0022-538X/82/100098-09$02.00/0
Copyright©1982, American Society for Microbiology
Vol. 44, No. 1
Characterization of
a
Temperature-Sensitive
Defect of
Enterovirus
Type 70
NAOKAZUTAKEDA,*KIKUKOMIYAMURA, REISAKU KONO,ANDSHUDOYAMAZAKI
Central Virus Diagnostic Laboratory, National Institute of Health, Musashimurayama, Tokyo 190-12,Japan
Received 8March 1982/Accepted8 June 1982
The
mechanism of the failure of enterovirus
type70toreplicateatanonpermis-sive temperature
(39°C)
wasinvestigated,
and the following resultswereobtained.(i) Viral RNA synthesis
was notobserved
at39°C
in LLC-MK2cells,
inaccordance
withourprevious findings with primary
monkey kidney cells(Miya-muraet
al.,
Intervirology
9:206-213,1978). (ii)
Shutoff of hostcellmacromolecu-lar
synthesis by virus infection
was asefficient
at39°C
as at a permissivetemperature
(33°C). This inhibitory effect similarly
occurredevenin thepresenceof
guanidine hydrochloride. (iii) Viral protein synthesis
proceededin
vivoatthenonpermissive
temperature,and the
rateof
the protein synthesiswashigherthanthat
atthe
permissive
temperatureunder the conditions in which
sufficient viralmRNA
had been accumulated. This
wasalso confirmed
by analyzing theintracellular proteins synthesized
atthe nonpermissive
temperature by sodiumdodecyl sulfate-polyacrylamide gel electrophoresis, which identified
them asvirus-specific proteins. (iv) When infected cells
wereincubated
at39°C
and thentransferred
to33°C, viral RNA synthesis took place
evenin the
presence ofcycloheximide. (v) Furthermore, in experiments performed with
anin vitro
cell-free
assay system,viral
polymerase activity
wasfound in the membrane-bound
preparation extracted from infected cells which had been incubated
at39°C
in thepresence or
absence of
guanidine hydrochloride. These results indicate that
earlytranslation of mRNA proceeds normally
atthe nonpermissive
temperature andthat the
temperature-sensitive defect resides in the transcriptional
stage.Enterovirus type 70
(EV70), a causative agent
of acute
haemorrhagic
conjunctivitis
(14), has
atemperature-sensitive
nature,
ashas been shown
by the
fact that the wild strains grow best
at33°C
and
do not grow
at39°C
(21,
22).
This
character-istic of
the
virus is
particularly
interesting
be-cause
the
conjunctiva,
the
temperature
of which
is
relatively low,
is the
primary
site for
replica-tion of this virus in natural
infections.
Moreover,
the
virus
seems not togrow
extensively
in
theintestinal
tractand is
noteasily
isolated from it
(29). These
findings
distinguish
the
virus
frommany other
enteroviruses; thus,
EV70 is
consid-ered
as anaturally
occurring
temperature-sensi-tive virus (22).
Inprevious studies,
the
sedimen-tation
profile
of
the RNAextracted from
EV70-infected
primary monkey
kidney
cells
showed
that
no
virus-specific
RNAreplication
occurred
at
the
nonpermissive
temperature
(23).
In
the
present
study,
weattempted
toinvesti-gate
moreprecise
mechanisms
toregulate
viralgrowth at the
nonpermissive
temperature. The
location of
thesite
of
thetemperature-sensitive
lesion in viral
replication
will also be
discussed.
MATERIALSANDMETHODS
Chemicals. Guanidine hydrochloridewas obtained
from Research PlusLaboratories, Inc., Denville, N.J.
Actinomycin D (AMD) was purchased from Makor
ChemicalsLtd.,Jerusalem,Israel. Bovineserum
albu-min fraction V, ATP,CTP,andGTPwerefromSigma
ChemicalCo., St. Louis, Mo.; agarwas from Difco
Laboratories, Detroit, Mich.; andNonidet P-40 was
from Shell ChemicalCo., Houston,Tex.Polyethylene
glycol (Carbowax 6000), trichloroacetic acid,
cyclo-heximide, acrylamide,N,N'-methylenebisacrylamide,
sodiumdodecyl sulfate(SDS),
N,N,N',N'-tetrameth-ylethylenediamine, ammonium persulfate, Tris, and
glycine were purchased from Wako Pure Chemical
Industries, Ltd., Osaka, Japan.Phosphoenolpyruvate
and pyruvate kinasewerepurchasedfromBoehringer
MannheimBiochemicals,Mannheim,West
Germany.
Dithiothreitol wasfrom Seikagaku Kogyo
Co., Ltd.,
Tokyo, Japan.
L-[355]methionine
(1,080 to 1,350 Ci/mmol), 14C-methylated
protein mixture, and[5,6-3H]uridine
(40Ci/mmol) wereobtained from theRa-diochemicalCentre, Amersham, England.
"4C-amino
acid mixture (50
mCilmatom)
and [3H]UTP (45 Ci/mmol) were purchased from New England Nuclear
Corp.,Boston, Mass.
