Proteolytic Activity through Mutational Analysis
Valeria Lulla, Liis Karo-Astover, Kai Rausalu, Andres Merits, Aleksei Lulla
Institute of Technology, University of Tartu, Tartu, Estonia
Semliki Forest virus (genusAlphavirus) is an important model for studying regulated nonstructural (ns) polyprotein processing. In this study, we evaluated the strictness of the previously outlined cleavage rules, accounting for the timing and outcome of each of three cleavages within the ns polyprotein P1234, and assessed the significance of residues P6 to P4 within the cleavage sites using an alanine scanning approach. The processing of the 1/2 and 3/4 sites was most strongly affected following changes in resi-dues P5 and P4, respectively. However, none of the mutations had a detectable effect on the processing of the 2/3 site. An analysis of recombinant viruses bearing combinations of mutations in cleavage sites revealed tolerance toward the cooccurrence of native and mutated cleavage sites within the same polyprotein, suggesting a remarkable plasticity of the protease recognition pocket. Even in a virus in which all of the cleavage sequences were replaced with alanines in the P6, P5, and P4 positions, the processing pattern was largely preserved, without leading to reversion of cleavage site mutations. Instead, the emergence of second-site mu-tations was identified, among which Q706R/L in nsP2 was confirmed to be associated with the recognition of the P4 position within the modified cleavage sites. Our results imply that the spatial arrangement of the viral replication complex inherently contributes to scissile-site presentation for the protease, alleviating stringent sequence recognition requirements yet ensuring the precision and the correct order of processing events. Obtaining a proper understanding of the consequences of cleavage site manipulations may provide new tools for taming alphaviruses.
A
lphaviruses (familyTogaviridae) are plus-stranded RNA vi-ruses that infect vertebrate hosts and insect vectors. Semliki Forest virus (SFV) and Sindbis virus (SINV) are the best-studied members of the genus. The genome of alphaviruses is approxi-mately 11.5 kb long and contains two open reading frames. Struc-tural proteins are expressed from subgenomic RNA (sgRNA) and are dispensable for viral RNA replication. Depending on the viral species and strain, nonstructural (ns) proteins are expressed in the form of one (P1234) or two (P123 and P1234) polyproteins, which are subsequently processed into mature nsP1 to nsP4 (1,2).A polyprotein precursor(s) is absolutely required for replicase complex formation and cannot be replaced with individually ex-pressed nsPs (3). Virus replication begins with the synthesis of minus-strand RNA, coinciding with the formation of the physical structures of the replicase complexes, which are membrane in-vaginations termed spherules. Spherules contain viral RNA repli-cation intermediates (double-stranded RNAs [dsRNAs]) and rep-licase complex proteins (4). Spherules initially form at the plasma membrane of the cell and can be translocated into the cytoplasm, where fusion with endosomes results in the formation of virus replication organelles— cytopathic vacuole type I (CPV-I) struc-tures (4,5). Replication events and, possibly, spherule formation are accompanied by gradual and well-regulated P1234 processing. The P1234 polyprotein cleavage “rules,” which were first de-scribed more than 2 decades ago, ensure that the polyprotein is first processed into P123 and nsP4 (6,7), forming the early repli-case, which is capable of synthesizing the minus strand of RNA (3, 8–10). This cleavage is the only processing event that is absolutely required for virus infectivity (11,12). P123 is subsequently pro-cessed into nsP1 and P23, which is a reaction that takes place incis
(13). P23 is then processed intransinto nsP2 and nsP3 (13,14). The last reaction proceeds extremely rapidly, and the P23 inter-mediate cannot normally be detected in cells infected with wild-type (wt) SFV or SINV (6,11,15). Mature nsP1 to nsP4 arrange
the late replicase, which synthesizes new genomic RNAs and sgRNAs (10,16). Significant amounts of nsP3 (17), nsP2 (18), and nsP1 (19) localize to different compartments of the cell, where they likely exhibit a number of specific functions that are different from those involved in RNA replication.
nsP2 is the sole proteolytic enzyme responsible for the process-ing of P1234 of SFV (20,21). The alphaviral ns protease presum-ably functions as a molecular switch that first releases nsP4 to activate the minus-strand RNA synthetic capability of the viral replicase and, upon subsequent P123 processing, mediates con-formational changes that are necessary for the progression to plus-strand RNA synthesis (9,16). Disturbance of these events results in serious defects. Viral mutants with a hyperactive form of nsP2 are reportedly not infectious (22). Viruses with mutations block-ing cleavage between nsP1 and nsP2 (here the 1/2 site; similar designations are used for other sites as well) and/or cleavage of the 2/3 site display diminished replication levels and temperature-sensitive (ts) phenotypes. In the case of SINV, these defects can be compensated for by second-site adaptive mutations in nsP4 (12, 14,16). Changes in P123 processing may also result in reduced cytotoxic properties of viral vectors (23), a reduced ability to sup-press Jak/STAT signaling (24), and changes in neurovirulence (25).
Previous studies by our group addressing the processing re-quirements for individual cleavage sites within the SFV ns
poly-Received1 June 2013Accepted5 July 2013 Published ahead of print17 July 2013
Address correspondence to Aleksei Lulla, [email protected].
V.L. and L.K.-A. contributed equally to the study.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01485-13
on November 7, 2019 by guest
http://jvi.asm.org/
protein revealed that the 3/4 site is efficiently recognized intrans
with a short sequence of approximately 6 amino acid (aa) residues preceding the scissile bond, with the P4 amino acid residue play-ing the most important role (11) (conventionally, the P-side des-ignates the region of the cleavage site located upstream of the scissile bond, while the P=-side indicates the region located down-stream of it [26]). However, it is unlikely that the P4 residues in the cleavage sites within the alphavirus ns polyprotein serve as a uni-versal decisive factor that determines substrate recognition. Hence, 2/3 site cleavage was found to be the outcome of a specific macromolecular assembly that positions the cleavage site region into the active site of the protease, whereas the amino acid com-position of the 2/3 site itself is of lesser importance (14,27). It can therefore be concluded that alphaviral protease recognition of the two different sites relies on fundamentally different approaches. However, all three cleavage sites within the viral ns polyprotein should ultimately fit into the same recognition pocket of the pro-tease and become efficiently cleaved in the context of the function-ing replication complex, thus suggestfunction-ing that a common principle for their recognition should exist.
Despite significant progress in the understanding of alphavirus protease functionality, it is still not entirely clear what controls the timing of cleavage events within P1234 or how premature cleav-ages are prevented. In this study, we aimed to further elaborate the unifying molecular principles of substrate recognition to explain the sequential order of the proteolytic processing of the alphaviral ns polyprotein. Based on our previous results, we postulated that the recognition of each cleavage site represents a combination of specific sequence recognition and cleavage site presentation im-posed by macromolecular assembly-dependent positioning that does not require advanced sequence recognitionper se. To assess the importance of each of these components, we employed tar-geted alteration of the cleavage site sequences, expecting that, un-der conditions of unfavorable sequence-dependent substrate rec-ognition of a particular cleavage site within the polyprotein, assembly-dependent substrate presentation to the protease will reveal its contribution. To highlight the importance of particular amino acid residues involved in P1234 processing, we first ana-lyzed the region encompassing P6, P5, and P4 (P6-P4 region) of each cleavage site through alanine scanning. Next, SFV mutants containing potential processing-hampering changes in one, two, or all three cleavage sites were created and analyzed. This ap-proach was aimed at clarifying whether the architecture of the replication complex would compensate for defects in primary se-quence recognition and continue to force substrate accommoda-tion in the recogniaccommoda-tion pocket of the protease. We also expected that mutations in critical regions such as the cleavage sites would necessitate virus evolution, thus revealing the genetic interactions between particular protease domain residues and the correspond-ing substrate residues that are meant to interact with each other.
It was found that mutant viruses tolerated changes in cleavage sites surprisingly well. The order of the appearance of the correct cleavage products remained largely unchanged, implying a signif-icant extent of plasticity and tolerance in the cleavage process. This suggests that the macromolecular assembly-driven processes can compensate for the lack of specific sequence recognition and con-tribute to correct processing. Importantly, a reduction of the effi-ciency of processing in a majority of mutants was not sufficient to cause compensatory changes. A change revealing a genetic inter-action between residue 706 within the protease domain and the P4
residue in the cleavage site was found only for virus with all cleav-age site mutations. Interestingly, selected viruses with combina-tions of mutacombina-tions in all three cleavage sites were genetically stable. They were unable to infect mosquito cells but efficiently infected different mammalian cells. However, these viruses were defective in suppression of the interferon (IFN) response, implying the po-tential of a cleavage site mutagenesis approach for creation of attenuated virus variants.
