0022-538X/84/110314-06$02.00/0
Copyright C 1984,American Society for Microbiology
Specificity of Initiation of Plus-Strand
DNA
by Rous
Sarcoma Virus
JULIE K. SMITH, ANITACYWINSKI, AND JOHN M. TAYLOR*
The InstituteforCancer Research, FoxChase Cancer Center, Philadelphia, Pennsylvania 19111 Received3May1984/Accepted19July 1984
Wepreviously reported that inthe endogenous reaction of Roussarcomavirusdisrupted by melittin, plus-strand DNAinitiates on a smalloligonucleotide primer and that this initiationcanbe reconstructed in vitro in reactions containing purifiedminus-strand DNA as template, viral RNA as a source of primer, and reverse transcriptase (Smith et al., J. Virol. 49:200-204, 1984). Further studies on thespecificity of initiation in the endogenous reaction have shown thefollowing. (i)Theprimerwas12nucleotides inlength. Its sequence began with a5'pyrimidine, followed by 11 purines, ending withrGrA-3'. This sequence was in agreement with the known plus-strand RNA sequence immediately upstream from the initiation site. Thus, the primer began one nucleotide5'tothe so-calledpolypurine tract that has beenfound on all retrovirus genomes. (ii) The transition point between RNA primer and DNA product wasprecisely located. Itwasbefore the end of the polypurine tract. Thus the polypurine tract, although essential for virus replication and probably aflagfor the priming event, did not define the limits of the RNA primer. After primer removal, the DNA had a 5' phosphate, consistent with generation by the viral RNase Hactivity. Thepriming specificityin reconstructed reactions was also examinedfurther, with thefollowing observations. (i) When the source of RNA primerwasprehybridized tothe templateviral DNA, the generation, utilization, and subsequent removal of primer were essentially the same as those observed in the endogenous reaction. In the absence of deliberate prehybridization, some specificity waslost.Therewerethen additional locationsforthe5' end of theprimeraswellasthetransition point between RNA primer and DNA. (ii) Purine-rich oligoribonucleotides created by RNase A digestion of viral RNA could prime strong-stop plus DNA, but again with the loss ofspecificity relative to that in the endogenous reaction. (iii) The 5' end of the minus-strand DNA template was not requiredfor initiation of strong-stop plus DNA.Therefore, thespecificityof initiation didnotdepend uponanintramolecular interaction requiring the two inverted repeat sequences that flank the long terminal repeat.
During the replication of retroviruses a viral enzyme, reverse transcriptase, copies the viral RNA genome into a double-stranded DNA intermediate (reviewed in reference 27). Minus-strand DNA (complementary to the viral RNA genome) is synthesized first, using a cellular tRNA as the primer(5). The growing minus-strand DNA isthe template for the synthesis of plus-strand DNA. The first plus-strand DNA seenisadiscrete species called strong-stop plus DNA (14, 29). Strong-stop plus DNA is initiated at a precise location(13). Foraviansarcomavirus, thisspecies isca.340 nucleotidesin length (29).
Wepreviously demonstrated that in the endogenous reac-tion of avian sarcoma virus virions permeabilized with melittin, the strong-stop plus DNAis initiated on an oligori-bonucleotide primer(21). The specific initiation isthe result of three precise nucleolytic events: two to generate the discrete ends oftheprimerandathirdtoremovethe primer afteruse.
We also showed that strong-stop plus initiation can be reconstructedwithpurifiedcomponents: minus-strandDNA astemplate,high-molecular-weightviral RNAas asourceof primers,and reversetranscriptase (21). Champouxetal. (3) have used similar reconstructed reactions to show that RNase A-digested viral RNA can provide primers for the initiation of strong-stop plus DNA by Moloney murine leukemia virus.
Ithas been suggested thatdegradation of the viral RNAby RNase H, an activity of the reverse transcriptase, could provide primers for plus-strand DNA synthesis (27, 29). Such a mechanism has been demonstrated in the reverse transcription of homopolymers(31). Inaddition,RNA-DNA
* Correspondingauthor.
junctions have been detected in the total plus-strand DNA synthesized in areconstructed reaction (17). Our data on the specificity of initiation, as presented here, are consistent with such a role forRNase H.
Inall retroviruses examined,the initiation site of strong-stop plus DNA isadjacent to a series ofpurinenucleotides in the viral RNA(28).Thispolypurinetract(PPT) isrequiredin cis for retroviral replication (22). Thus the circumstantial evidence is that the PPTplaysarole in theprimingof plus-strand DNA synthesis. Our studies have determined the actual relationship between this PPT and the RNA species usedasthe primer.