Cells, virus,and infection.LLC-MK2 cells,an
estab-98
on November 10, 2019 by guest
http://jvi.asm.org/
VOL.44, 1982
lished cell line derived from rhesus monkey kidney
cells(12), and Bristol-HeLa cellswere propagatedat
37°C in Eagle minimal essentialmedium (MEM)
sup-plementedwith5% calf serum. To prepare seed virus,
cells grown in roller bottles (growth area,
approxi-mately 800cm2)wereinfected with the standard strain
of EV70, J670/71, and incubated at33°C (22). Virus
fluid wasroutinely concentrated by8% polyethylene
glycolin 0.5 MNaCl, and a multiplicity of infection of
about 500 PFU was employed. In all experiments,
virusadsorption was performed for 30 min at 4°C and
another 30 min at37°C. "Hoursafter infection" used
in thefigures refers to the elapsed times after the end
of the adsorption period. MEM without serum was
used for the culture medium after infection unless
otherwise specifically described. Culture bottleswere
immersed in a waterbath, and temperature was
main-tained within an errorof less than +0.05°C.
Infectivity assay. Toquantitatevirusyields in
infect-ed cells, onlycell-associated virus wasmeasured by
plaque assay on confluent monolayers of LLC-MK2
cells. Two or three dishes(Falcon tissue culture dish
3002) were employed for each serial 10-fold virus
dilution. The inoculated cultures were incubated in
MEM containingO.9o agar(Difco) at33°C ina
hu-midified atmosphere of 5%CO2.After 2days, the agar
overlay was removed, the cell monolayers were
stained with0.1% crystal violet in
20%o
ethanol, andplaques were counted.
Measurements of RNA and protein syntheses. (i) Viral RNAsynthesis.Toestimate therateof viral RNA
synthesis, infected LLC-MK2 cell monolayers (5x106
cells)wereincubated in 5.0 ml of MEMcontaining5%
fetal calfserumand 10 ,ug of AMD per mltoblock host
cellular RNAsynthesis (32). At therequiredtime, the
cells were pulse-labeled with 2 to 10 ,uCi of
[5,6-3H]uridine per0.5 mlfor 30 min. The incorporation
wasterminated byadding 0.5 ml of cold MEM
contain-ing1,OOOx concentrations of unlabeled uridine. After
three cycles of washing with the samemedium, the
cellsweresolubilized with 1.5 mlof cold TNE (0.01 M
Tris[pH7.4], 0.1 MNaCl,0.001 MEDTA)containing
0.5% SDS. Radiolabeled viral RNAwas precipitated
withanequalvolumeof cold10%trichloroacetic acid
onice for 60 min and thenwascollectedonglass fiber
filters(WhatmanGF/C). After drying, the filterswere
submerged in toluene-based fluors, andradioactivity
wascounted with aPackard scintillation
spectropho-tometer.
(il)
Viralprotein synthesis. Tomeasureviralproteinsynthesis, infected HeLa cell monolayers (3 x
106
cells)werecovered with 5.0 mlof MEMcontaining 2.5
,ugof AMD per ml and5%fetal calfserumandwere
incubated in the presence of 2 mMguanidine
hydro-chloride during the first 3 h after infection (1, 30).
During this period, host cellular protein synthesiswas
shutoff by virusinfection, andnewviral RNA
synthe-sis was inhibited. After the removal of guanidine
hydrochloride, the incubation was continued in the
absence ofguanidine hydrochloride to allow newly
synthesized viralRNA toaccumulate in the cells.At
therequired time, the cellswerewashed three times
withMEMwithoutmethionine and labeled with 5 ,Ci
of
["S]methionine
per ml. At the end of 30 minofpulse-labeling, 1.5 mlof coldMEMcontaining 300x
concentrations of unlabeled methioninewasaddedto
the cultures toterminate theincorporation. The cells
99
wereimmediatelychilled onice andsubjectedtothree
cycles offreezing and thawing. Theradioactivity
in-corporatedintotrichloroaceticacid-insoluble fractions
was collected and measured in a liquid scintillation
counter. ForSDS-polyacrylamidegelelectrophoresis
(SDS-PAGE), infected HeLa cell monolayers were
similarly pulse-labeled for 30 min with 20 ,uCi of
[355]methionine. Inpulse-chaseexperiments, infected
HeLa cells were incubated and pulse-labeled in a
suspension cultureasdescribed in thelegendtoFig.5.
(iii)CellularRNA andprotein syntheses. Toestimate
the rates of cellular RNA and protein syntheses,
infected LLC-MK2 cell monolayers (5 x 106 cells)
were incubated with 5 ml of MEM. At therequired
time, the cells were washed three times with cold
Earle balanced saltsolution andweregiven 0.5 mlof
Earle balanced salt solutioncontaining0.4p.Ciof
[5,6-3H]uridineand 0.1 ,uCi of
"4C-amino
acidmixture. Atthe end of15 min ofpulse-labeling, the cells were
quickly chilled inanice water bath, and 1.5 ml of cold
MEMcontaining 1,000x concentrations of unlabeled
uridine was added to terminate the incorporation.
After three cyclesof freezing and thawing,
trichloro-aceticacid-precipitableradioactivitywasdetermined.
Mock-infected control culturesweresimilarly treated,
exceptthatviruswasomitted.