MATERIALS AND METHODS
Cells and media.BHK-21 cells were grown in Glasgow’s minimal essen-tial medium (GMEM; Gibco) containing 10% fetal calf serum (FCS), 2% tryptose phosphate broth (TPB), 20 mM HEPES (pH 7.2), 100 U/ml pen-icillin, and 0.1 mg/ml streptomycin. HS633T and L929 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; PAA Laboratories GmbH) containing 10% heat-inactivated FCS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. HEK-Blue cells (InvivoGen) were grown in DMEM containing 10% heat-inactivated FCS, 50 U/ml penicillin, 50 g/ml streptomycin, and 100g/ml Normocin (InvivoGen). B16-Blue cells (InvivoGen) were grown in RPMI medium (PAA) containing 10% heat-inactivated FCS, 50 U/ml penicillin, 50g/ml streptomycin, and 100 g/ml Normocin. All cells were grown in a humidified incubator at 37°C under 5% CO2.
Bacterial strains.Escherichia colistrains DH5␣and XL10 (Gibco) were used for the propagation of plasmids. Plasmids containing infectious cDNAs (icDNAs) of SFV4 were propagated by using SOY medium (Gibco) containing 0.05 mg/ml ampicillin and glucose to a final concen-tration of 0.4%.
Recombinant polyprotein construction andin vitrotranslation.All of the point mutations were generated by using plasmids containing se-quences encoding the 1/2, 2/3, or 3/4 site regions of SFV4 via PCR-based mutagenesis. The mutated fragments were subsequently introduced into the coding sequence of SFV4 P123, P1234, or P1^2^34 in the pTM1 vector (14,28), utilizing the available restriction sites. The resulting clones were verified by sequencing and designated 4A1, 5A1, P123-6A1, and P123-1AAA (mutations in the 1/2 site); P123-4A2, P123-5A2, P123-6A2, and P123-2AAA (mutations in the 2/3 site); P1234-4A3, P1234-6A3, and P1234-3AAA (mutations in the 3/4 site); and P1^2^34-4A3, P1^2^34-6A3, and P1^2^34-3AAA (mutations in the 3/4 site of the polyprotein, where cleavages of the 1/2 and 2/3 sites were blocked by the mutation of P2 Gly residues to Ile).
A P4 Arg¡Glu mutation in the 3/4 site was introduced via site-di-rected mutagenesis, and the corresponding clone was designated P1234-34RE. Combinations of AAA mutations in the 1/2 and 2/3 sites or all three of the cleavage sites were obtained by subcloning the corresponding frag-ments from P1234-1AAA, P123-2AAA, and P1234-3AAA; the resulting clones were designated P1234-1⫹2AAA and P1234-3⫻AAA. The second-site mutations Q706R and Q706L, identified in the nsP2 regions of plaque-purified isolates of SFV4-3⫻AAA and SFV4-34RE, were intro-duced into P1234-3⫻AAA or P1234-34RE through site-directed mu-tagenesis and subcloning, and the obtained clones were designated P1234-3⫻AAA-Q706R and P1234-34RE-Q706L, respectively.
In vitrotranslation reactions were carried out by using the TNT-cou-pled T7 rabbit reticulocyte lysate system (Promega) according to the man-ufacturer’s protocol. Reaction mixtures (10 l) containing 4Ci of [35S]methionine (PerkinElmer) and 0.5g of plasmid DNA were incu-bated for 1 h at 30°C. In experiments to study the processing of polypro-teins with mutations in the 3/4 site, the proteasome inhibitor MG132 (final concentration, 10M) was added to the reaction mixture to prevent the degradation of nsP4. Thereafter, translation was stopped by adding cycloheximide at a final concentration of 1 mM. Incubation was subse-quently continued for 1 h under the same conditions, after which 10 ng of RNase A was added, and the mixture was incubated for another 5 min. Translation/processing products were separated via SDS-PAGE (1/10 of the lysate volume per lane) and visualized by using a Typhoon imager (GE
on November 7, 2019 by guest
http://jvi.asm.org/
Healthcare). Western blot analyses (in this case, two-fifths of the lysate volume was used per lane) were performed by using a rabbit polyclonal antiserum against nsP4 and a horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody; the signal was visualized by using the ECL im-munoblot detection kit (GE Healthcare).
Recombinant virus construction.icDNA clones with selected muta-tions were generated by transferring appropriate fragments from P123-1AAA, P123-2AAA, P1234-3AAA, P1234-1⫹2AAA, and P1234-3⫻AAA into the pSP6-SFV4 vector (29), utilizing the available restriction sites. The resulting plasmids were verified through sequencing and designated pSFV4-1AAA, pSFV4-2AAA, pSFV4-3AAA, pSFV4-1⫹2AAA, and pSFV4-3⫻AAA (seeFig. 2A). The pSFV4-34RE, pSFV4-3⫻AAA-Q706R, and pSFV4-34RE-Q706L plasmids were subsequently obtained by replac-ing fragments of pSP6-SFV4 with the correspondreplac-ing sequences from P1234-34RE, P1234-3⫻AAA-Q706R, and P1234-34RE-Q706L. pSFV4-1^2^3 was generated by replacing a fragment of pSP6-SFV4 with the corresponding sequence of P1^2^34, and to obtain pSFV4-1^2^3-E452A, the codon corresponding to Glu residue 452 in nsP4 was replaced with the codon for Ala. Capped transcripts, prepared with SP6 polymerase after linearization of the plasmids with SpeI, were used for the transfection of BHK-21 cells via electroporation. An infectious center assay (ICA) was performed as previously described (30). The primary virus stocks were collected following incubation for 24 h at 37°C, titrated by using a plaque assay on BHK-21 cells, and used in subsequent experiments.
Virological methods.Secondary stocks of parental SFV4 and mutated viruses were obtained as follows. Confluent BHK-21 cells grown in 100-mm dishes were infected with primary viral stocks at a multiplicity of infection (MOI) of 0.1 and then incubated for 1 h, after which cells were washed once with phosphate-buffered saline (PBS) and covered with complete GMEM containing 2% FCS. After 24 to 48 h of incubation at 37°C, the medium was collected and filtered, and viral titers were then determined by using a plaque assay on BHK-21 cells. The procedure was repeated to obtain subsequent passages. Viral growth curves were deter-mined in BHK-21 cells infected by primary viral stocks at an MOI of 5.
To adjust the titers of SFV stocks obtained for BHK-21 cells to the less susceptible HS633T and L929 cells, BHK-21 cells grown in a 6-well plate were infected with 50 PFU per well of SFV(3H)4-EGFP, which releases the enhanced green fluorescent protein (EGFP) marker protein from its ns region (31). HS633T and L929 cells grown in the same manner were infected with an identical amount of virus. The cells were covered with GMEM containing 2% FCS and 0.9% Bacto agar (Becton, Dickinson) and incubated at 37°C for 72 h. The foci of EGFP-positive cells were counted for all three cell lines. The number of foci formed by SFV(3H)4-EGFP in HS633T and L929 cells was 5 and 20 times lower, respectively, than that in BHK-21 cells. The obtained values were employed to recalculate titers of all viral stocks for HS633T and L929 cells.
Virus isolates were obtained via plaque purification, which was carried out as follows. Plaque-titrated viral primary stocks were used to infect confluent monolayers of BHK-21 cells such that approximately 10 to 20 plaques per 35-mm dish were expected to appear. Following infection, the cells were overlaid with GMEM containing 2% FCS and 0.9% Bacto agar and incubated at 37°C for 48 to 120 h, depending on the growth charac-teristics of the mutant virus involved. Afterwards, neutral red dye in 1% agar was added, and the plates were incubated for a further 2 to 8 h. The plaques were cut out, transferred onto a BHK-21 monolayer on a 24-well tissue culture plate, and incubated in GMEM containing 2% FCS. Viral stocks were collected following the observation of cytopathic effects or at 120 h postinfection (p.i.).
The presence of the introduced mutations was verified by reverse tran-scription-PCR (RT-PCR) and sequencing. For these procedures, viral RNA was extracted from each stock by using the RNeasy minikit (Qiagen) and then reverse transcribed by using the First-Strand cDNA Synthesis kit (Fermentas) and PCR amplified by using appropriate pairs of SFV-spe-cific primers. To identify second-site mutations, a set of PCR fragments
covering the complete SFV genome was sequenced from at least two in-dividual plaque-purified isolates for each mutated genome.
Metabolic labeling and immunoprecipitation.Pulse-chase labeling was carried out as described previously (11). Briefly, BHK-21 cells were infected with the corresponding viruses at an MOI of 100. At 3 h p.i., the cells were starved in methionine-free medium for 30 min and then labeled with 4.5Ci [35S]methionine (PerkinElmer) for 15 min. In the chase samples, the pulse was followed by a chase for 10 or 60 min in the presence of an excess of unlabeled methionine. The cells were then lysed by boiling in 1% SDS, and the material was divided into two equal amounts and subjected to immunoprecipitation analysis using combinations of anti-sera raised against the ns proteins (nsP1 plus nsP2 and nsP3 plus nsP4). The precipitated proteins were denatured by heating in Laemmli buffer, separated via SDS-PAGE, and visualized by using a Typhoon imager (GE Healthcare).
