalov, and C. M. Rice.2000. Efficient initiation of
HCV RNA replication in cell culture. Science290:1972–1975.
16.Boni, C., A. Penna, G. S. Ogg, A. Bertoletti, M. Pilli, C. Cavallo, A. Cavalli, S. Urbani, R. Boehme, R. Panebianco, F. Fiaccadori, and C. Ferrari.2001. Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology 33:963–971.
17.Brunetto, M. R., M. M. Giarin, F. Oliveri, E. Chiaberge, M. Baldi, A. Alfarano, A. Serra, G. Saracco, G. Verme, H. Will, et al.1991. Wild-type and e antigen-minus hepatitis B viruses and course of chronic hepatitis. Proc. Natl. Acad. Sci. USA88:4186–4190.
18.Buckwold, V. E., B. E. Beer, and R. O. Donis.2003. Bovine viral diarrhea virus as a surrogate model of hepatitis C virus for the evaluation of antiviral agents. Antivir. Res.60:1–15.
19.Bukh, J.2004. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology39:1469–1475.
20.Carman, W. F., W. Boner, G. Fattovich, K. Colman, E. S. Dornan, M. Thursz, and S. Hadziyannis.1997. Hepatitis B virus core protein mutations are concentrated in B cell epitopes in progressive disease and in T helper cell epitopes during clinical remission. J. Infect. Dis.175:1093–1100. 21.Carman, W. F., A. R. Zanetti, P. Karayiannis, J. Waters, G. Manzillo, E.
Tanzi, A. J. Zuckerman, and H. C. Thomas.1990. Vaccine-induced escape mutant of hepatitis B virus. Lancet336:325–329.
22.Cavanaugh, V. J., L. G. Guidotti, and F. V. Chisari.1998. Inhibition of hepatitis B virus replication during adenovirus and cytomegalovirus infec-tions in transgenic mice. J. Virol.72:2630–2637.
23.Chang, C., G. Enders, R. Sprengel, N. Peters, H. E. Varmus, and D. Ganem. 1987. Expression of the precore region of an avian hepatitis B virus is not required for viral replication. J. Virol.61:3322–3325.
24.Chang, K. M., B. Rehermann, J. G. McHutchison, C. Pasquinelli, S. South-wood, A. Sette, and F. V. Chisari. 1997. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J. Clin. Investig.100:2376–2385.
25.Chen, H. S., M. C. Kew, W. E. Hornbuckle, B. C. Tennant, P. J. Cote, J. L. Gerin, R. H. Purcell, and R. H. Miller.1992. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. J. Virol.66:5682–5684.
26.Chen, M. T., J. N. Billaud, M. Sallberg, L. G. Guidotti, F. V. Chisari, J. Jones, J. Hughes, and D. R. Milich.2004. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA101:14913–14918.
27.Cheney, I. W., V. C. Lai, W. Zhong, T. Brodhag, S. Dempsey, C. Lim, Z. Hong, J. Y. Lau, and R. C. Tam. 2002. Comparative analysis of anti-hepatitis C virus activity and gene expression mediated by alpha, beta, and gamma interferons. J. Virol.76:11148–11154.
28.Chisari, F. V.2000. Rous-Whipple Award Lecture. Viruses, immunity, and cancer: lessons from hepatitis B. Am. J. Pathol.156:1117–1132. 29.Chisari, F. V., and C. Ferrari.1995. Hepatitis B virus immunopathogenesis.
Annu. Rev. Immunol.13:29–60.
30.Chisari, F. V., C. A. Pinkert, D. R. Milich, P. Filippi, A. McLachlan, R. D. Palmiter, and R. L. Brinster. 1985. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science230:1157–1160. 31.Cooper, S., A. L. Erickson, E. J. Adams, J. Kansopon, A. J. Weiner, D. Y.
Chien, M. Houghton, P. Parham, and C. M. Walker.1999. Analysis of a successful immune response against hepatitis C virus. Immunity10:439– 449.
32.Crotta, S., A. Stilla, A. Wack, A. D’Andrea, S. Nuti, U. D’Oro, M. Mosca, F. Filliponi, R. M. Brunetto, F. Bonino, S. Abrignani, and N. M. Valiante. 2002. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med.195:35–42. 33.Disson, O., D. Haouzi, S. Desagher, K. Loesch, M. Hahne, E. J. Kremer, C.
Jacquet, S. M. Lemon, U. Hibner, and H. Lerat.2004. Impaired clearance of virus-infected hepatocytes in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology126:859–872.