Preparation ofcytoplasmicextracts and acetone
pre-cipitation ofradiolabeled polypeptides. Monolayersor
suspensionsofradiolabeled HeLa cellswerechilled in an icewaterbath and washed three times with cold isotonic salt solution (0.01 M Tris [pH 7.4], 0.14 M
NaCl, 0.015 MMgCl2).The solution containing 0.5%
Nonidet P-40 wasadded, and the cellswereplacedon
ice for 20 minto besolubilized (26). Theextract was
thencentrifuged at 800 xgfor 5 minat4°C to remove
nuclei, and thesupernatantwasstoredat-70°C until
it was to be used. Seven to eight volumes of cold
acetone was added to the radiolabeled cytoplasmic
extracts, and the mixtureswerekeptat -20°C
over-night. The precipitatewascollectedbycentrifugation
at10,000x gfor 20 min, air dried, and then dissolved
inthesample buffer. Thelysatewas boiledfor 3 min
before electrophoresis (5).
SDS-PAGE.Electrophoresis wascarriedoutwitha
discontinuous gel system by the method of Laemmli
(15). For fluorography, gel slabs were fixed in 30%
methanol-10%acetic
acid-60o
water, treatedby themethodof Bonner and Laskey (3)orimpregnated with
EN3HANCE
(NewEngland NuclearCorp.), dried invacuo, and exposed to Sakura X-ray film for an
appropriateperiodat-70°C.
Preparation ofthe viral polymerase complex. The
crude viral replication complexwas prepared by the
method of Yin and Knight (33). Cells were removed by
scrapingfrom the culture flasks and thenwere
collect-edby low-speedcentrifugation(600xg)for5min, and
the cell pelletwasfrozenat-70°C. Approximately4x
108
frozencellsweresuspended in 10 ml of cold buffercontaining0.05 MTris (pH 7.2),0.002MMgCI2, and 0.1 MNaCl. Thecellswereallowedtoswell for 10 min
onice and were ruptured with 15 strokes in a
tight-fittedglassDouncehomogenizer. Unbroken cells and
nucleiwereremovedbycentrifugation at 800x gfor 5
min, and the supernatantfraction was centrifugedat
30,000 xgfor 20 min. The pellet was suspended ina
buffer containing 0.05 M Tris (pH 8.0) and 0.01 M
NaClat afinal concentration of50,ugoftotalprotein
on November 10, 2019 by guest
http://jvi.asm.org/
per 50
RI
and served as the viral polymerase complex.The protein concentration was determined by the
method of Lowry et al. (18).
Assay for polymerase activity in theceil-freesystem.
A50-ilsample of the polymerase complex was mixed with an equal volume of the reaction mixture contain-ing the followcontain-ing constituents: 100 mM Tris buffer (pH
8.0); 40 mM KCI; 10mM
MgC92;
2.5 mMphosphoenol-pyruvate; 13 mMdithiothreitol; 10 ,ug of AMD per ml;
50,ugof pyruvate kinase per ml; 0.3 mM each of
ATP,
CTP, and GTP; and 10,Ciof[3H]UTPper ml (cold
UTP was notincluded in the reaction mixture because
the addition of 10 ,uM UTP did not affect the results substantially). The mixtures were incubated at 33°C in
a water bath. After the desired time, the incubation
was terminated by spotting the mixture onto
DEAE-cellulose disks (Whatman DE 81) which were then
soaked for 5 min at room temperature in 5%Na2HPO4.
The wash was repeated four times more inNa2HPO4
and twice each in water and ethanol, and the dried
disks were counted in a toluene-based scintillation fluid.
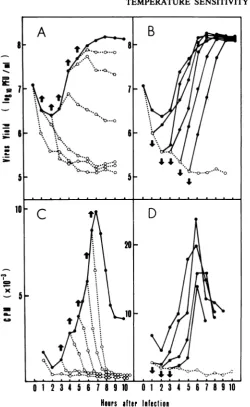
RESULTS
Effect of temperature shift on viral growth and
viral RNA synthesis. To investigate the effect of
temperature on
EV70 replication, temperature
shift
experiments were carried out. The infected
cells
wereshifted from 33
to39°C
orvice
versaat
various times after
infection, and the virus
yields
and
viral RNA
synthesis
were
determined
(Fig.
1). When the
infected
cells
wereincubated
at
33°C, virus grew
exponentially
after
a2-h
latent
period, and one cycle
of
viral growth
wasaccomplished within 7 h after infection. No viral
replication
occurred
if the
shift-up
was
done
during the first 2 h after
infection,
and
if the
shift-up
wascarried
outlater than 2
h after
infection, some viral growth was observed
dur-ing the next
1 hafter the shift but soon leveled
off
(Fig.
1A). The RNA
synthesis rapidly
de-creased
and ceased within
1h
after the shift
(Fig.
1C).
Inshift-down
experiments,
the cultures
were
transferred from 39 to 33°C. The viral
growth
immediately
started and reached
amaxi-mum.
The virus continued
togrow
at asimilar
rate when
the
temperature
wasshifted down
within
5h after infection
(Fig. 1B).
When theshift-down
wasmade
within 3 h after
infection,
viral RNA synthesis started at a
similar
rateafter
a 1- to2-h
lag and reached
amaximum
6 to7 h
after
infection,
irrespective of
thetime
of
thetemperature shift
(Fig.