IFN-␣/assay.Two 35-mm plates of HS633T cells at 70% confluence were infected at an MOI of 20 at 37°C. After 1 h of incubation, inoculums were removed, and cells were washed once with PBS and overlaid with growth medium. At selected time points, samples were obtained from the first plate by collecting 20% of the growth medium and replacing it with the same amount of fresh medium. For the second plate, all growth medium was harvested and replaced with fresh medium at each time point. Aliquots of samples collected from the second plate were analyzed in a plaque assay with BHK-21 cells. For alpha/beta interferon (IFN-␣/) activity measurements, the infectious virus present in the collected sam-ples was inactivated via UV irradiation by using a UV cross-linker (5 min at 1,000J/cm2). The concentrations of IFN-␣/in the samples were measured by using HEK-Blue IFN-␣/sensor cells (InvivoGen), which express a secreted alkaline phosphatase (SEAP) marker under the control of the ISG54 promoter, according to the manufacturer’s protocol. SEAP activity was detected with QUANTI-Blue (InvivoGen) staining using a Tecan plate reader at 655 nm. The IFN-␣2 standard employed for nor-malization of the results was obtained from PBL Biomedical Laboratories. The same assay was performed by using mouse L929 cells, except that these cells were infected at an MOI of 5, and the released IFN-␣/was quantified by using B16-Blue IFN-␣/ sensor cells (InvivoGen) and mouse IFN-␣1 (PBL Biomedical Laboratories) as a standard. A UV-inac-tivated SFV4 sample derived from BHK-21 cells (IFN-␣/-negative) was used as a negative control to assess the possibility of IFN induction by inactive virus.
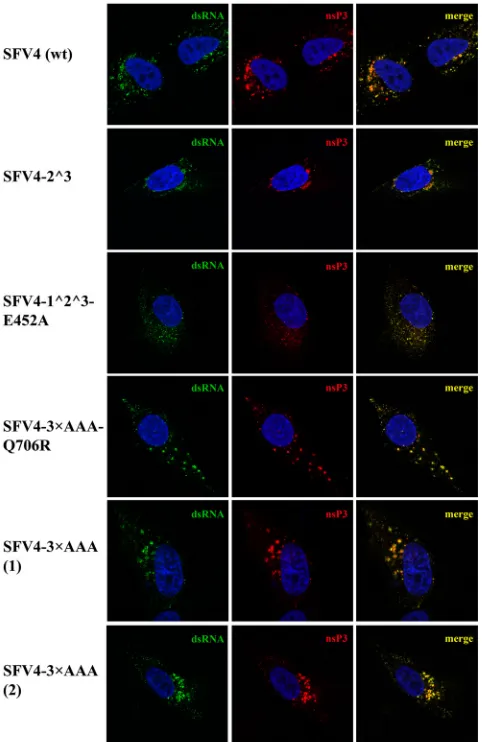
Immunofluorescence microscopy.HS633T cells were grown on cov-erslips and infected with SFV4 or mutant forms of SFV4 at an MOI of 20 at 37°C. At 8 h p.i., the cells were fixed with 4% paraformaldehyde, quenched with 50 mM NH4Cl, permeabilized with 0.1% Triton X-100, and costained by using rabbit nsP3 antiserum and a mouse anti-dsRNA antibody (J2; Scicons) as primary antibodies. Anti-rabbit Alexa-568- and anti-mouse Alexa-488-conjugated goat antibodies (Molecular Probes) were used as secondary antibodies. The nuclei were counter-stained with 4=,6-diaminidino-2-phenylindole (DAPI). Immunofluores-cence images were obtained by using an LSM710 confocal microscope (Zeiss).
RESULTS
The minimal length of the P-side of the polypeptide required for efficient processing of the 3/4 site of SFV4 was previously found to be 6 aa residues, among which the P3 (Ala) and P2 (Gly) residues are conserved in all three cleavage sites (11). The existing data regarding the specific roles of these 6 aa residues in polyprotein processing are rather limited. It has been shown that no variation in P2 Gly residues is tolerated at any cleavage site (20). Addition-ally, it has been demonstrated for intransprocessing of the 3/4 site that the amino acid residues at positions P4, P3, and P1 tolerate little variation, whereas the requirements for the P5 position are much less strict (11). To verify the importance and roles of the
on November 7, 2019 by guest
http://jvi.asm.org/
nonconserved P6, P5, and P4 residues at different cleavage sites in the processing of the ns polyprotein, an alanine scanning ap-proach was used.
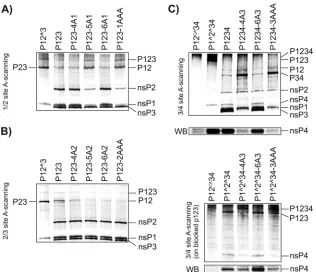
Processing of thein vitro-translated mutant polyproteins. The changes in polyprotein processing dynamics were first as-sayed by using constructs in which the amino acid residues at P6, P5, or P4 or at all three positions (P6-P4) at the 1/2 or 2/3 site of P123 were replaced with alanines. It was revealed that the intro-duction of an Ala residue at the P5 position in the 1/2 site left a major portion of P12 uncleaved, whereas the same residue at the P4 or P6 position did not affect 1/2 site processing (Fig. 1A). As expected, the introduction of Ala residues at the P6-P4 positions had the same effect on P123 processing, as did the P5 Ala substi-tution alone (Fig. 1A). In agreement with our previous findings (14), similar scans performed for the 2/3 site failed to reveal any clear changes in cleavage efficiency (Fig. 1B). The observed reduc-tion of the amount of P12, as detected in some experiments in-volving mutant P123 polyproteins (Fig. 1B), was not consistent and was most likely unrelated to the cleavage of the 2/3 site.
The processing of the 3/4 site may occur in multiple ways. First, it may be processed within full-length P1234; in this case, cleavage may take place either incisor intrans. Second, it may be processed
intransin the P34 intermediate, which is typical of the late stage of
SFV infection. Finally, the 3/4 site can be processed by P1234 and by any of its processing intermediates containing the nsP2 region (20). Therefore, the role of the P6 and P4 positions in 3/4 site processing was assayed in the context of both cleavage-competent (P1234) and cleavage-deficient (P1^2^34) polyproteins. The analysis carried out by using the P1234-derived substrates re-vealed that, unlike in the case of the 1/2 site, P4 Ala results in very inefficient cleavage at the 3/4 site and causes accumulation of the P34 processing intermediate (Fig. 1C, top). These data are in ac-cordance with results that we previously obtained by using a pu-rified protease and short substrates (11). In contrast, similar to the substitutions at the P5 position (11), the introduction of the P6 Ala residue had no detectable effect (Fig. 1C), indicating that the P6 position has a minor or no effect on 3/4 site processing. This result was confirmed directly by using the P1234-3AAA substrate. The processing of this mutant polyprotein was found to be indis-tinguishable from that of P1234-4A3 (Fig. 1C). The experiment using the cleavage-deficient P1^2^34-derived substrates revealed that the defect in 3/4 site processing caused by P4 Ala did not result from decreased processing of the P34 intermediate but from the defect in 3/4 site processing in general: in assays involving FIG 1Alanine scanning of the P6-P4 region of the 1/2, 2/3, and 3/4 sites within SFV4 ns polyproteins. Polyproteins were translatedin vitroin the presence of [35S]methionine. The reactions were stopped by adding cycloheximide, and the autoprocessing products of the polyproteins were separated via SDS-PAGE and visualized with a Typhoon imager. The presence of nsP4 was also revealed by Western blotting. The positions of the polyproteins and their cleavage products are shown on the right, and the substrate names are given at the top of the lanes. (A) Processing of polyproteins with mutations in the 1/2 site. P12^3 contains a Gly¡Val mutation in the P2 position of the 2/3 cleavage site. In P123-4A1, P123-5A1, and P123-6A1, the P4, P5, or P6 position of the 1/2 site contains Ala residues. In P123-1AAA, all of the amino acid residues of the P6-P4 region of the 1/2 site are replaced with Ala residues. (B) Processing of polyproteins with mutations in the 2/3 site. In P123-4A2, P123-5A2, and P123-6A2, the P4, P5, or P6 position of the 2/3 site contains Ala residues. In P123-2AAA, all of the amino acid residues of the P6-P4 region of the 2/3 site are replaced with Ala residues. (C) Processing of polyproteins with mutations in the 3/4 site. The proteasome inhibitor MG132 (final concentration, 10M) was added to the reaction mixture to prevent the degradation of nsP4. (Top) In P1234-4A3 and P1234-6A3, the amino acid residues at the P4 and P6 positions of the 3/4 site, respectively, are replaced with Ala residues. In P1234-3AAA, all of the amino acid residues in the P6-P4 region of the 3/4 site are replaced with Ala residues. (Bottom) The same mutations analyzed in the context of P1^2^34. P12CA34 indicates the polyprotein containing the C478A mutation in the active center of the nsP2 protease, preventing the cleavage of the polyprotein. A Western blot (WB) generated by using antiserum against nsP4 is presented below both panels. The experiment was repeated twice; data from one reproducible experiment are shown.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.135.451.65.337.2]P1^2^34, the 3/4 site was fully processed, whereas severely re-duced processing was observed for both P1^2^34-4A3 and P1^2^34-3AAA (Fig. 1C, bottom). These defects not only re-sulted in increased amounts of unprocessed polyproteins but also reduced the amount of nsP4 released. As nsP4 is an unstable pro-tein (32) and is therefore difficult to detect via autoradiography, its presence was also confirmed by Western blotting using the
samein vitrotranslation mixtures (Fig. 1C, bottom lines of both
panels). The results of these assays clearly confirmed that the pres-ence of the P4 Ala residue in the 3/4 site results in a reduction of released nsP4.