34.Doherty, P. C.1993. Immune exhaustion: driving virus-specific CD8⫹T cells to death. Trends Microbiol.1:207–209.
35.Eckels, D. D., H. Zhou, T. H. Bian, and H. Wang.1999. Identification of antigenic escape variants in an immunodominant epitope of hepatitis C virus. Int. Immunol.11:577–583.
36.Erickson, A. L., Y. Kimura, S. Igarashi, J. Eichelberger, M. Houghton, J. Sidney, D. McKinney, A. Sette, A. L. Hughes, and C. M. Walker.2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity15:883–895. 37.Failla, C., L. Tomei, and R. De Francesco.1994. Both NS3 and NS4A are
required for proteolytic processing of hepatitis C virus nonstructural pro-teins. J. Virol.68:3753–3760.
38.Ferrari, C., G. Missale, C. Boni, and S. Urbani.2003. Immunopathogenesis of hepatitis B. J. Hepatol.39(Suppl. 1):S36–S42.
39.Foy, E., K. Li, R. Sumpter, Jr., Y. M. Loo, C. L. Johnson, C. Wang, P. M. Fish, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr.2005. Control
of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. USA102:2986–2991. 40.Foy, E., K. Li, C. Wang, R. Sumpter, Jr., M. Ikeda, S. M. Lemon, and M.
Gale, Jr.2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science300:1145–1148.
41.Franco, A., L. G. Guidotti, M. V. Hobbs, V. Pasquetto, and F. V. Chisari. 1997. Pathogenetic effector function of CD4-positive T helper 1 cells in hepatitis B virus transgenic mice. J. Immunol.159:2001–2008.
42.Fredericksen, B., G. R. Akkaraju, E. Foy, C. Wang, J. Pflugheber, Z. J. Chen, and M. Gale, Jr.2002. Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol.15:29–40. 43.Frese, M., T. Pietschmann, D. Moradpour, O. Haller, and R.
Barten-schlager.2001. Interferon-alpha inhibits hepatitis C virus subgenomic RNA replication by an MxA-independent pathway. J. Gen. Virol.82:723–733. 44.Frese, M., V. Schwarzle, K. Barth, N. Krieger, V. Lohmann, S. Mihm, O.
Haller, and R. Bartenschlager.2002. Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology35:694– 703.
45.Fujita, T., Y. Kimura, M. Miyamoto, E. L. Barsoumian, and T. Taniguchi. 1989. Induction of endogenous IFN-alpha and IFN-beta genes by a regu-latory transcription factor, IRF-1. Nature337:270–272.
46.Gale, M. J., Jr., M. J. Korth, N. M. Tang, S. L. Tan, D. A. Hopkins, T. E. Dever, S. J. Polyak, D. R. Gretch, and M. G. Katze.1997. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology230:217– 227.
47.Ganem, D., and A. M. Prince.2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med.350:1118–1129. 48.Girones, R., and R. H. Miller.1989. Mutation rate of the hepadnavirus
genome. Virology170:595–597.
49.Giuggio, V. M., H. L. Bonkovsky, J. Smith, and A. L. Rothman.1998. Inefficient recognition of autologous viral sequences by intrahepatic hepa-titis C virus-specific cytotoxic T lymphocytes in chronically infected subjects. Virology251:132–140.
50.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice.1993. Characterization of the hepatitis C virus-encoded serine pro-teinase: determination of proteinase-dependent polyprotein cleavage sites. J. Virol.67:2832–2843.
51.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice. 1993. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. USA90:10583–10587.
52.Grakoui, A., N. H. Shoukry, D. J. Woollard, J. H. Han, H. L. Hanson, J. Ghrayeb, K. K. Murthy, C. M. Rice, and C. M. Walker.2003. HCV per-sistence and immune evasion in the absence of memory T cell help. Science 302:659–662.
53.Gruener, N. H., F. Lechner, M. C. Jung, H. Diepolder, T. Gerlach, G. Lauer, B. Walker, J. Sullivan, R. Phillips, G. R. Pape, and P. Klenerman. 2001. Sustained dysfunction of antiviral CD8⫹T lymphocytes after infec-tion with hepatitis C virus. J. Virol.75:5550–5558.
54.Guidotti, L. G., K. Ando, M. V. Hobbs, T. Ishikawa, L. Runkel, R. D. Schreiber, and F. V. Chisari.1994. Cytotoxic T lymphocytes inhibit hepa-titis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc. Natl. Acad. Sci. USA91:3764–3768.