1D).
Shutoff effect on host
cell
macromolecular syn-thesis.Circumstantial evidence suggests
that theinhibition of
hostprotein and
RNAsyntheses
inpicomavirus-infected
cells
resultsfrom
aviral
gene-specific
effect(s), although its molecular
mechanism is
notcompletely understood (4, 8,
11, 19).
Toexamine whether this viral function
could
occur atthe
early stage of
infection,
theshutoff
effect
onmacromolecular
synthesis
of
infected cells was investigated. EV70-infected
and
mock-infected cells were pulse-labeled with
[3H]uridine and
14C-amino
acid
mixture at 1-h
intervals after infection to monitor cellular RNA
and
protein syntheses. The shutoff effect was
evaluated by the rate of
inhibition estimated as
the
incorporated radioactivities in infected cells,
expressed as a percentage
of
those of
mock-infected cells (Fig. 2). The RNA inhibition was
50 to
60%
at
6 to 7 h
after infection, although the
kinetic curves of the inhibition were not exactly
the same at
39 and at
33°C. The inhibition of
cellular
protein
synthesis
reached 70 to
80%6
at 6
to 7 h
after infection at both temperatures. In
one
experiment
on
cellular
protein
synthesis,
guanidine
hydrochloride, a selective inhibitor of
picornavirus RNA synthesis, was added to the
culture medium after infection
(Fig.
2B).
The
shutoff
effect was similarly observed in the
pres-ence
of the drug. These results suggest that
some
of the early events preceding viral RNA
synthesis
could occur
even at39°C.
Translation of
viral mRNAat the
nonpermis-sive
temperature. It is
generally supposed that
one
of the processes
preceding
RNA
replication
in
picornaviruses is early
translation (17). If this
process is
blocked at 39°C, then subsequent viral
replication may not occur.
Therefore,
viral
pro-teins
synthesized
in
vivo
weremonitored
to
examine whether viral RNA
canfunction
asmRNA at
39°C. First, infected cells were
prein-cubated
at33°C
in the
presence
of
guanidine
hydrochloride to eliminate cellular
protein
syn-thesis, and then the guanidine hydrochloride
was
washed
off, after which the cells
wereincubated in the absence of
guanidine
hydro-chloride
toaccumulate sufficient viral mRNA
and
weretransferred
again
to39°C.
Viral
protein
synthesis
wasthen
compared by measuring
[
5S]methionine
taken
upby
cells
at39 and
33°C
(Fig.
3). Viral
protein synthesis proceeded
at39°C
aswell
as, or evenbetter
than,
it did
at33°C
regardless
of the
presence orabsence of
guanidine
hydrochloride (Fig. 3).
The
subse-quent
decrease in
therateof
protein synthesis
at39°C
wasprobably
due
todefects in viral
RNAsynthesis
atthis
temperaturesince the kinetic
curve
followed the
samepattern
asthat
ob-served
at33°C
in
the presenceof
guanidine
hydrochloride.
These results
indicate
thatthe
translation of
viral
mRNAproceeds
atthe
non-permissive
temperature.To
make
certain
thatthe translation
products
formed
at39°C
werevirus-specific proteins,
extracts
from infected cells
wereanalyzed
by
SDS-PAGE. Infected cells
wereincubated
at33°C for
3h in
the presenceof
guanidine
hydro-chloride and continued
toincubate
at33°C
in the
absence
of
guanidine hydrochloride.
During
that
time,
large
amountsof viral
mRNAmight
have
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
-U
x-co
'0
1
°2°3
6 7
8
g°I9 1
2
01 23456178910 0123456180910
[image:4.496.123.374.62.470.2]Hours
after Infection
FIG. 1. Effectof temperature shift on viral growth and viral RNAsynthesis. LLC-MK2cellmonolayers(106
cells)grownin a tissue culture flask
(5-cm2
growth area) were infected with theJ670n1strain and incubated forvarious times before they were transferred from the permissive temperature (33°C) to the nonpermissive
temperature
(39°C) orvice versa. The temperature was shifted up (A andC)ordown(B and D) at thetimesindicatedbythearrows.Tomeasurethe virusyield, duplicatecellcultures from eachtemperature groupwere
collectedat1-hintervals,washed threetimes,andsubjectedtothreecyclesoffreezingandthawing.Virusyields
weredeterminedby plaqueassay onLLC-MK2cell monolayersasdescribed in the text (A and B). Viral RNA
synthesiswasmonitoredby 30-min pulse-labelingof infected cells with
[5,6_3H]uridine
atintervals of 0.5or1hasdescribedinthetext
(C
andD). Thetotalradioactivity incorporatedintoanacid-precipitable
fraction fromSDS-solubilized cells wasplotted.Eachcircle represents the middle of thelabeling
period.
Symbols:0,
incubatedat33'C; 0,
incubatedat39°C.been accumulated in the
cells,
sothat
amaximal
rate
of viral
protein synthesis
could be
expected.
The
cultures
werethen
transferred
to39°C
at4or5
h
after the removal of
guanidine
hydrochlo-ride.
Subsequently,
virus-specific proteins
werepulse-labeled for 30
minat30,
60, and 120
minafter the shift and
wereanalyzed by SDS-PAGE
(Fig.