Taken together, these experiments revealed that the impacts of the P6-P4 positions on the recognition and processing of each cleavage site in P1234 are different: P4 represents the major deter-minant of the processing of the 3/4 site, while P5 serves this func-tion for the 1/2 site, and none of these posifunc-tions have any detect-able effect on the processing of the 2/3 site.
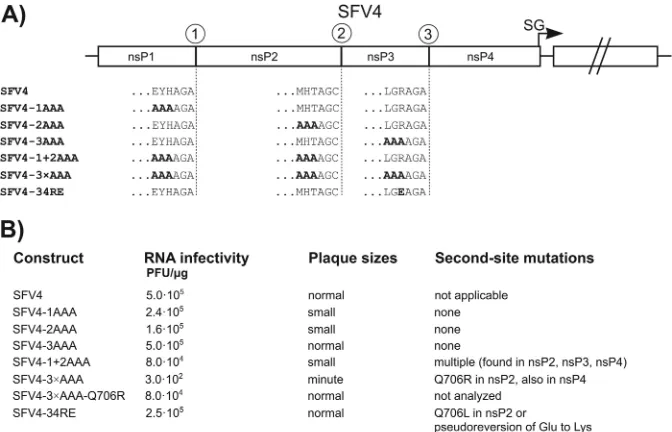
Infectivity of mutant RNA genomes and analysis of second-site mutations.To analyze the effect of these mutations on virus-infected cells, triple mutations of each cleavage site were intro-duced into an SFV4 icDNA clone (29); schematic representations of the obtained mutant genomes and their abbreviations are shown inFig. 2A. Corresponding RNAs were electroporated into BHK-21 cells, and their infectivities were examined by using ICA. This analysis revealed that alanines at the P6-P4 positions of any single cleavage site had little or no effect on the infectivity of the corresponding RNA genome (Fig. 2B). All of the mutant viruses replicated to a high titer and were similar to the parental virus, with the exception of plaques formed by 1AAA and SFV4-2AAA, which were smaller than those formed by SFV4 (Fig. 2B). In the case of SFV4-1AAA, this property can be attributed to the detected reduced rate of 1/2 site processing (Fig. 1A). However, for SFV4-2AAA, these results were unexpected, because no
appar-ent defect of 2/3 site processing was detected for the correspond-ing polyprotein (Fig. 1B). Therefore, the effect could be attributed either to minor changes that were undetectable in the cell-free system or to other defects that are important for virus infection but unrelated to 2/3 site processing. Consistent with the high in-fectivity of the corresponding transcripts, sequencing of the mu-tated regions by using either virus stocks collected directly from transfected cells or those passaged five times in BHK-21 cells did not reveal any reversions or pseudoreversions of the mutated se-quences. Furthermore, the growth kinetics of these viruses were very similar to those of a wt virus (Fig. 3), and complete sequenc-ing of several plaque-purified virus isolates did not reveal any potential compensatory changes in distant regions of their ge-nomes. Thus, it can be concluded that in cell culture, the presence of neutral Ala residues in the P6-P4 region of any single cleavage site was well tolerated.
Changes in a single cleavage site can reduce the speed of its processing but would unlikely alter the overall processing pattern. Therefore, two additional viral mutants were constructed, one containing P6-P4 alanines at the first two cleavage sites (SFV4-1⫹2AAA) and another in which all three sites contained Ala res-idues in the P6-P4 positions (SFV4-3⫻AAA) (Fig. 2A). The ICA revealed that the infectivity of the SFV4-1⫹2AAA RNA tran-scripts was reduced 6-fold compared to that of SFV4, while over a 1,000-fold reduction of infectivity was observed for transcripts corresponding to SFV4-3⫻AAA. In addition, the rescued viruses produced small (SFV4-1⫹2AAA) or minute (SFV4-3⫻AAA) plaques in BHK-21 cells (Fig. 2B). SFV4-1⫹2AAA maintained its characteristic phenotype (small plaques and reduced final titers) when it was passaged five times in BHK-21 cells, and sequencing failed to detect any reversions or pseudoreversions in the mutated P6-P4 positions or in any other regions of the cleavage sites. Se-quencing of the full genomes of several plaque-purified isolates of FIG 2Analysis of the effect of selected mutations in the context of the SFV4 genome. (A) Schematic representation of the parental SFV4 genome and the SFV4-1AAA, SFV4-2AAA, SFV4-3AAA, SFV4-1⫹2AAA, SFV4-3⫻AAA, and SFV4-34RE genomes. The sequences of the P6-P1 region of each cleavage site are given with the one-letter amino acid code; the mutated residues are presented in boldface type. The arrow indicates the position of the subgenomic (SG) promoter. (B) The infectivity ofin vitro-synthesized viral RNAs, the comparative plaque sizes determined in the ICA, and second-site mutations identified in the selected plaque-purified virus isolates are shown.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.124.460.66.282.2]SFV4-1⫹2AAA revealed point mutations in the nsP2, nsP3, and nsP4 regions for some of the isolates but not all of them; thus, no universal second-site mutation was revealed. Whether any of the detected nonconserved changes affected the growth of this res-cued virus remains unknown, as their effects (if present at all) were too small to be detected by using the approaches described above. In contrast, the 1,000-fold decrease in infectivity revealed for the transcripts of SFV4-3⫻AAA (Fig. 2B) strongly indicated the possibility of the appearance of the compensatory changes in the viral progeny. Indeed, while the viral titers increased very slowly in cultures transfected with SFV4-3⫻AAA transcripts (data not shown), the collected virus stock displayed relatively normal growth characteristics: compared to SFV4, its accumulation was only slightly delayed (1 to 2 h), and the final titers were reduced by no more than 10-fold (Fig. 3). These characteristics were main-tained after the stock was passaged five times in BHK-21 cells, indicating that changes in the viral genome required to rescue and/or to increase its infectivity had already taken place during viral release and propagation in the RNA-transfected cells. The sequence analysis of the SFV4-3⫻AAA stocks collected from transfected cells or obtained after five passages showed that all of the introduced mutations were maintained. Sequencing of the complete genomes of two individual plaque-purified isolates of SFV4-3⫻AAA revealed several second-site mutations in regions encoding nsP4 and nsP2. While the changes found in the nsP4 region were different between analyzed isolates, the change de-tected in nsP2 (Q706R) was common for both isolates. Taking
into account the location of this mutation in the protease domain, the Q706R mutation represented a likely candidate for a second-site compensatory change. This possibility was analyzed directly by introducing this mutation back into pSFV4-3⫻AAA. The ICA performed by using transcripts from this construct (termed SFV4-3⫻AAA-Q706R) revealed that the Q706R mutation resulted in a ⬎200-fold increase of RNA infectivity (Fig. 2B), strongly support-ing the conclusion that this ssupport-ingle change in the protease region was indeed the major factor restoring the infectivity of the SFV4-3⫻AAA mutant.