55.Guidotti, L. G., P. Borrow, M. V. Hobbs, B. Matzke, I. Gresser, M. B. Oldstone, and F. V. Chisari.1996. Viral cross talk: intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver. Proc. Natl. Acad. Sci. USA93:4589–4594.
56.Guidotti, L. G., T. Ishikawa, M. V. Hobbs, B. Matzke, R. Schreiber, and F. V. Chisari.1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity4:25–36.
57.Guidotti, L. G., B. Matzke, H. Schaller, and F. V. Chisari.1995. High-level hepatitis B virus replication in transgenic mice. J. Virol.69:6158–6169. 58.Guidotti, L. G., R. Rochford, J. Chung, M. Shapiro, R. Purcell, and F. V.
Chisari.1999. Viral clearance without destruction of infected cells during acute HBV infection. Science284:825–829.
59.Heise, T., L. G. Guidotti, V. J. Cavanaugh, and F. V. Chisari.1999. Hep-atitis B virus RNA-binding proteins associated with cytokine-induced clear-ance of viral RNA from the liver of transgenic mice. J. Virol.73:474–481. 60.Heise, T., L. G. Guidotti, and F. V. Chisari. 2001. Characterization of nuclear RNases that cleave hepatitis B virus RNA near the La protein binding site. J. Virol.75:6874–6883.
61.Heise, T., L. G. Guidotti, and F. V. Chisari.1999. La autoantigen specifi-cally recognizes a predicted stem-loop in hepatitis B virus RNA. J. Virol. 73:5767–5776.
62.Hijikata, M., N. Kato, Y. Ootsuyama, M. Nakagawa, and K. Shimotohno. 1991. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci. USA 88:5547–5551.
63.Honda, A., M. Hatano, M. Kohara, Y. Arai, T. Hartatik, T. Moriyama, M. Imawari, K. Koike, O. Yokosuka, K. Shimotohno, and T. Tokuhisa.2000. HCV-core protein accelerates recovery from the insensitivity of liver cells
on November 8, 2019 by guest
http://jvi.asm.org/
to Fas-mediated apoptosis induced by an injection of anti-Fas antibody in mice. J. Hepatol.33:440–447.
64.Hoofnagle, J. H.2002. Course and outcome of hepatitis C. Hepatology 36:S21–S29.
65.Hosono, S., P. C. Tai, W. Wang, M. Ambrose, D. G. Hwang, T. T. Yuan, B. H. Peng, C. S. Yang, C. S. Lee, and C. Shih.1995. Core antigen mutations of human hepatitis B virus in hepatomas accumulate in MHC class II-restricted T cell epitopes. Virology212:151–162.
66.Hu, Z., Z. Zhang, E. Doo, O. Coux, A. L. Goldberg, and T. J. Liang.1999. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J. Virol.73:7231–7240.
67.Ikeda, M., M. Yi, K. Li, and S. M. Lemon.2002. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol.76:2997–3006.
68.Ishikawa, T., D. Kono, J. Chung, P. Fowler, A. Theofilopoulos, S. Kakumu, and F. V. Chisari. 1998. Polyclonality and multispecificity of the CTL response to a single viral epitope. J. Immunol.161:5842–5850.
69.Isogawa, M., M. D. Robek, Y. Furuichi, and F. V. Chisari.2005. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 79:7269–7272.
70.Kakimi, K., L. G. Guidotti, Y. Koezuka, and F. V. Chisari.2000. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med.192:921–930.
71.Kakimi, K., T. E. Lane, S. Wieland, V. C. Asensio, I. L. Campbell, F. V. Chisari, and L. G. Guidotti.2001. Blocking chemokine responsive to gam-ma-2/interferon (IFN)-gamma inducible protein and monokine induced by IFN-gamma activity in vivo reduces the pathogenetic but not the antiviral potential of hepatitis B virus-specific cytotoxic T lymphocytes. J. Exp. Med. 194:1755–1766.
72.Kaneko, T., T. Moriyama, K. Udaka, K. Hiroishi, H. Kita, H. Okamoto, H. Yagita, K. Okumura, and M. Imawari.1997. Impaired induction of cyto-toxic T lymphocytes by antagonism of a weak agonist borne by a variant hepatitis C virus epitope. Eur. J. Immunol.27:1782–1787.
73.Kapadia, S. B., A. Brideau-Andersen, and F. V. Chisari.2003. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. USA100:2014–2018.