4). Protein bands from both temperatures
were
identical, and almost all cleavage
products
of the viral
proteins
weredetectable
exceptthe
two
capsid proteins
VP2and
VP4.These data
not
only confirm the
occurrenceof
virus-specific
protein synthesis
at39°C
butalso
suggestthat
some
cleavages
evenproceed
atthe
nonpermis-sive
temperature. The latter
possibility
wasfur-ther
investigated
(seebelow).
44,
on November 10, 2019 by guest
http://jvi.asm.org/
0
1
2 3 4 5 6
7
01 2
3 4
56
7
Hours
after
Infection
FIG. 2. Inhibitory effect of EV70 infection on cellularRNA and protein syntheses. The J670/71
strain-infectedormock-infectedLLC-MK2 cell monolayerswereincubated with MEMateither 33or39°C.Atvarious
timesafterinfection, the cultureswerepulse-labeled for15min withamixture of[5,6-3H]uridine and
"4C-amino
acidmixture. One culturegroupwasincubatedat39°C in thepresenceofguanidinehydrochloride (2 mM). The
radioactivitiesincorporated into cellular RNAandproteins ininfected cellswereplottedaspercentagesof the
correspondingvalues inmock-infectedcells. Each circle indicates the middle of thelabeling period. (A) Cellular
RNAsynthesis at39°C(0)and33°C (X); (B)cellularprotein synthesisat39°C (0), 33°C (0),and39°C inthe
presenceofguanidine hydrochloride (O).
Cleavage of viral polypeptides at the
nonper-missive temperature. To examine the cleavage
process at 39°C in detail, viral proteins synthe-sized at 33°C in HeLa cells were pulse-labeled for 5
min
with [35S]methionine at5 h after the removal of guanidine hydrochloride and thenwere chased in the presence of an excess amount of the unlabeled amino acid atboth 33 and39°C for the periods indicatedinthelegend
to Fig. 5. Figure 5 shows comparative gel
pat-terns ofvirus-specific proteins after the chase.
At bothtemperatures, theintensity of
nonstruc-tural viral proteins of high molecular weight larger than NCVP2 decreased gradually. Con-versely, NCVP7c increased obviously at both
temperatures. Since the nonstructural protein NCVP4,acandidate for the viral RNA
polymer-ase, wasalready detectable before the chase, the
cleavage to NCVP4 at 39°C could not be
ex-plained from this experiment.
However,
it seemed that the cleavage to NCVP4 at 39°C proceeded normally because the decrease in NCVP2, a precursor peptide of NCVP4 andNCVP7c (13,28), and the concomitant increase inNCVP7cwereobvious. Thedifference is clear
inthe band ofa virion structural protein, VP2,
whichwasevidentat33°C butnotat39°C. The absence of VP2at39°C would be explainedbya
lack of RNA synthesis at thistemperature,
be-causedenovoviralRNAsynthesiswasrequired
forits encapsidation into progenyvirus,
result-ing in the cleavage of VPO to VP2and VP4 (2, 25).
In vivo synthesis of viral RNA polymerase at
the nonpermissive temperature. Viral protein synthesis was observed at 39°C if the viral
mRNAhad been accumulatedinthe cells (Fig. 4). This result suggests that input virion RNA
canalsofunctionasmRNAat39°Csothat viral
RNApolymerase is produced at this tempera-ture. Intracellular synthesis of viralpolymerase
at the nonpermissive temperature during the
first 2 hafter infection was monitoredby
mea-suring the subsequent synthesis of viral RNA
after the shift-down in infected cells in the presenceof
cycloheximide,
which should block100
F
A
CD
w-cC
C.
= C=
_2
'a
0
C\
50
F
J. VIROL.
B
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.496.94.408.57.351.2]TEMPERATURE SENSITIVITY OF EV70 103
10
0 1 2 3 4 5 Hirs after Remval of6Oaaidi,e-Il
FIG. 3. Translation of viral mRNA. HeLa cell
monolayerswereinfected with theJ670M1 strain and
were incubated in the presence ofguanidine
hydro-chloride (2 mM) at33°C for 3 h. The five groups of
cultures weresubsequentlywashedwellto removethe
guanidine hydrochloride (0 h after the removal of
guanidinehydrochloride) andincubated at33°C with-out guanidine hydrochloride, except for group E, which was similarly treated but was further incubated
with guanidinehydrochloride after being washed and
which served as a control. After 2 h (arrow), each
group received thefollowingtreatment: A,shifted up
to 39°C and incubated without guanidine
hydrochlo-ride; B, incubated at33°C without guanidine
hydro-chloride; C, guanidine hydrochloride addedagain to
the culture medium and incubated at 39°C; D,
guani-dinehydrochlorideaddedagaintothe culturemedium
and incubated at 33°C; E, incubated at 33°C in the
presence ofguanidine hydrochloridefrom 0 to 5 h. At
various times indicated in the figure, the cultures were
pulse-labeledwith[35S]methioninefor 30min,and the
radioactivity incorporated into an acid-precipitable
fraction was measured as described in the text.
de
novosynthesis
of viral
polymerase
at33°C
(Fig.