The Q706R mutation in nsP2 was detected only in the case of SFV4-3⫻AAA and not for SFV4-1⫹2AAA. The major difference between these viruses is the presence of P4 Ala in the 3/4 site in SFV4-3⫻AAA (Fig. 1C). This observation suggested that the Q706R mutation in nsP2 compensated for the introduction of an unfavorable P4 Ala residue into the 3/4 site. Q706 in nsP2 is im-mediately adjacent to the M707 residue, which has been predicted to be one of the protease subsites that is responsible for the recog-nition of the P4 position (33). Curiously, a previous analysis also suggested that the charged P4 residue of the 3/4 site in the ns polyproteins of many alphaviruses is likely involved in an inter-molecular interaction via the formation of a salt bridge with an oppositely charged S4 subsite residue within the substrate recog-nition pocket of the protease (e.g., P4 Arg of the SFV 3/4 site presumably establishes contacts with Asp711 of SFV4 nsP2 [33]). To directly test whether there were genetic interactions involving the P4 residue of the 3/4 site, we constructed another recombinant virus, SFV4-34RE, in which the Arg residue in the P4 position of the 3/4 site was replaced with a Glu residue. Despite the expected severe defect due to amino acid charge reversal, we found that the genome of the SFV4-RE site exhibited an initial infectivity that was only 2-fold lower than that of the genome of wt SFV4. This is similar to SFV4-3AAA and to viruses containing Arg¡Thr or Arg¡His changes in the P4 position of the 3/4 site (11): all these changes had little or no effect on the infectivity of SFV4. However, unlike the above-mentioned changes, P4 Glu was poorly toler-ated: in approximately 50% of the rescued viruses, a sion of Glu to Lys occurred. Most importantly, this pseudorever-sion was not the only way in which the virus overcame the observed defect: in several clones, P4 Glu was maintained, and its effect was compensated for in a manner similar to that of SFV4-3⫻AAA; i.e., the same residue in the protease recognition pocket was changed, but in this case, Gln706 was replaced with Lys (Fig. 2B). Together with the data obtained from the analysis of the SFV4-3⫻AAA isolates, this finding strongly supports the role of residue 706 of nsP2 in the recognition of the P4 residue within the 3/4 site.
Effects of combined mutations on the processing ofin vitro -translated polyproteins.To determine how the mutations pres-ent in the SFV4-1⫹2AAA, SFV4-3⫻AAA, and SFV4-34RE ge-nomes affect ns polyprotein processing in a cell-free system, the processing of P1234-1⫹2AAA, P1234-3⫻AAA, and P1234-34RE was analyzed as described above. These assays revealed that com-pared to wt P1234, the levels of fully processed nsP1 and nsP2 were considerably reduced in the case of P1234-1⫹2AAA, whereas the levels of nsP3 and nsP4 were not. Importantly, changes in the levels of the processing intermediates were also observed: the lev-els of P123 and P12 were increased, while that of P34 was consid-erably reduced (Fig. 4). With the exception of the latter result, these effects were expected and can be attributed to the nonopti-FIG 3One-step growth curves of rescued viruses. BHK-21 cells were infected
with P1 stocks of SFV4, 1AAA, 2AAA, 3AAA, SFV4-1⫹2AAA, and SFV4-3⫻AAA at an MOI of 5. Aliquots of the culture media were collected at 4, 6, 8, 10 12, 18, and 24 h p.i., and the viral titer in the aliquots was analyzed via plaque titration on BHK-21 cells. The symbols used to rep-resent the growth curve of each virus are given. The results of one out of two
reproducible experiments are presented for clarity.
on November 7, 2019 by guest
http://jvi.asm.org/
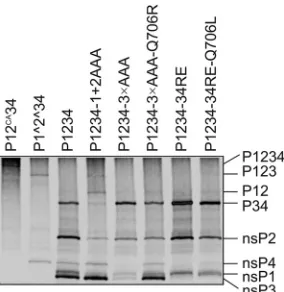
[image:6.585.79.248.62.335.2]mal P5 Ala at the 1/2 site (Fig. 1A). However, the reduced level of P34 was rather unexpected. One explanation for this effect might be that after P123 processing has been slowed down, the switch to the late cleavage pattern (in which P1234 is cleaved by nsP2 into P12 and P34), driven by the accumulation of free nsP2, did not occur. Alternatively, processing of the 3/4 site incis, which is pos-sible only in P1234 (stabilized by P5 Ala in the 1/2 site) and not in the P34 substrate, was activated. In either case, due to the intro-duced mutations, in addition to the altered rate of cleavage at individual sites, the overall P1234 cleavage pattern was changed.
In contrast to P1234-1⫹2AAA, remarkably diminished levels of all fully processed ns proteins were observed for P1234-3⫻AAA. This was especially evident for nsP3 and nsP4 and was correlated with the corresponding increase in the level of unpro-cessed P34; both of these effects are likely due to the presence of the Ala residue in the P4 position of the 3/4 site. In this mutant, P123 and P1234 were also detectable (Fig. 4[to see these polypro-teins clearly, overexposure of the gel was required]), although P123 was present at a slightly lower level than in the case of P1234-1⫹2AAA. Thus, both the P5 Ala in the 1/2 site and the P4 Ala in the 3/4 site are responsible for the observed processing defects, which are, in some cases, opposite each other (P5 Ala in the 1/2 site reduces the levels of the P34 intermediate, whereas P4 Ala in the 3/4 site increases them). Thus, processing of polyproteins with multiple defects revealed a more complex phenotype (Fig. 4) than could be deduced from the effects of individual mutations (Fig. 1), confirming that the processing of the 1/2 site and of the 3/4 site are not isolated events but clearly influence each other.
As the very low infectivity of SFV4-3⫻AAA most likely re-sulted from a disturbed processing pattern, it was important to verify the effect of the identified Q706R compensatory change on the processing of P1234-3⫻AAA. When this mutation was intro-duced into the P1234-3⫻AAA expression construct, the process-ing pattern of thein vitro-translated polyprotein became nearly identical to that of wt P1234 (Fig. 4). These data point to the conclusion that the infectivity of the SFV4-3⫻AAA mutant was
restored through reestablishing the normal, early (P1234 ¡
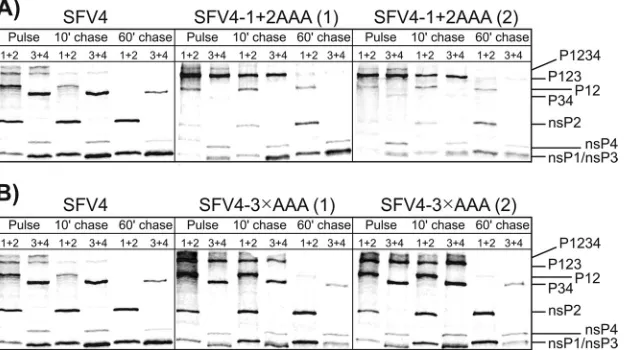
P123⫹nsP4, leading to replicase complex formation) polypro-tein processing. In addition, the effect of the Q706L mutation in nsP2 was analyzed in the context of P1234-34RE. First, the P4 Glu residue markedly reduced the processing of the 3/4 site but did not have any apparent effect on the processing of the other sites. The processing efficiency of the 3/4 site of P1234-34RE was slightly increased by introducing the Q706L mutation (note the appear-ance of free nsP4; in contrast, free nsP3 with an RE mutation at the C terminus cannot be clearly detected, as it comigrates with nsP1 [Fig. 4]), again confirming that amino acid residue 706 of nsP2 is involved in the recognition of the P4 residue of this cleavage site. Effects of combinations of mutations on P1234 processing in virus-infected cells.The processing of P1234 with P6-P4 alanine residues at the 1/2 and 2/3 sites or at all three cleavage sites was studied in infected BHK-21 cells through a pulse-chase experi-ment. For both mutants, two viral stocks, originating from indi-vidual plaque-purified viruses, were analyzed. In the case of SFV4-3⫻AAA, both stocks also contained the Q706R second-site mutation in nsP2, as an analysis of SFV4-3⫻AAA without com-pensatory changes was impossible due to the immediate emer-gence of second-site mutations (see above). This analysis clearly revealed that the processing of P1234 in all of the analyzed isolates was quite similar to that in parental SFV4: all of the individual nsPs were detected in the pulsed samples, and unprocessed pre-cursors were almost completely absent after a 1-h chase (Fig. 5). The almost identical processing patterns revealed for the two in-dividually plaque-purified stocks of SFV4-1⫹2AAA indicate that the dissimilar second-site mutations did not affect polyprotein processing. As observed in the case ofin vitro-translated P1234-1⫹2AAA, the major difference in these mutants compared to their wt counterpart was the increased stability of the P123 and P12 processing intermediates, which were both detectable in the chased samples (Fig. 5A). This effect likely resulted from the de-layed processing of the 1/2 site caused by the P5 Ala residue. Again, a clear reduction of P34 levels was evident in all of the samples, including those obtained immediately after the pulse (Fig. 5A). This effect is caused by the stabilization of the P123 portion of the ns polyprotein, which results in the processing of the 3/4 site at the stage of the P1234 precursor, and P34 is accordingly formed at lower levels. Both isolates of the SFV4-3⫻AAA mutant showed a more wt-like phenotype than did SFV4-1⫹2AAA, most likely due to the presence of the Q706R mutation in nsP2. The increased stability of the P123 precursor, which was present at a greater abundance in both pulsed and short-chase samples, was the only difference observed compared to wt SFV4 (Fig. 5B), and no aber-rantly or incompletely processed products (within the limits of accuracy of this analysis) were detected.