MATERIALSANDMETHODS
Virus, cells, and reagents. The Prague A strain of Rous sarcomaviruswasderivedfrom the transformedquailclone Q-PrA-4 (19). The virus was propagated in quail embryo fibroblastsaspreviouslydescribed(7). Restriction enzymes wereusedaccording totheinstructionsofthe manufacturer (Bethesda ResearchLaboratories), with the addition of 0.5 U of Ribonucleasin (Bolton Biologicals) per
,ll.
Reverse transcriptase, purified from avianmyeloblastosis virus,was generously provided bytheOffice ofProgramResources and Logistics, VirusCancerProgram, Beltsville, Md.PlasmidpSRA2,which contains Roussacromavirus DNA (Schmidt-Ruppin A strain) inserted into pBR322, was ob-tained from W. DeLorbe and co-workers (4).
DNA synthesis and analysis. Conditions for the synthesis and analysis of strong-stop plus DNA from endogenous reactions and from reconstructed reactions were as previ-ously described(21) andarepresented brieflyhere.
In the endogenous reactions, pelleted virions were per-meabilized with melittin and incubated with deoxyribonucle-314
on November 10, 2019 by guest
http://jvi.asm.org/
Hpa Sph Eco
Hr
H_-
/ <DNA-" A I | I§ - DNA+
I /f I ~I II
-570 0 200 400
NUCLEOT
IDESFIG. 1. Features of the viralDNAaround the origin of strong-stop plusDNA. DNA- represents theminus-strand template, and DNA+ represents the twosizes ofstrong-stop plus DNA. Positions
of restriction enzyme sites used in this study are based on the
nucleotidesequence dataofSchwartz et al. (20). Hpa,HpaII;Sph,
SphI; Eco, EcoRI. Alsoshown are the inverted repeat sequences
(>ol) and the RNA primers used to initiate each DNA strand
(w)-otide triphosphates. Labeled DNA products were made by including[a-32P]TTP inthereactionmixture. Afterdigestion with eitherEcoRIorSphI, the DNA product was denatured and hybridized in solution to an excess of DNA from a recombinant M13 bacteriophage which containedtheminus strand ofthe long terminal repeat (LTR) (25). The hybrid molecules were isolated by rate zonal sedimentation. This provided a partial purification of those labeled molecules containing sequences relatedtostrong-stop plusDNA. This DNA wasthenanalyzed on an80-cmgel of 6% acrylamide-urea. Acloned fragmentof DNAwhich containedtheorigin of strong-stop plus DNA (an EcoRI-PvuII fragment of pSRA2) was submitted to
base-specific
cleavages described by Maxam andGilbert(11) and servedas asize standardon the high-resolutionacrylamide
gels.For the reconstructed reactions we used as a template minus-strand DNA synthesized in an endogenous reaction. Thehigher-molecular-weight DNAspecies wereisolatedby two cycles of rate zonal sedimentation through alkaline sucrose. This DNA was incubated with viral RNA and reversetranscriptase. Labeled DNA products were made by theaddition of
[(x-32P]TTP
tothe reaction. Labeled species related to strong-stop plus DNA were partially purified and thenanalyzed
by gel electrophoresis as described above. Unlabeledreaction products wereglyoxalated (12) and sub-jected to electrophoresis on slabgelsof
2% agarose. After transferto nitrocellulose (23), sequences related to strong-stopplus oritscomplementweredetected withprobes made from recombinant M13 DNAs containing inserted LTR sequences (9, 25).RESULTS
Sizeandsequence of theRNA primerinendogenous
reac-tions. In our
previous studies,
the RNAprimer
on plus-strand DNA appeared to be uniform in size (21). The followingstudies wereundertakentodetermine whether the primers werepredominantly
ofasingle
sequenceand,
if so, to compare that sequence to thepublished
sequenceofthe viral DNA (20). To do this wepurifiedthoselabeledspecies
fromanendogenous
reaction
thatwererelatedtostrong-stop plus DNA. Amap ofthe DNA around theorigin
of strong-stopplusand thelocationsof the restrictionsitesused in this study are shown inFig.
1. This DNA wasdigested
with EcoRI andanalyzed
on anacrylamide gel (Fig.