6). Immediate viral RNA
synthesis
wasobserved if
the temperature
wasshifted down
at2
h after
infection,
evenin the
presenceof
cycloheximide,
but
noviral
RNAsynthesis
wasobserved if the
temperature wasmaintained
at39°C.
This indicates that the viral RNA
polymer-ase was
synthesized
at39°C and could function
at
33°C but
not at39°C.
In
vitro
assay of viral polymeraseactivity
of
infected cells. The
synthesis of viral polymerase
in
the early
stageof infection
atthe
nonpermis-sive
temperature wasdemonstrated in
vivo in
the
previous
experiment
(Fig.
6). To
confirm it
further,
we attempted to use a cell-free assaysystem
in which the viral
polymerase
activity in
various
cellextracts was compared in vitro bythe
kinetic
curvesof
incorporation
of[3H]UMP
into
RNAproducts. The
J670/71
virus-infected
cells
wereincubated
for
1.5 h atthe
permissive
or
the
nonpermissive
temperaturebefore they
were
harvested
toprepare
the crudereplication
complex. Guanidine hydrochloride
wasadded
tosome
cultures
toinhibit de
novosynthesis of
viral
mRNA so thatthere might exist onlyinput
virion RNA which can function as mRNA to
translate viral polymerase.
A
warmth-adapted
clone, which had beenartificially induced by serial passages of the prototypeJ670/71 strain at an elevated tempera-ture to grow at 39°C, was used as a positive control for theproduction of viral polymerase at
390C.
Thereplication complex extracted from cells infected with the J670/71 strain at 330C (Fig.
7A)
and that from cells infected with the
warmth-adapted clone
at390C (Fig. 7C)
revealedcompa-rable kinetics of polymerase
activity.
It shouldbe noted that the
polymerase activity
of the complex extracted from cellssimilarly
infected but incubated in the presence of guanidinehy-A
_
B
C
D E
F G H I
J
1-_.
___
_. 49_
~VP]
VP
2VP4
FIG. 4. SDS-PAGE of virus-specific proteins
formed in EV70-infected cells at the nonpermissive
temperature. Infected HeLa cell monolayers were
incubated at33°Cinthe presence ofguanidine
hydro-chloride for 3 h, and the cultures were washed to
removeguanidinehydrochloride and were again
incu-bated with MEM withoutguanidine hydrochloride at
33°C.Then, some groups were shifted up to39°Cat 4h
(lanes E, F, andG)and 5 h(lanes H andI)after the
removalofguanidine hydrochloride. The cultures of
each temperature group werepulse-labeledfor 30 min
with [35S]methionine at intervals of 0.5 or 1 h. The
cytoplasmic extracts were analyzed forvirus-specific
proteins by SDS-PAGE. Electrophoresis was
per-formed onresolving gel slabs (170 by 150 by 1.2 mm)
for 6 h at 140 V. The gels were fluorographed as
described inthe text. Lanes A to D, Incubated and
pulse-labeled at 33°C at 4, 5, 6, and 7 h after the
removal ofguanidine hydrochloride; lanes E to G,
shiftedup to39°C at 4 h after the removal of guanidine
hydrochloride and pulse-labeled at 30, 60, and 120 min
after the shift; lanes H to I, shifted up to 39°C at 5 h
after the removal of guanidine hydrochloride and
pulse-labeled at 30 and 60 min after the shift; lane J,
"4C-labeled
virions oftheJ670/71 strain.VOL.44,1982
*, C
' E
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.496.52.194.56.197.2] [image:6.496.261.436.258.449.2]A B C D E F G H J K L M K .1 ...
92.51( Ti
69K_m _ il_f __ -"
46K_W
VDt
VP314.3O
FIG. 5. Pulse-chase analysis of the formation of
EV70 viral proteins. HeLacells (3 x 10'cells) were
mixed with 1.0 ml of a suspension of theJ670/71 strain
andwereincubated at 4°C for 30 min, then at 37°C for
30 min, for virus adsorption. The cell suspensions
werecentrifuged to remove unadsorbed virus, and the
cellpellet was suspended in 30 ml of MEM containing
AMD. Incubation wasperformed for 3 h in the
pres-enceof guanidine hydrochloride and another 5 h in the
absence ofguanidine hydrochloride with gentle
stir-ring. The cells were washed by centrifugation with
MEM without methionine, and precipitated cells were
then pulse-labeledfor 5 min in 1.0 ml of MEM
contain-ing100
p.Ci
of[(5SI
methionine. Afterwards,thecellswere chilled immediately in an ice water bath and
dilutedwith 10 ml of MEMcontaining300x
concen-trationsof unlabeled methionine. At this time, 1.0 ml
of thecell suspension was takenasaprechase sample.
Half (5.0 ml) of the remaining cell suspension was
transferred to39°C,and the other half was held at 33°C
and then both cell suspensionswerechased.