Thus, the differences in the processing pattern observed in the
in vitrotranslation/processing reaction (Fig. 4) were also fully
re-produced in the context of virus infection (Fig. 5). When consid-erable disturbances were caused by the combination of introduced mutations (such as 3⫻AAA [Fig. 4]), the correct (or almost cor-rect) processing pattern was reestablished through the most cost-effective method: the introduction of a single second-site muta-tion capable of compensating for multiple defects.
Multiplication of SFV4-3ⴛAAA viruses is restricted in many cell lines, while formation of CPV-I structures is apparently not affected. ns polyprotein processing is undoubtedly one of the most basic processes in the alphavirus infection cycle; therefore, FIG 4Effects of combinations of cleavage site mutations and the identified
second-site mutations on the processing ofin vitro-translated P1234 polypro-teins. The analysis was performed essentially as described in the legend ofFig. 1, and MG132 (final concentration, 10M) was used to prevent the degrada-tion of nsP4. Noncleavable P12CA34 and partially cleavable P1^2^34 (the pro-cessing of which results in P1^2^3 and free nsP4) were used as controls. The positions of the polyproteins and the products of the cleavage reactions are shown on the right. The experiment was repeated twice; data from one repro-ducible experiment are shown.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.93.235.66.212.2]its pattern is likely determined by different viral (11,13,14) rather than cellular factors. However, this fact does not exclude the pos-sibility that compensatory changes acquired in one cell type do not have the same effect on other cell types, as was recently reported for mutants of Venezuelan equine encephalitis virus (VEEV) (34), as basic functions, such as the formation of the active replicase complex, are not the only functions associated with the ns proteins of alphaviruses. In addition, the restoration of basic functions through second-site changes may occur at the cost of other, more specific functions of these proteins.
To investigate this possibility, we used SFV4-3⫻AAA viruses (two original plaque-purified isolates [Fig. 5A] and the rescued SFV4-3⫻AAA-Q706R) to infect different cell types. First, it was found that these mutants were not able to productively infect cells of mosquito origin, C6/36 (data not shown). This finding is in line with the observation that many mutations with a mild or no effect on vertebrate cells seriously inhibit the replication of alphaviruses in cells of their invertebrate vectors (12). Next, we observed that these mutant viruses were able to multiply in several cell lines of mammalian origin, including Huh7 human hepatocarcinoma cells (data not shown), indicating that unlike what was observed for VEEV nsP3 mutants (34), the effects of compensatory changes in the nsP2 protease were not specific for BHK-21 cells. However, in many of the tested cell lines, such as mouse L929 and human HS633T cells, the propagation of these mutant viruses was se-verely restricted.
HS633T cells were chosen to examine whether the mutant vi-ruses were capable of forming replication organelles in these cells. HS633T cells were found to be considerably less susceptible to wt SFV4 infection than BHK-21 cells: approximately 5 infectious units of SFV4 for BHK-21 cells corresponded to 1 infectious unit for HS633T cells. Furthermore, we were not able to synchronously infect all HT633T cells growing on a tissue culture plate at any reasonable MOI. Even at an MOI of 20, SFV4 was able to infect only approximately 95% of the HS633T cells. Both the
SFV4-3⫻AAA and SFV4-2^3 isolates, when used under these condi-tions, were able to infect 30 to 40% of the HS633T cells. Thus, the relative infectivity of the mutant viruses in HS633T cells was much lower than that of wt SFV4. This finding indicates that the com-bination of the mutations in the processing sites of the ns poly-protein and in nsP2 affected the infectivity of SFV4 for HS633T cells to a greater extent than for BHK-21 cells.
However, the immunofluorescence analysis revealed that in those HS633T cells where the mutant viruses managed to establish an infection, large nsP3- and dsRNA-containing vesicular struc-tures (representing 85 to 95% of all nsP3-containing complexes), presumably representing CPV-Is containing the SFV replicase complexes, were formed. These structures, with the exception of a slightly increased size, were similar to those observed in cells in-fected by wt SFV4 (Fig. 6), indicating that the formation of virus replication organelles was not affected and that other factors were responsible for limiting the infectivity of the mutant viruses. To study this phenomenon in greater detail, the SFV4-2^3 mutant, which is unable to process P23 (14), and the SFV4-1^2^3 mutant, which shows a complete lack of processing of the P123 polypro-tein, were also analyzed. Thein vitrotranscripts of the latter mu-tant were found to display extremely low infectivity in BHK-21 cells, and rescued SFV4-1^2^3 failed to reach titers higher than 1⫻103PFU/ml. To overcome this problem, an adaptive muta-tion, previously described for SINV (12), was introduced into the icDNA clone. This mutation, which changes Glu452 in nsP4 (cor-responding to the Glu451 residue in SINV nsP4) to an Ala residue, increased the infectivity of thein vitrotranscripts of the construct, now designated SFV4-1^2^3-E452A, to a level of 100 PFU/g RNA and allowed the rescued virus to multiply in BHK-21 cells to a titer of 4.5⫻107PFU/ml. Apparently, both SFV4-1^2^3 and SFV4-1^2^3-E452A also required additional second-site muta-tions to replicate to the indicated levels. A common mutation located within nsP1 (P336T) was indeed identified in plaque-pu-rified isolates of both of these viruses; analysis of its functional FIG 5Processing of the ns polyproteins in BHK-21 cells infected with wt SFV4, SFV4-1⫹2AAA, or SFV4-3⫻AAA. Cells were infected with wt SFV4 and two independently plaque-purified isolates of SFV4-1⫹2AAA and SFV4-3⫻AAA [designated (1) and (2)] and labeled with a 15-min pulse of [35S]methionine, followed by a 10- or 60-min chase with an excess of cold methionine. The proteins were extracted from the infected cells, denatured with SDS, and analyzed via immunoprecipitation and SDS-PAGE. The viruses used in the assays are indicated at the top of each panel, and the origins of the samples (pulse or chase) are shown below. The numbers directly above each lane denote the combination of antibodies against SFV ns proteins used for immunoprecipitation. The positions of the SFV ns proteins and their polyprotein precursors are indicated on the right. (A) Analysis of ns polyprotein processing for plaque-purified isolates of SFV4-1⫹2AAA. (B) Analysis of ns polyprotein processing for plaque-purified isolates of SFV4-3⫻AAA.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.139.451.69.244.2]significance was beyond of scope of the current study. Similar to other analyzed mutant viruses, SFV4-1^2^3-E452A was severely restricted in its ability to infect HS633T cells. However, in this case, a specific phenotype that was different from that of SFV4 and other mutants was also revealed through immunofluorescence microscopy. In infected HS633T cells, SFV4-1^2^3-E452A was able to induce the formation of visibly smaller dsRNA-containing foci (Fig. 6). Similar effects were detected in BHK-21 cells infected by these viruses (data not shown), indicating that the observed phenotypes of the replication organelles of mutant viruses were not cell type specific.
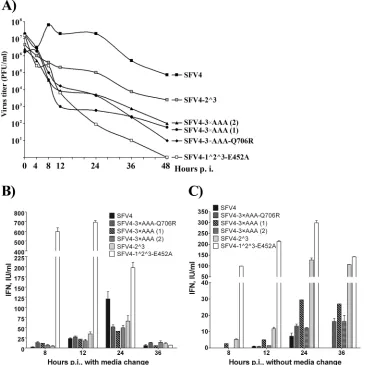
Infection of HS633T cells by viruses with mutations in cleav-age sites induces a type I interferon response that restricts the spread and multiplication of mutant viruses. Next, we con-structed one-step growth curves for SFV4, the SFV4-3⫻AAA iso-lates, SFV4-2^3, and SFV4-1^2^3-E452A in HS633T cells using a
high initial MOI of 20. Consistent with our previous observations, only SFV4 was able to produce infectious progeny in quantities exceeding the amount of virus used to infect the cells (Fig. 7A). In contrast, for SFV4-1^2^3-E452A, a steady decline of virus titers was observed throughout the experiment. Due to the large amount of virus used to infect the cells, we could not determine whether the virus detected during the course of infection was gen-eratedde novoin the infected cells or represented residual virus from the infection medium. Such behavior was not surprising, as HS633T cells display an intact interferon system (35), and an anal-ogous mutant of SINV exhibits severe growth restriction in IFN-␣/-positive cells (12). The growth curves for the remainder of the mutants were located between two described extremes but were more similar to that of SFV4 (although the titers were much lower) (Fig. 7A). This finding is consistent with a low level of production of infectious progeny; in addition, noticeable cyto-toxic effects were observed in SFV4-2^3- and wt SFV4-infected HS633T cells at 24 h p.i. However, the mutants did not reach high titers, indicating that their spread in the HS633T cell culture was restricted.