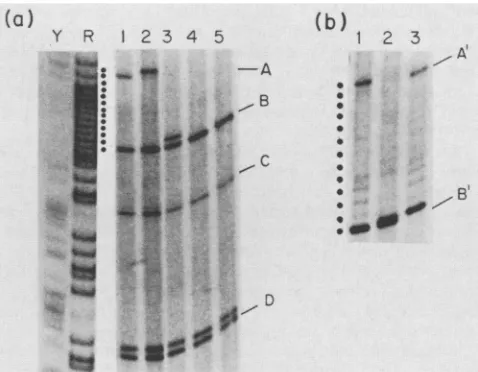
2a, lane 2). Asdescribedpreviously (21), species C andDcorrespond to the 3' EcoRI fragments of strong-stop plus DNA with or without the 18 nucleotides copied from the minus-strand tRNA primer. The labeled band which is one nucleotidelarger than band D represents strong-stop plus DNA in which only a single base has been copied from the tRNA primer. Species A and B correspond to the 5' EcoRI fragments with or without the oligoribonucleotide primer. Relative tothesingle-nucleotide calibration provided by the adjacent sequencing ladder, the primer was reproducibly measured tobe 12 bases inlength (Fig. 2; unpublisheddata). To confirm this size assignment and to characterize the sequence oftheprimer, samplesweredigestedtocompletion with base-specific RNases. RNase A, which cuts 3' to pyrimidines, reduced the primer length by one nucleotide (Fig. 2a,lane1). RNaseT1,which cuts 3' torG,reducedthe primer toone nucleotide in length (Fig. 2a, lane 3). RNase U2, which cuts 3' to rA, and alkali, which hydrolyzes all ribonucleotides, both removed the entire primer (Fig. 2a, lanes 4 and 5, respectively). From these digestions the sequenceof the RNAprimerwasdeducedtobe 5'-Y(R)9GA-3' where Y = T or C and R = A or G. This sequence is consistent with the known RNA sequenceof the viralRNA (20)immediately upstream from the initiation site.
(a)
Y R l 2 3 4 5
.N.A
*4s
_"
-A
_: s..~~
: a ,B~~~
(b)
b)1
2 3
AI
*T
0
0
0
.so
.
S
*_
FIG. 2. Acrylamide gel electrophoresis ofstrong-stop plus DNA
from endogenous reactions. Radiolabeled DNA synthesized by
melittin-disrupted virions wasdigested withEcoRI (a) orSphI (b)
and then hybridized in solutionto an excess ofrecombinant M13
DNA containing inserted LTR minus-strand sequences. Hybrids wereisolatedby sedimentation on a 5 to20%sucrose gradient. The
fractions enriched forstrong-stopplusDNA weresubjected to the treatments indicated and thenresolvedon80-cm gelsof6%
acryl-amide-urea. The dots indicate single nucleotide spacings, and
labeled bands are asdescribed inthe text. (a)EcoRI digestion. At
the left is a sequencing ladder generated from cloned viral DNA
containing the origin ofstrong-stop plus DNA which was 3'-end
labeledattheEcoRI site. Sequencing reactions were performed by
the methodofMaxamand Gilbert (11). Lanes: Y, (T + C)-specific
reaction; R, (A + G)-specific reaction; 1, product digested with RNase A (10 mM Tris [pH 7.6], 1mM EDTA, 10 mM NaCl; total
reaction volume,100,ulincluding5 ,ugoftRNA and 10
p.g
of RNase A[Sigma Chemical Co.]; 30 min at 37°C); 2, untreated product; 3,product digestedwith RNase
T,
(samereaction buffer as RNase A, 10 U of T1 [Calbiochem-Behring]; 30 min at 37°C); 4, productdigested with RNAse U2 (50 mM sodium acetate [pH4.7], 2 mM
EDTA;totalreactionvolume, 100p.1 including 5 ,ug of tRNA and5
Uof U2[Sankyo];20min at50°C);5, product hydrolyzed with alkali
(0.3N NaOH; 10min at 100°C). Bands A toD refertolane 2. (b)
SphIdigestion. Lanes: 1, untreatedproduct; 2, producttreatedwith phosphatase (10 mM Tris [pH 8.0], 120 mM NaCl; total reaction volume, 100
,u1
with 125 U of bacterial alkaline phosphatase ([Bethesda Research Laboratories];60 min at65°C);3, producthydrolyzedwith alkali(0.3NNaOH; 10min at 100°C).A' and B'refertolane 1.on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.59.294.75.160.2] [image:2.612.315.554.283.469.2]Removal of the RNA primer in endogenous reactions. To examine the 5' terminus more closely, the strong-stop plus DNA was digested with SphI. This enzyme cuts ca. 100 bases from the 5' end (Fig. 1), releasing fragments A' andB', which are smaller and hence better resolved than the corre-sponding fragments (A and B) released byEcoRI (Fig. 2b, lane 1). The migration of the fragments was measured by scanning the autoradiograms with a densitometer and then measuring the distance between thevarious peaks relative to the single nucleotide spacing. When the sample was first treatedwith alkali, band A' disappeared, as expected, anda novel species appeared which migrated slightly slowerthan band B'(Fig. 2b, lane 2). The difference in migrationrelative to B' was equivalent to one third of a single nucleotide increment. The identity of the novel species became appar-ent when the sample was treated with alkalinephosphatase before electrophoresis (Fig. 2b, lane 3). Band B' then disappeared, and only the novel species remained. We interpreted these results to mean that the naturalremovalof the primerin the endogenous reaction leaves a 5'phosphate on strong-stop plus DNA. Incontrast, removal of theprimer with alkali leaves a 5' OH, resulting in a species with a relatively slower electrophoretic mobility. The ability of a terminal phosphate to increase mobility has been previously described (24). RNase H is known to produce oligoribonu-cleotides with a 5'-terminal phosphate (2, 30). This experi-ment showed that the strong-stop plus DNA also has a 5' phosphate after primer removal.