Periodi-cally, 1.0-ml samples were removed to prepare the
cytoplasmic extracts which were then analyzed by
SDS-PAGE and fluorographed as described in the
legend to Fig.4. LaneA, Molecular weight marker
proteinsat200,000 (200K,myosin),92.5K
(phosphor-ylase b),69K (bovine serum albumin), 46K
(ovalbu-min), 30K (carbonic anhydrase), and 14.3K
(lyso-zyme); lane B, 1C-labeled virions of the J670/71
strain; lane C, sample before chase; lanes D to H,
samples chasedat33°C for 10, 30, 60, 90, and 120 min,
respectively; lanes ItoM,samples chasedat39°Cfor
10, 30, 60, 90, and120min, respectively.
that
produced in the
infected cells which
wereincubated
atthe
permissive
temperaturein the
presence
of
guanidine hydrochloride,
aninhibi-tor
of viral mRNA
synthesis (Fig. 7B). The
results indicate that the
translation proceeds
normally
atnonpermissive
temperatureby
using
input virion
RNA asmRNA.
DISCUSSION
EV70 is
apathogenic, naturally
occurring,
temperature-sensitive
virus. Since
manyfresh
isolates from the
eyesof
acutehaemorrhagic
conjunctivitis patients
have
been shown
to pos-sessthe
temperature-sensitive
nature astheir
common
characteristic,
this
propertyhas
been
considered
tobe
associated
with
a processthrough
which the virus
acquires pathogenicity
for human
conjunctivae
(22).
Therefore,
molecu-lar
biological studies of the intracellular
eventsassociated
with the
temperature-sensitive
defect
in
viral
replication will
provide
aninteresting
experimental model
toelucidate
amechanism
for the
appearanceof
naturally
occurring
tem-perature-sensitive
virus
or amechanism
by
which viruses acquire the
ability
toreplicate
better
atlow
temperaturesand become
patho-genic
in
low-temperature
tissues.
In
picomavirus
infection,
viral RNA
synthesis
is
preceded
by
serial
processesof the
early
replication
steps,i.e.,
viral
adsorption
tocells,
subsequent penetration
and
uncoating of
the
virus, and early
translation
of its
input
RNA
(17).
Our
presentstudy,
aswell
asthe data
A
x
C-.. .\\ B
O
...O...0 2 3 4 5 6 7
1 2345617
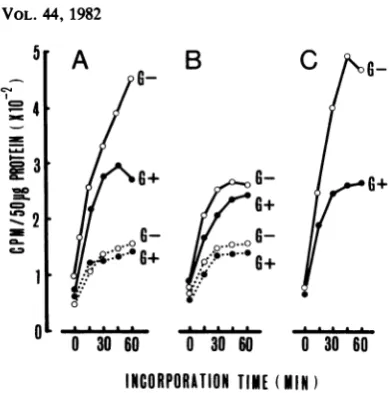
drochloride was low but clearly demonstrated
by the increasing incorporation of [3H]UMP into RNAasafunction of thereaction time in vitro.
The data indicate thatourin vitro assaysystem
is sensitive enough to detect such amounts of viralpolymeraseas weresynthesized in the cells
by utilizing only input virion RNAas the
tem-plate. Thus, the replication complex prepared from cells which had been infected with the J670/71 strain and incubated at the
nonpermis-sive temperature was found to contain viral
polymerase activity inanamountequivalentto
Hours after Infection
FIG. 6. Detectionofvirus-specificRNA
polymer-ase activity by the use ofan in vivo assay system.
LLC-MK2 cell monolayers were infected with the
J670/71 strain and incubated at 39°C. At 2 h after
infection(arrow),somecultureswereshifted downto
33°Cin the absence(A)orpresence(B) of
cyclohexi-mide (10 ,ug/ml) and were pulse-labeled with
[5,6-3H]uridine
for 30 minat1-hintervals. Theremainingcultures were incubated and labeled at 39°C in the
absence ofcycloheximide(C).Theradioactivity
incor-poratedintoanacid-precipitablefractionwascounted
byscintillation spectrometry.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.496.60.254.74.245.2] [image:7.496.307.407.434.565.2]TEMPERATURE SENSITIVITY OF EV70 105
I
C
G_
c=4
0
30 60
0
30 60
0
30
60
INCORPORATION
TIME
(MIN
FIG. 7. Detection of RNApolymerase activity ina
cell-freesystemin thecrudereplication complex from
cellsincubated for 1.5 h after infection. LLC-MK2 cell
monolayers grown in flasks were infected with the
J670/71 strainorwithitswarmth-adaptedmutant(see
text)atamultiplicity of infection of approximately 40.
After virus adsorption for 1 h, half of the cultures
received 20 mlof MEMcontaining5 sLg of AMDper
ml, and the other half received MEM containing the
sameconcentration of AMD and 200,ug ofguanidine
hydrochloride permland all cultures wereincubated
at the temperatures described below. At 1.5 h after
infection, the cells were collected and disrupted to
makethereplication complexatafinalconcentration
of 50 ,ug of totalprotein per50 ±l. Forassaying the
polymerase activity, each100-,ulreaction mixture
con-taining 50 ,u ofreplication complexwasincubatedat
33°Cin awaterbath. Samples weretakenat15-min
intervals, andthe totalradioactivityincorporatedinto
the RNAproductwasdeterminedasdescribedinthe
text. The replication complexes wereprepared from
cells treated under various conditions: infected with
the J670/71strain andincubatedat33°C (A)or39°C (B)
orinfected with thewarmth-adapted clone and
incu-batedat39°C (C). Mock-infected culture controls for
eachtemperaturewereincludedin(A) and (B). Graphs
represent preparations from cells incubated in the presence (0)orabsence (0) ofguanidine
hydrochlo-ride, made with virus-infected(-)ormock-infected
(. )cells.