As the type I interferon response is known to restrict the spread of alphavirus infection in mammalian cells, the production and release of IFN-␣/into the growth medium were analyzed (Fig. 7B). Infection with SFV4-1^2^3-E452A caused IFN-␣/release as early as 4 h p.i. (data not shown); for the remainder of the mutant viruses, IFN-␣/release was detected at 8 h p.i. (Fig. 7B). However, the release of IFN-␣/by SFV4-1^2^3-E452A-infected cells reached much higher levels at this and later times of infection. In contrast, the release of IFN-␣/from SFV4-infected cells was barely detectable at 8 h p.i. (it should be noted that in this exper-iment, there were at least 2.5-fold more wt SFV4-infected cells than cells infected with any mutant virus). This observation is consistent with the ability of wt alphaviruses to efficiently suppress the type I IFN response (36,37). Over the next 16 h, the produc-tion of type I IFN by cells infected with SFV4-3⫻AAA isolates and SFV4-2^3 continued to increase. Interestingly, cells infected by SFV4 also started to produce interferons in amounts that rivaled those generated in response to the mutant viruses (except for SFV4-1^2^3-E452A) by 12 h p.i. and exceeded them by 24 h p.i. Subsequently, IFN-␣/production rapidly declined and was al-most undetectable at 36 h p.i. For wt SFV4 and, to a lesser extent, SFV4-2^3, this decline was associated with the death of the in-fected cultures; no excessive cell death was detected for cultures infected with other mutant viruses.
The most straightforward explanation for the data presented in Fig. 7Bis that for all of the mutant viruses, IFN-␣/was produced mostly by the initially infected cells, indicating that all of these viruses display some defect in their ability to suppress the IFN response. This defect is greatest by far for SFV4-1^2^3-E452A. In contrast, SFV4 is capable of efficiently shutting down IFN-␣/ production in infected cells: at 8 h p.i. (which corresponds to the most efficient release of virions from wt SFV4-infected cells [Fig. 7A]), only very small amounts of IFN-␣/were consistently de-tected (Fig. 7B). This IFN may originate from cells infected by aberrant viruses, which are always present in any preparation of RNA viruses, or from cells that are for some reason resistant to SFV infection. The biological significance of such small amounts of IFN for alphavirus infection was recently demonstrated: they are able to prime noninfected cells, which will then respond to infection with rapid and very powerful type I IFN production FIG 6Localization of virus replication organelles formed by wt and mutant
SFV4 variants in infected HS633T cells. HS633T cells grown on coverslips and infected with the indicated viruses at an MOI of 20 were fixed at 8 h p.i. Cells were permeabilized and costained with an nsP3 polyclonal antibody and then incubated with a secondary Alexa-568-conjugated antibody and an anti-dsRNA J2 antibody, followed by incubation with a secondary Alexa-488-con-jugated antibody; the nuclei were counterstained with DAPI. Immunofluores-cence images of a representative cell(s) are shown; both single and merged channels are presented.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.44.285.68.439.2](36). Thus, it is very likely that the IFN detected in wt SFV4-infected cell cultures at 12 and 24 h p.i. was produced by cells primed by small amounts of IFN and subsequently infected by SFV4 progeny.
However, if this assumption is correct, a similar behavior should be expected for SFV4-2^3, which was also cytotoxic and consistently produced at least 10 times more infectious virions than any other mutant (Fig. 7A). Since this was not observed (Fig. 7B), we reasoned that replacing all of the medium at each time point also removed the majority of progeny virions and, thus, prevented or delayed the spread of the mutant virus in-fection. Therefore, the experiment was also performed in a such way that only one-fifth of the growth medium was col-lected at each time point. The measurement of IFN-␣/ pro-duction under this setup indeed revealed much more robust production of IFN-␣/ by SFV4-2^3-infected HS633T cells (Fig. 7C), clearly supporting our hypothesis. At the same time, the amounts of IFN-␣/released by cells infected by any other mutant virus were somewhat reduced. The same trend was
observed in the case of wt SFV4. The most plausible explana-tion for the latter finding is that because the high titer of infec-tious progeny was not removed, secondary infection took place under higher-MOI conditions and led to a more rapid shutdown of IFN production in cells infected by viral progeny. In addition, the data on IFN-␣/induction revealed that the seemingly small differences in the design of this experiment (complete replace-ment of medium versus collection of smaller samples) had a sur-prisingly large impact on IFN induction (compareFig. 7BandC). Thus, if only a single type of experimental setup is used, the resul-tant data should be interpreted carefully.
Finally, we carried out the same experiment using mouse L929 cells. The overall sensitivity of the assay was lower in this case, most likely because L929 cells were much less efficiently infected by SFV4, especially by mutant forms of the virus. Nevertheless, the same tendencies observed in HS633T cells (Fig. 7B) were also re-corded in L929 cells (data not shown), indicating an important role of the IFN response in limiting the infection of these cells by mutant SFV4 variants.
FIG 7Growth of wt and mutant viruses and IFN-␣/release in infected HS633T cells. HS633T cells were infected at an MOI of 20, and after 1 h, cells were washed once with PBS and covered with growth medium. Samples were collected in two different ways: all of the growth medium was collected at the indicated time points and replaced with fresh medium (A and B), or an aliquot of 20% of the growth medium was removed and replaced with the same amount of fresh medium (C). (A) Infectious virus production. The titers in the obtained stocks were determined by using plaque assays on BHK-21 cells. (B and C) The IFN-␣/levels in the collected samples were measured by using a HEK-Blue IFN-␣/cell-based assay. The data represent the mean values and standard deviations from triplicate assays.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.111.476.69.434.2]DISCUSSION
The apparent specificity of viral enzymes makes them attractive targets for antiviral chemotherapy. Thus, obtaining a proper un-derstanding of the mechanisms underlying the regulation of viral enzyme activity may potentially lead to the development of drugs with improved specificity and affinity, whereas estimating the ex-tent of the plasticity of enzyme functionality may allow for predic-tion of the probability and means of emergence of drug-resistant viral mutants. Although subsites of the protease recognition pocket are believed to be mainly responsible for substrate recog-nition, viral strategies for initially expressing protein machinery components as polyproteins (38,39) and physically isolating the replication process of viral genomes using virus-induced organ-elles predispose to the establishment of a “closed system” with respect to viral enzymes acting on their substrates (40). In such a case, even if error-prone genome replication leads to amino acid substitutions in the protease domain or in the regions surround-ing the cleavage positions, it can be envisioned that a viral protease may still find an intended scissile bond within a polyprotein thanks to the guidance provided by architectural features of the multicomponent virus replication complex. In a condensed envi-ronment, all its constituents (including cellular factors) can be expected to influence the conformation of protease and accessi-bility of the substrate. Not surprisingly, notions regarding the in-fluence of conformational or cellular factors on polyprotein pro-cessing can be tracked in publications addressing almost every virus in which protease activities have been studied. Both the mac-romolecular context (41–45) and cleavage site-adjacent sequences clearly extending out of the immediate substrate recognition pocket (46,47) have often been found to be implicated in differ-ences in the rate of processing or particular order of cleavage events. Additionally, a number of recent reports suggest that rep-licated RNA functioning as part of the viral holoenzyme may also influence the ability of a viral protease to process substrates (48– 51). Therefore, simplification of viral polyprotein processing for research studies using short peptide substrates, recombinant pro-tease fragments, and defined experimental conditions may lead to the existing fine-tuning mechanisms being overlooked (11,21).
In this study, we attempted to evaluate the level of alphaviral protease tolerance of modified cleavage regions in the natural con-text of the ns polyprotein. We hypothesized that substrate recog-nition in the viral polyprotein is a result of both the sequence-dependent accommodation of the cleavage peptide into the active-site pocket of the protease and, at a higher level, being a macromolecular assembly-guided event (14,27). In such a case, if the information contained in the amino acid residues that are sufficient for substrate recognition is eliminated, the structural design of the replication complex should alleviate the deleterious effects of the insufficient affinity of the cleavage site sequences, thus revealing the extent of assembly-dependent substrate target-ing in the case of each of three particular sites within the polypro-tein.
Quite unusually for standard protease-substrate relationships, but in accord with our initial hypothesis, we found that substitu-tion with alanine at any single posisubstitu-tion in the P6-P4 region or even throughout almost the entire recognition sequence (except for the absolutely conserved P2 Gly) did not prevent any of the three sites in the ns polyprotein from being cleaved. Even more strikingly, the introduction of AAA mutations in individual cleavage sites did
not compromise the infectivity of the corresponding viral RNA. Only combinations of AAA mutations in the 1/2 and 2/3 sites and, ultimately, in all three cleavage sites affected viral RNA infectivity. This led to the selection of compensatory mutations, but not (pseudo)reversions in altered cleavage site sequences, also result-ing in largely preserved polyprotein processresult-ing efficiencies and processing patterns. These results provide direct experimental ev-idence that cleavage site sequences play a supporting, but not vital, role in deciding whether cleavage will take place in the context of the viral replicase complex, highlighting that other higher-order mechanisms are involved in the maintenance of the correct pro-cessing pattern for the SFV4 ns polyprotein. In at least several cases (Fig. 4and5), these mechanisms clearly dominated over the recognition of short cleavage site sequences. The nature of these mechanisms and their universality among different alphaviruses are currently a focus of interest in our laboratory.