Specificity of initiation in reconstructed reactions. Studies were undertaken to compare the specificity of priming and plus-strand initiation observed in endogenous reactions to that obtained in reconstructed reactions with purified com-ponents: reverse transcriptase, minus-strand DNA as tem-plate, and viral RNA as a source of primers. The products from such reconstructed reactions, with or without alkali treatment to remove the primer, are shown in Fig. 3, lanes 1 and 2. Similar to the products ofendogenous reactions, we observed the bands previously designated A, B, and D. Band C was absent. This was expected since in the endogenous reaction band Ccorresponds to the 3' end of strong-stop plus DNA with an additional 18 nucleotides copied from the minus-strand tRNA primer. The minus-strand template used in the reconstructed reactions had been treated with both RNase A and alkali so species C could not bemade. For the products of an endogenous reaction, bands A and B were homogeneous (Fig. 2). However, for the reconstructed reac-tions a sizeheterogeneity was observed in both bands A and B (Fig. 3, lanes 1 and 2). There were a different number of extra species around band A as compared with band B, and weinterpreted this to mean that the heterogeneity was due to differences in both the size of the RNA primer and the position of the RNA-DNA junction. These heterogeneities were significantly reduced in reconstructed reactions in which the template DNA and source of RNAprimers were hybridized before synthesis (Fig. 3, lanes 3 and 4).
We havepreviously shown that an RNase A digest of viral RNA is an efficient source of primers for the synthesis of strong-stop plus DNA in reconstructed reactions (21). This wasexpected since RNase A only cleaves at pyrimidines and since the sequence upstream from the initiation site of strong-stop plus DNA is made up of purines, thePPT.Figure 4dshows the RNA oligomer that would be generatedfrom an RNase A digestion of viral sequences around thestrong-stop plus DNA origin. Further experiments were undertaken to characterize the RNA species that were actually used as primers and to determine where the plus-strand DNA was
1 2 3 4
.,.17
5 6
-A
[image:3.612.383.503.76.260.2]B
FIG. 3. Acrylamide gel electrophoresisofstrong-stopplusDNA from reconstructed reactions. Minus-strand viral DNAcontaining 0.6ng of LTRminus-strandsequences wasincubatedinareaction mixturecontainingreversetranscriptase andviralRNA as indicat-ed. Labeled products related to strong-stop plus DNA were en-riched for and analyzed on acrylamide gels as described in the legendtoFig. 2. Lanes: 2, 180 ng of viral RNA(containing6.6 ng of LTR sequences)added; 1, sampleas inlane2 excepttreatedwith alkali(0.3 NNaOH; 100°C; 10 min); 3, 180 ngofviral RNAadded, prehybridized(to aCrtof 1 mol -s/liter)toDNAtemplate;4,sample
asinlane 3except treated withalkali; 5,180 ngofviralRNAadded, predigestedwith RNase A; 6, sample asin lane 5 except treated with alkali. Bands labeled A and Bare asdescribed in the text, andthe dots represent single nucleotide spacings fromasequencing ladder runin anadjacentlane, as inFig. 2.
initiated in such a reconstructed reaction (using RNase A-digested viral RNA as a source of primers) relative to an endogenous reaction. As before, labeled strong-stop plus-containing DNA products were isolated, digested with EcoRI, andsubjected toelectrophoresis on polyacrylamide gels without or with aprevious alkali treatment(Fig.3, lanes 5 and6). The resulting bands were compared to anadjacent sequencing ladder of cloned DNA, as in Fig. 2A. The indicated band B in lane 6 corresponds to the single DNA initiation site, after primerremoval, as seen in the endoge-nous reactions (Fig. 2a). Clearly in thisreconstructed reac-tion DNA synthesis initiated at twoadditional sites, appar-ently located onenucleotide oneither side of thatsite. The indicated band A in lane 5, with RNAprimer still associated, showed the samedegree ofheterogeneityasband B(Fig.3). We interpreted this as evidence that the primers were predominantly of a single size, 11 bases, and that the heterogeneity around bands A and B was due to two new positions for the location of the RNA-DNA junction. A comparison of lanes 5 and 6 also suggests that RNase H removesthe RNAprimers moreefficiently fromincorrectly initiated molecules than from those initiated atthe correct location (Fig. 3). The significance of this finding is being furtherinvestigated.