74.Koike, K., K. Moriya, K. Ishibashi, Y. Matsuura, T. Suzuki, I. Saito, S. Iino, K. Kurokawa, and T. Miyamura.1995. Expression of hepatitis C virus envelope proteins in transgenic mice. J. Gen. Virol.76:3031–3038. 75.Koike, K., K. Moriya, K. Ishibashi, H. Yotsuyanagi, Y. Shintani, H. Fujie,
K. Kurokawa, Y. Matsuura, and T. Miyamura.1997. Sialadenitis histolog-ically resembling Sjogren syndrome in mice transgenic for hepatitis C virus envelope genes. Proc. Natl. Acad. Sci. USA94:233–236.
76.Lanford, R. E., B. Guerra, H. Lee, D. R. Averett, B. Pfeiffer, D. Chavez, L. Notvall, and C. Bigger.2003. Antiviral effect and virus-host interactions in response to alpha interferon, gamma interferon, poly(I)-poly(C), tumor necrosis factor alpha, and ribavirin in hepatitis C virus subgenomic repli-cons. J. Virol.77:1092–1104.
77.Larkin, J., M. Clayton, B. Sun, C. E. Perchonock, J. L. Morgan, L. D. Siracusa, F. H. Michaels, and M. A. Feitelson.1999. Hepatitis B virus transgenic mouse model of chronic liver disease. Nat. Med.5:907–912. 78.Lauer, G. M., E. Barnes, M. Lucas, J. Timm, K. Ouchi, A. Y. Kim, C. L.
Day, G. K. Robbins, D. R. Casson, M. Reiser, G. Dusheiko, T. M. Allen, R. T. Chung, B. D. Walker, and P. Klenerman. 2004. High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology127:924–936.
79.Lauer, G. M., and B. D. Walker.2001. Hepatitis C virus infection. N. Engl. J. Med.345:41–52.
80.Lechner, F., N. H. Gruener, S. Urbani, J. Uggeri, T. Santantonio, A. R. Kammer, A. Cerny, R. Phillips, C. Ferrari, G. R. Pape, and P. Klenerman. 2000. CD8⫹T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur. J. Immunol.30:2479–2487. 81.Lechner, F., D. K. Wong, P. R. Dunbar, R. Chapman, R. T. Chung, P.
Dohrenwend, G. Robbins, R. Phillips, P. Klenerman, and B. D. Walker. 2000. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med.191:1499–1512.
82.Lee, Y. I., G. M. Hur, D. J. Suh, and S. H. Kim.1996. Novel pre-C/C gene mutants of hepatitis B virus in chronic active hepatitis: naturally occurring escape mutants. J. Gen. Virol.77:1129–1138.
83.Lerat, H., M. Honda, M. R. Beard, K. Loesch, J. Sun, Y. Yang, M. Okuda, R. Gosert, S. Y. Xiao, S. A. Weinman, and S. M. Lemon.2002. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology122:352–365.
84.Li, K., E. Foy, J. C. Ferreon, M. Nakamura, A. C. Ferreon, M. Ikeda, S. C. Ray, M. Gale, Jr., and S. M. Lemon.2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adap-tor protein TRIF. Proc. Natl. Acad. Sci. USA102:2992–2997.
85.Lin, C., B. D. Lindenbach, B. M. Pragai, D. W. McCourt, and C. M. Rice. 1994. Processing in the hepatitis C virus E2-NS2 region: identification of p7 and two distinct E2-specific products with different C termini. J. Virol. 68:5063–5073.
86.Liu, J., E. Shue, K. L. Ewalt, and P. Schimmel.2004. A new gamma-interferon-inducible promoter and splice variants of an anti-angiogenic human tRNA synthetase. Nucleic Acids Res.32:719–727.
87.Lohmann, V., J. O. Koch, and R. Bartenschlager.1996. Processing path-ways of the hepatitis C virus proteins. J. Hepatol.24:11–19.
88.Lohmann, V., F. Korner, A. Dobierzewska, and R. Bartenschlager.2001. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol.75:1437–1449.
89.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bar-tenschlager.1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science285:110–113.
90.Machida, K., K. Tsukiyama-Kohara, E. Seike, S. Tone, F. Shibasaki, M. Shimizu, H. Takahashi, Y. Hayashi, N. Funata, C. Taya, H. Yonekawa, and M. Kohara.2001. Inhibition of cytochrome c release in Fas-mediated sig-naling pathway in transgenic mice induced to express hepatitis C viral proteins. J. Biol. Chem.276:12140–12146.
91.Maini, M. K., C. Boni, G. S. Ogg, A. S. King, S. Reignat, C. K. Lee, J. R. Larrubia, G. J. Webster, A. J. McMichael, C. Ferrari, R. Williams, D. Vergani, and A. Bertoletti.1999. Direct ex vivo analysis of hepatitis B virus-specific CD8⫹T cells associated with the control of infection. Gas-troenterology117:1386–1396.