Nevertheless, the amino acid composition of the cleavage sites clearly plays its own role. Alanine scanning revealed that, consis-tent with previous studies (11), P4 Arg is the main determinant of the processing of the 3/4 site. At the same time, it was found that substitution of P5 Tyr at the 1/2 site had a significant effect on processing. Interestingly, the defect resulting from alanine substi-tution was most profound for the 3/4 site, suggesting that the sequence-dependent component of substrate recognition is more important for this particular site. However, the ability to tolerate unfavorable substitutions within the 3/4 site, such as charge re-moval or reversal, suggests that the presentation of the 3/4 site in replication complex assembly, in combination with the plasticity of the substrate recognition pocket of the protease, allows it to overcome severe substrate sequence defects. Curiously, it was pre-viously noted that cleavage sites in different alphaviruses display a different susceptibility to being cleaved when presented as short sequences encompassing regions around a scissile bond. In the case of VEEV, all of the protease sites can be cleaved into a short form, whereas in SINV, none of the sites can be recognized in a short format (52), although in polyproteins, all of them are effi-ciently processed (6). SFV represents an intermediate variant ex-hibiting a 2/3 site not recognizable in the short form and different cleavage efficiencies at the 1/2 and 3/4 sites (11,21,52). We suggest that our finding that the ns polyprotein of SFV was still correctly processed despite the introduced defects in the cleavage sequences may provide a hint that the same strategy of assembly-dependent recognition is employed to a greater or lesser extent in the case of SINV or VEEV, respectively. Moreover, we propose that such a substrate recognition mode lies at the heart of the processing reg-ulation mechanism: only proper structural changes reflecting the progress of viral replication can validate the macromolecular as-sembly-driven positioning of the scissile site, thus providing nu-merous possibilities for cleavage control. Remarkably, mutational analysis also revealed cross talk between 1/2 and 3/4 site process-ing, which is consistent with a mixed (intra- and intermolecular) mode of cleavage of the 3/4 site: the intramolecular mode evi-dently takes precedence in the early stages of infection, relying on replicase assembly, and thus, substrate sequence defects can be tolerated, whereas the intermolecular mode of cleavage dominates later in infection and is more dependent on the cleavage sequence and the progress of nsP2 to maturity (controlled mostly by 1/2 cleavage).
For the purpose of developing successful protease inhibitors, it is useful to know which cleavage will be most efficiently
on November 7, 2019 by guest
http://jvi.asm.org/
pressed and which stage of the viral infection process might be most affected if a particular cleavage site remains uncleaved. In this context, we recognize that, at least in the case of SFV4, poly-protein cleavage events relying on macromolecular assembly will likely be relatively tolerant of inhibition due to an intrinsically high concentration of the substrate and protease in a preas-sembled complex. In addition, this and previous studies have re-vealed that alphaviruses can replicate even when cleavage of the 1/2 and 2/3 sites is completely blocked by mutations, and only minor changes in polymerase, achieved through changing one or a few amino acid residues, are required to restore viral fitness to an acceptable level (12,53). In this study, we demonstrated a func-tional relationship between the P4 residue and amino acid residue 706 of nsP2 of SFV. Such information has obvious value for the validation of predictions made on the basis of the crystal structure of a protease and simulations of molecular dynamics (33,54). Importantly, this connection became apparent only following the manipulation of the most sequence-dependent 3/4 cleavage site, whereas changes in the sequences of the other cleavage sites were ignored by the virus, thus demonstrating the limitations of such an approach for further analysis of genetic interactions.
In accord with observations that even small perturbations in a well-balanced viral life cycle can significantly influence viral cyto-toxic effects and dissemination capabilities (12, 25, 55, 56), we found that viruses with modified cleavage sites and accompanying compensatory mutations exhibited a reduced ability to suppress interferon production and/or signaling. Thus, although the pro-cessing profile of the ns polyproteins of SFV4 AAA mutants re-mained largely unaffected and the generation of virus replication organelles was apparently preserved (Fig. 6), subtle differences in the timing of the release of the processing products may play a significant role in the success of viral infection. Notably, many studies addressing viral proteases from different viruses have re-vealed their involvement in the subversion of the cellular machin-ery (57, 58) or counteraction against immune defense mecha-nisms through cleaving certain cellular targets (59). Remarkably, nsP2s of the Old World alphaviruses have been shown to induce the degradation of Rpb1, leading to the cessation of cellular tran-scription; however, this degradation is evidently not mediated by the proteolytic activity of nsP2 (60).
Although the currently available literature provides no exam-ples of alphaviral proteolytic targeting of host proteins, it would be surprising if alphaviruses do not exhibit such activity. Thus, al-though the repertoire of potential alphaviral protease targets within host cells is unknown at present, it is tempting to speculate that a compensatory mutation in the vicinity of the protease rec-ognition pocket could potentially change the intransspecificity profile of the protease. This specificity profile change could lead to an inability to employ viral proteolytic activity to adjust the cellu-lar environment to meet the requirements for efficient virus rep-lication, either through the neutralization of cellular restriction factors that normally limit viral infection (61,62) or through the modification of cellular accessory proteins to facilitate progres-sion to the next stage of replication (63,64). If so, the described approach shows the potential for creating viruses with a restricted phenotype for the purpose of vaccine development. In this con-text, our finding that viruses with modified proteolytic sites are unable to productively infect mosquito-derived cells also fulfills an important selection criterion applied to a potential antiarbovi-rus vaccine candidate. Furthermore, it is important to note that
despite the successive propagation of SFV4-3⫻AAA virus vari-ants, their ability to produce infectious virions remained lower than that of the wt virus. Therefore, the described mutants appear to be genetically trapped, such that despite an enormous potential to select for compensatory modifications, these changes appar-ently do not provide a sufficiappar-ently great advantage to revert to the wt profile (65). This finding suggests that a longer sequence of mutation selection events is required, which is improbable under the conditions of active cellular defense. For vaccine candidate selection, it would be interesting to explore the potential of a cleavage site modification approach in combination with the in-troduction of mutations restricting viral polymerase fidelity, which would be expected to further limit the evolutionary poten-tial of the virus.
In conclusion, our findings can be used to further explain the extent of the promiscuity of the alphaviral protease and concom-itant nonobvious advantages for the virus as well as allow us to make predictions about other distinguishing functional proper-ties and behavioral aspects of alphaviral proteases that can be ex-perimentally tested. The possibility of using targeted mutagenesis at cleavage sequences to promote alteration of viral protease sub-strate specificity to limit or eliminate the ability of the virus to attack critical host targets also appears to be promising as a new route for the rational design of attenuated viral variants with re-duced cytotoxicity and replication restricted to certain cell types. We believe that such knowledge may become important for de-signing drugs and vaccines targeting alphaviruses, which are cur-rently only in the nascent stages.
ACKNOWLEDGMENTS
We thank Galina Halus for excellent technical assistance.
This work was supported by Estonian Science Foundation grants 7501, 7407, and 9421; target financing project SF0180087s08; and the European Union through the European Regional Development Fund via the Center of Excellence in Chemical Biology.
REFERENCES
1.Strauss JH, Strauss EG.1994. The alphaviruses: gene expression, repli-cation, and evolution. Microbiol. Rev.58:491–562.
2.Kääriäinen L, Ahola T.2002. Functions of alphavirus nonstructural pro-teins in RNA replication. Prog. Nucleic Acid Res. Mol. Biol.71:187–222. 3.Lemm JA, Rümenapf T, Strauss EG, Strauss JH, Rice CM. 1994.
Polypeptide requirements for assembly of functional Sindbis virus repli-cation complexes: a model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J.13:2925–2934.
4.Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, Frolov I. 2010. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol.84:11679 –11695.
5.Spuul P, Balistreri G, Kääriäinen L, Ahola T.2010. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest virus replication complexes from the plasma membrane to modified lyso-somes. J. Virol.84:7543–7557.
6.De Groot RJ, Hardy WR, Shirako Y, Strauss JH.1990. Cleavage-site preferences of Sindbis virus polyproteins containing the non-structural proteinase. Evidence for temporal regulation of polyprotein processing in vivo. EMBO J.9:2631–2638.
7.Shirako Y, Strauss JH.1990. Cleavage between nsP1 and nsP2 initiates the processing pathway of Sindbis virus nonstructural polyprotein P123. Virology177:54 – 64.
8.Lemm JA, Rice CM.1993. Assembly of functional Sindbis virus RNA replication complexes: requirement for coexpression of P123 and P34. J. Virol.67:1905–1915.
9.Lemm JA, Rice CM.1993. Roles of nonstructural polyproteins and cleav-age products in regulating Sindbis virus RNA replication and transcrip-tion. J. Virol.67:1916 –1926.