RoleofthetemplateDNA inspecificplus-strand initiation. Inthe above studies with endogenous reactions, and even with reconstructed reactions, the size of the RNA primers and thelocationof the RNA-DNAjunctionwereremarkably specific. Thefollowingexperimentwasundertaken to test an hypothesis of how the viral minus-strand DNA template might contribute tothis specificity.
The viral LTR is essentially a double-stranded copy of
on November 10, 2019 by guest
http://jvi.asm.org/
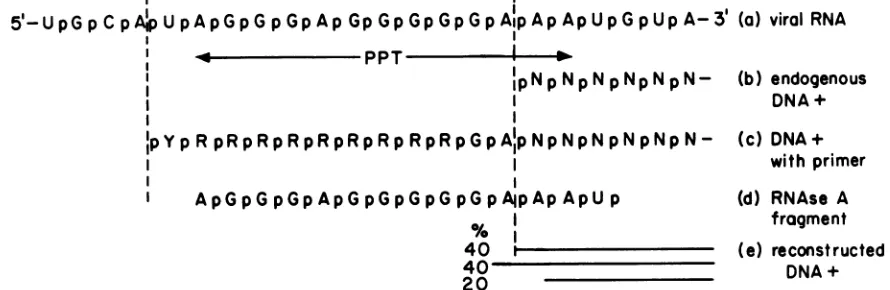
5'-UpGpCpAipUpApGpGpGpAp
GpGpGpGpGpAlpApApUpGpUpA-3'
(a) viral RNAI
PPT
IIIpNpNpNpNpNpN-IpYp
RpRpRpRpRpRpRpRpRpGpA'pNpNpNpNpNpN-A pRGp p p p p p p p G pAlp NAp p p
I ApGpGpGpApG pGpG pGpG pApAp ApU p
40 1
(b) endogenous DNA+
(c)
DNA+ withprimer
(d)
RNAse
A fragment(e)
reconstructed DNA+FIG. 4. Features of the nucleotide sequence around theoriginof strong-stop plus DNA. N, Nucleotide; Y, pyrimidine; R, purine. (a) Published sequenceof viral RNA (20);(b) deduced5'end of strong-stopplusDNA,assynthesizedin theendogenous reaction; (c)sequenceof theRNAprimer attached to strong-stop plus DNAfromanendogenousreaction; (d)RNaseAfragmentthat would be released from viral RNA inthe regionincluding strong-stopplus DNA; (e)interpretation of the three initiation sites observed in reconstructed reactions that used RNaseA-digested viralRNAas a sourceofprimerswithindicated relative amounts,asdeducedby densitometry (asfromFig. 3, lane6). strong-stop plus DNA. It is flanked by a short inverted
repeat sequence (IR) which for Rous sarcoma virus is 15 bases in length with three mismatches (20). Thus on the minus-strand DNA template, a copy of the IR is located precisely at the initiation site of strong-stop plus DNA. A copy complementary to this is present at the 5' end of the template(Fig. 1). This suggested that a possible mechanism for the specific primer generation and utilization might involve an interaction between these two IR elements. To test this hypothesis we digested a sample of endogenously synthesizedDNAwith HpaII beforepurifyingthefragment containingtheoriginof strong-stopplusDNA on analkaline sucrose gradient. Such a digestion should remove the 5' terminusof the minus-strandtemplate, includingonecopyof the IR (Fig. 1). We compared the activity of this DNA template relative to undigested minus-strand template in reconstructed reactions. Unlabeled reaction products were analyzedonagarosegels. After Southern transfer(23), LTR-specific probes were used to detect template and product DNA (Fig. 5). The three lanes labeled (-) arefrom recon-structed reactions with the undigested minus-strand tem-plate, and those labeled (+) are from reactions with the HpaII-digested template. An M13 probe specific for LTR minus-strand sequences was used to show the size ofthe minus-strand DNAtemplate.TheundigestedDNAtemplate was large and heterogeneous, but afterHpaIIdigestion was reducedtoadiscretebandof850bases,thepredicted size of theHpaII fragment-containingLTRsequences(Fig.5,lanes 1). Lanes 2and 3showwhat was detected with anM13probe specific for LTR plus-strand sequences in reconstructed reactionswith no addedprimers (lanes 2)orin reactions with viral RNA added as a source of primers (lanes 3) (Fig. 5). The mainplus-strand product fromtheundigested template wasstrong-stopplusDNA, whereasthe majorproductfrom the HpaII-digestedtemplatewas260bases (Fig. 5, lanes3). This was the size predicted for correct initiation of strong-stop plus DNA and for elongation to the HpaII site. In summary, this experiment did not detect any role for an interaction between the IR sequences in the specific initia-tion ofstrong-stopplus DNA.