92.Maini, M. K., S. Reignat, C. Boni, G. S. Ogg, A. S. King, F. Malacarne, G. J. Webster, and A. Bertoletti.2000. T cell receptor usage of virus-specific CD8 cells and recognition of viral mutations during acute and persistent hepatitis B virus infection. Eur. J. Immunol.30:3067–3078.
93.Marusawa, H., M. Hijikata, T. Chiba, and K. Shimotohno.1999. Hepatitis C virus core protein inhibits Fas- and tumor necrosis factor alpha-mediated apoptosis via NF-B activation. J. Virol.73:4713–4720.
94.McClary, H., R. Koch, F. V. Chisari, and L. G. Guidotti.2000. Inhibition of hepatitis B virus replication duringSchistosoma mansoniinfection in trans-genic mice. J. Exp. Med.192:289–294.
95.McClary, H., R. Koch, F. V. Chisari, and L. G. Guidotti.2000. Relative sensitivity of hepatitis B virus and other hepatotropic viruses to the antiviral effects of cytokines. J. Virol.74:2255–2264.
96.Milich, D. R., J. E. Jones, J. L. Hughes, J. Price, A. K. Raney, and A. McLachlan.1990. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc. Natl. Acad. Sci. USA87: 6599–6603.
97.Missale, G., A. Redeker, J. Person, P. Fowler, S. Guilhot, H. J. Schlicht, C. Ferrari, and F. V. Chisari. 1993. HLA-A31- and HLA-Aw68-restricted cytotoxic T cell responses to a single hepatitis B virus nucleocapsid epitope during acute viral hepatitis. J. Exp. Med.177:751–762.
98.Mizushima, H., M. Hijikata, S. Asabe, M. Hirota, K. Kimura, and K. Shimotohno.1994. Two hepatitis C virus glycoprotein E2 products with different C termini. J. Virol.68:6215–6222.
99.Moradpour, D., V. Brass, E. Bieck, P. Friebe, R. Gosert, H. E. Blum, R. Bartenschlager, F. Penin, and V. Lohmann.2004. Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication. J. Virol.78:13278–13284.
100.Moradpour, D., C. Englert, T. Wakita, and J. R. Wands.1996. Character-ization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology222:51–63.
101.Moriya, K., H. Fujie, Y. Shintani, H. Yotsuyanagi, T. Tsutsumi, K. Ishi-bashi, Y. Matsuura, S. Kimura, T. Miyamura, and K. Koike.1998. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med.4:1065–1067.
102.Moriya, K., H. Yotsuyanagi, Y. Shintani, H. Fujie, K. Ishibashi, Y. Mat-suura, T. Miyamura, and K. Koike.1997. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol.78:1527–1531. 103.Moriyama, T., S. Guilhot, K. Klopchin, B. Moss, C. A. Pinkert, R. D. Palmiter, R. L. Brinster, O. Kanagawa, and F. V. Chisari.1990. Immuno-biology and pathogenesis of hepatocellular injury in hepatitis B virus trans-genic mice. Science248:361–364.
104.Nayersina, R., P. Fowler, S. Guilhot, G. Missale, A. Cerny, H. J. Schlicht, A. Vitiello, R. Chesnut, J. L. Person, A. G. Redeker, et al.1993. HLA A2 restricted cytotoxic T lymphocyte responses to multiple hepatitis B surface antigen epitopes during hepatitis B virus infection. J. Immunol.150:4659– 4671.
105.Nikolich-Zugich, J., M. K. Slifka, and I. Messaoudi.2004. The many im-portant facets of T-cell repertoire diversity. Nat. Rev. Immunol.4:123–132. 106.Ogata, N., H. J. Alter, R. H. Miller, and R. H. Purcell.1991. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA88:3392–3396.
107.Okuda, M., K. Li, M. R. Beard, L. A. Showalter, F. Scholle, S. M. Lemon, and S. A. Weinman.2002. Mitochondrial injury, oxidative stress, and anti-oxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology122:366–375.
108.Pasquetto, V., L. G. Guidotti, K. Kakimi, M. Tsuji, and F. V. Chisari.2000. Host-virus interactions during malaria infection in hepatitis B virus trans-genic mice. J. Exp. Med.192:529–536.
109.Pasquetto, V., S. F. Wieland, S. L. Uprichard, M. Tripodi, and F. V.