DISCUSSION
Thesestudies have shownthat in the synthesisof strong-stop plus DNA in the endogenous reaction of disrupted
virionsseveral remarkably specific events are involved (Fig. 4b and c). (i) Onlyone size of primerwas seen, indicating precise5' and3' ends. (ii) Thesiteofinitiation ofDNAwas alwaysatthesamenucleotide. (iii)Theprimerwas apparent-ly removedin asingle step, since intermediatescontaininga partial primerwere notdetected. Furthermore,thesequence oftheprimerwasbothuniform andconsistent withthe RNA sequence (20)expected upstreamfrom the origin of strong-stopplusDNA.
These specificities of the endogenous reaction were also achieved in reconstructed reactions with purified compo-nents.The necessaryspecificitiesthuscouldbeprovided by thetemplateDNA,thesourceofRNAprimers, andreverse transcriptase. Apparently there was no requirement for cellular or additional viral factors, such as specific nucleic acidbinding proteins. Thisdoes not excludethe possibility that invivo otherfactors could play anauxiliary role. The observed in vitro use ofRNA primers, in the absence of ribonucleotide precursors, has also ruled out de novo syn-thesis ofprimers, for example,by aprimase.
Thecombined evidence indicates that theRNAprimer is generatedfrom the viralRNAby theaction ofthe RNase H activitypresent in the viralreversetranscriptase.The main argument is that it was the only nuclease available in the reconstructed reaction.RNase Hdegradesthe RNAportion ofDNA-RNA hybrids (15). The avian retrovirus RNase H generally releases oligoribonucleotides of10 to12 bases with a 5'-terminal phosphate (2, 30), in agreement with our observations. However, the documented properties of the viral RNase H are not sufficient to explain the specificities wehave observed. Itmaybethat RNase Hismorespecific than has been previously described, or additional factors maybe involved. Onesuchfactorwould bethat we assay the functional combination of primer generation and primer utilization;RNase H must generate manyother oligonucleo-tides thatfailto act asprimers. Another factor could be the primary structure ofthe template. As others have noted, every retroviral genome contains a PPTin this region (28), and it is essential for infectivity (22). For another "retroid virus", cauliflower mosaic virus,there are two specific sites forplus-strand DNAinitiation, and both have a PPTin the immediate vicinity(6). Nevertheless, it isimportanttostress that thePPTmay have other roles, and as far as the initiation ofplus-strandDNAsynthesis, the PPT isatmost aflag.Itis
I I
I I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.86.531.77.222.2]Kb M
3.1 2.4
(-) (+)
1 23 123
___,
0.3 l F SS+
FIG. 5. Agarose gel electrophoresis of reconstructed reactions with undigested orHpaIl-digested templates. Reconstructed
reac-tions contained reverse transcriptase with or without added viral RNA.Reactionsindicatedby(-) hadhigh-molecular-weight
minus-strand DNA as a template. For reactions indicated by (+), the
template was the HpaII restriction fragment of viral DNA which containedtheoriginofstrong-stop plus DNA. Lanes: 1, sequences
detected with an M13 probe specific forthe LTR minus strand; 2,
sequences detected with an M13 probe specific for the LTR plus
strand; reaction run without added RNA; 3, sequences detected withanM13probe specific for LTRplus-strand sequences;reaction runwiththe additionofviral RNA. Attheleft is cloned viral DNA
(4) digestedwithEcoRI as anLTR size marker.
not theprimer.Theprimer begins onenucleotide outside the
5' end of the PPT and ends three nucleotides within the 3'
end(Fig. 4).Secondary structureof thetemplate mayyetbe
involvedinthespecificityofinitiation, by analogywithother replication origins (8), inparticular for thebacterial plasmid
Col El, inwhich both secondarystructure and anRNase H
determinetheinitiationsite(10, 26). Ourlimited studyshows only that there is no requirement for secondary structure
mediated by the two IR sequences on the template DNA.
The RNase H activity ofthe reverse transcriptase must
alsoberesponsible forthe efficientremoval ofRNAprimer. In reconstructed reactions, RNase H has been shown to
remove the avian tRNATrP primer for minus-strand DNA synthesis (18) and toremove RNaseA-derived
oligoribonu-cleotidesused asprimersforreversetranscriptionofmurine
leukemiavirussequences(3). Weobservedthat after primer
removala5'phosphate remainedontheDNA, analogousto the 5' phosphate left by RNase H on its RNA degradation
products (2).
Approximately 60% of the strong-stop plus DNA
mole-cules completelylacked5'-terminal ribonucleotides,
where-asthe remaindercontained thefull 12-baseprimer. This
all-or-nonepropertyof theprimerremovalis incontrast towhat has been described for RNA primer removal during the replication ofEscherichia coli (16) and simian virus40 (1). Why are 40% of the RNA primer molecules apparently spared from RNase H action underourreactioncondition?
Perhaps,asChampouxetal.have suggested, dissociation of
theprimer-product complexfromthe template DNAoccurs
sothatthe RNAprimerisnolongerasubstrateforRNase H (3).
We have reconstructed the initiationof plus-strand DNA synthesis withanRNase AdigestofviralRNA. Three sites ofinitiation ofDNA synthesiswere found:one the same as
occurs in the endogenous reactionand the othertwo adja-cent and one nucleotide away (Fig. 4e). We also detected three alkali-sensitive fragments that were 11 bases longer
thanthe threebandsseenafteralkali. Anobvious
interpreta-tionwasthatan11-baseRNAprimerwasusedateach of the threeinitiation sites. The RNase Afragment thatwould be
expected
from thisregion
of the viral RNA is 14baseslong
(Fig.
4d).
Before it could be used as aprimer
the 3'phosphate
wouldneedtobe removed.Also,
tobeconsistent with ourdata,
an additional two to four nucleotides would havetobe removed. Inasimilar studywithmurine leukemiavirus,
Champouxet al. detectedtworather than three DNA initiation sites (3). The three initiations we detectedbegan
withdeoxyadenosine
(Fig.
4e), and thiswas alsotrueforthetwo murine leukemia virus initiations. It is remarkable that of the many oligoribonucleotides released from viral RNA
by
RNaseA,
ratherthanRNase H,only
anarrow-size classwere utilized asprimers, and furthermore, the initiation site was almost coincident with that observed in
endogenous
reactions.
In summary, a variety of factors seem to influence the
specific
initiation of viral plus-strand DNA, but in vitro atleast,
all thefactors arecontributed by thetemplate,primer,
and enzyme. This is in marked contrast tothe
multicompon-ent systems known to berequired
for correct initiation ofprocaryotic
genomes and bacteriophages(8).
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants CC-22651, CA-06927-20, and RR-05539 from the National Institutes of Health; grantMV-7from theAmerican CancerSociety;and byan
appropriation from the Commonwealth of Pennsylvania. J.K.S.was supportedbyPublic Health Service PredoctoralTrainingGrant GM-07071-08tothe University ofPennsylvania.
Wethank W. Masonfor helpfuldiscussions. LITERATURE CITED
1. Anderson, S., and M. DePamphilis. 1979. Metabolism of Okaza-ki fragments during simian virus 40 DNA replication. J. Biol. Chem. 254:11495-11504.
2. Baltimore, D., and D. Smoler. 1972. Association ofan endori-bonuclease with the avian myeloblastosis virus deoxyribonucle-icacid polymerase. J. Biol. Chem. 247:7282-7287.
3. Champoux, J. J., E. Gilboa, and D. Baltimore. 1984. Mechanism of RNA primer removal by the RNase H activity of avian myeloblastosis virusreversetranscriptase.J.Virol. 49:686-691.
4. DeLorbe, W. J., P. A. Luciw, H. M. Goodman, H. E. Varmus,
andJ. M. Bishop. 1980. Molecular cloning and characterization of avian sarcoma virus circularDNAmolecules. J. Virol. 36:50-61.
5. Faras, A. J., J. M. Taylor, W. E. Levinson, H. M. Goodman,
and J.M. Bishop. 1973. RNA-directed DNA polymerase of
Roussarcoma virus: initiationof synthesiswith70SviralRNA astemplate. J. Mol. Biol. 79:163-183.
6. Hohn, T., M. Pietrzak, L. Dixon, I. Koenig, J. Penswick, B.
Hohn, and P. Pfeiffer. 1983. Involvement of reverse transcrip-tase in cauliflower mosaic virus replication, p. 28-33. In H. D. Robertson, S. H. Howell, M. Zaitlin, and R. L. Malmberg (ed.), Plantinfectious agents. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
7. Hsu, T. W., and J. M. Taylor. 1982. Single-stranded regionson
unintegrated avian retrovirus DNA. J. Virol.44:47-53. 8. Kornberg,A. 1980. DNA replication, p. 347-413. W. H.
Free-man& Co., San Francisco.
9. Mason, W. S., C. Aldrich, J. Summers, and J. M. Taylor.1982.
Assymetric replication of duck hepatitis B virus DNA inliver cells: free minus-strand DNA. Proc. Natl. Acad. Sci. U.S.A.
79:3997-4001.
10. Masukata, H., and J.-I. Tomizawa. 1984. Effects of point mutations on formation and structure of the RNA primer for Col El DNA replication. Cell 36:513-522.
11. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol.
65:499-560.
12. McMaster, G. K., and G. G. Carmichael. 1977. Analysis of
single- and double-stranded nucleic acids on polyacrylamide
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.132.257.75.197.2]andagarose gels by using glyoxal and acridine orange. Proc. Natl. Acad. Sci. U.S.A. 74:4835-4838.
13. Mitra, S. W., M. Chow, J. Champoux, and D. Baltimore. 1982. Synthesis of murineleukemia virusstrong stop DNAinitiatesat aunique site. J.Biol. Chem. 257:5983-5986.
14. Mitra, S. W., S. Goff, E. Gilboa, and D. Baltimore. 1979. Synthesis ofa 600nucleotide-longplus-strand DNAby virions of Moloney murine leukemia virus. Proc. Natl. Acad. Sci. U.S.A. 76:4355-4359.
15. Moelling, K., D. P. Bolognesi, H. Bauer, W. Busen, H. W. Plassman, and P. Hausen. 1971. Association of viral reverse
transcriptase with an enzyme degrading the RNA moiety of
RNA-DNAhybrids. Nat. New Biol. 234:240-243.
16. Ogawa, T., and T. Okazaki. 1984. Function ofRNAse H in
DNA replication revealed by RNAse Hdefective mutants of Escherichia coli. Mol.Gen.Genet. 193:231-237.
17. Olsen, J. C., and K. F. Watson. 1980. Avian retrovirus
RNA-directed DNAsynthesis by purifiedreverse transcriptase. Co-valentlinkage ofRNA toplus strandDNA. Biochem.Biophys. Res. Commun. 97:1376-1383.
18. Omer,C. A., and A. J. Faras. 1982. Mechanism ofrelease of the
avianretrovirus tRNATrPprimermolecule from viral DNA by
ribonuclease H during reversetranscription. Cell 30:797-805. 19. Sabran, J. L., T. W. Hsu, C. Yeater, A. Kaji, W. S. Mason, and
J. M. Taylor. 1979. Analysis of integrated avian RNA tumor virus DNA intransformedchicken, duck,and quailfibroblasts.
J.Virol.29:170-178.
20. Schwartz, D. E., R. Tizard, and W. Gilbert. 1983. Nucleotide
sequenceofRous sarcomavirus. Cell32:853-869.
21. Smith, J. K., A. Cywinski, and J. M. Taylor. 1984. Initiation of plus-strand DNA synthesis during reverse transcription ofan
avian retrovirusgenome. J.Virol.49:200-204.
22. Sorge, J., and S. H. Hughes. 1982. Polypurinetractadjacentto theU3 regionofthe Rous sarcomavirusgenomeprovidesa
cis-acting function.J. Virol. 43:482-488.
23. Southern, E. M. 1975. Detection ofspecific sequences among
DNAfragments separated by gel electrophoresis.J.Mol. Biol.
98:503-517.
24. Tapper, D. P., and D. A. Clayton. 1981. Altered mobility of polydeoxyribonucleotides in high resolution polyacrylamide gels duetoremoval of terminal phosphates.Nucleic AcidsRes.
9:6787-6794.
25. Taylor,J.M., A.Cywinski, and J. K. Smith. 1983. Discontinui-ties in the DNA synthesized byan avian retrovirus. J. Virol.
48:654-659.
26. Tomizawa, J.-I., and T. Itoh. 1982. The importance of RNA secondary structure in Col El primerformation. Cell
31:575-583.
27. Varmus, H., and R. Swanstrom. 1982. Replication of
retrovi-ruses, p. 369-512. In R. Weiss, N. Teich, H. Varmus, andJ.
Coffin (ed.),RNA tumorviruses, 2nded., ColdSpring Harbor Laboratory, Cold Spring Harbor,N.Y.
28. Varmus, H. E. 1983.Retroviruses, p.411-503. In J. A. Shapiro
(ed.), Mobile genetic elements. Academic Press, Inc., New
York.
29. Varmus, H. E., S. Heasley, H.-J. Kung, H. Opperman, V. C.
Smith,J. M. Bishop, and P. R. Shank. 1978.Kinetics of synthe-sis, structure, and purification of avian sarcomavirus-specific
DNAmade inthe cytoplasmof acutely infected cells. J. Mol. Biol. 120:55-82.
30. Verma, I. M. 1975. Studieson reverse transcriptase ofRNA tumor viruses. III. Properties of purified Moloney murine leukemia virus DNA polymerase andassociated RNase H. J.
Virol. 15:843-854.
31. Watson, K. F., P. L. Schendel, M. J. Rosok, and L. R. Ramsey. 1979. ModelRNA-directedDNAsynthesis by avian myeloblas-tosis virus DNA polymerase and its associated RNase H.
Biochemistry 18:3210-3219.