previously reported (22), confirms that the early
events,
including uncoating, proceed normally
in
EV70 infection
atnonpermissive
tempera-tures. It has been demonstrated by both the
immediate
rise in the intracellular synthesis ofviral
RNA after the shift-down to33°C
duringthe
courseof viral infection
at39°C
and moreevidently
by the shutoff of host cellmacromo-lecular
synthesis by viral infectionatthenonper-missive
temperaturesinceanexpression of viralmRNA
function is prerequisite
for the shutoff ofhost cell
macromolecular synthesis bypicorna-virus infection
(19).Furthermore, this
study indicates thatEV70
virion RNA can function as mRNA in vivo at
nonpermissive
temperatures to
synthesize
pre-cursor
proteins,
which
aresubsequently cleaved
into
specific components,
including
an RNApolymerase.
Despite
intracellular
synthesis
of
the RNA
polymerase, no RNA
synthesis
pro-ceeded unless the
incubation temperature
wasshifted down. Thus, we may reasonably
con-clude that the
early
translation of viral RNA
normally proceeds in
EV70-infected cells at
non-permissive
temperatures and that the
tempera-ture-sensitive
defect is located at the
transcrip-tional level.
Three
possibilities
will be
considered for
the
mechanism to inhibit RNA
synthesis
at
the
transcriptional level. The first
possibility
is that
viral RNA
polymerase could be inactive
intra-cellularly
atnonpermissive
temperatures. To
investigate
this
possibility,
we
tested the in vitro
polymerase
activity of
the
prototype
strain
J670/
71 at
39°C
and found that it
wasmuch
lower at
this temperature (data not
shown).
Thus, it
seemed
quite possible that temperature
sensitiv-ity of the prototype strain was due to its
heat-labile polymerase. However, we could not
con-clude that the
temperature-sensitive
defect
atthe
transcriptional level could be
simply
ex-plained
by an in vitro heat-labile property
of
the
replication complex because the
polymerase
complex of
ourwarmth-adapted
mutantalso
functioned better at
33°C
than
at
39°C
as
far as it
was
tested in vitro (data not shown). The second
alternative
is that a
hypothetical constituent(s)
of the
replication
complex, other than RNA
polymerase, which
would
participate
in
tran-scription,
might
be
temperature sensitive.
Third,
virion RNA could not function as the template
for the
synthesis
of the progeny
RNA.It
has
been recently
reported
that
anisolated
polymerase
of
poliovirus
requires
anoligouridy-lic acid
sequence as
its
primer
(10, 31) and that
VPg at the 5' end of
picornavirus
RNA
may
play
an
important role in the viral
replication
(9, 16,
24, 27). It has also been suggested that a host
cell
factor(s)
may be
involved in the process of
picornavirus
RNA
replication (6, 7, 20) and act
at
the
initiation step
of poliovirus
RNA
replica-tion
(6). Eventually, a
conclusive
answerfor
the
exact
mechanism of the
transcriptional defect in
EV70 infection will
need
more detailed
analyti-cal
studies on
thestructure
and function
of
viralas
well
ascellular
components which
participate
in the
process of RNA
replication.
ACKNOWLEDGMENTS
WearegratefultoRobert R.Wagner,University of Virgin-ia,for his critical reading of the manuscript and for useful discussion.
This work was supported in part by a grant-in-aid for scientific research from theMinistry of Education, Science, andCulture,Japan.
VOL.44,1982
e
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.496.50.245.52.249.2]LITERATURE CITED
1. Adler, R., and D. R. Tershak. 1974. Temperature-sensi-tive defect of type 2 poliovirus. Virology 58:209-218. 2. Baltimore, D., M. Girard, and J. E. Darnell. 1966.
As-pectsof thesynthesis of poliovirus RNA and the forma-tionofvirus particles. Virology 29:179-189.
3. Bonner, W. M., and R. A. Laskey. 1974. A film detection methodfortritium-labeled proteins and nucleic acids in polyacrylamide gels. Eur. J. Biochem. 46:83-88. 4. Crawford, N.,A. Fire,M.Samuel,P.A. Sharp, andD.
Baltimore.1981. Inhibition of transcription factor activity by poliovirus. Cell 27:555-561.
5. Cross, R. K., and B. N. Fields. 1976. Reovirus-specific polypeptides: analysis using discontinuous gel electropho-resis. J.Virol. 19:162-173.
6. Dasgupta,A., P. Zabel, and D.Baltimore. 1980. Depen-dence of theactivity of the poliovirus replicase on a host cellprotein.Cell19:423-429.
7. Dmitrieva, T. M., M. V. Schcheglova, and V. I. Agol. 1979.Inhibition of activityof encephalomyocarditis virus-inducedRNApolymerase by antibodies against cellular components.Virology92:271-277.
8. Ehrenfeld, E. 1982.Poliovirus-induced inhibition of host-cellproteinsynthesis. Cell 28:435436.
9. Flanegan, J.B., R. F. Pettersson,V.Ambros, M. J. Hew-lett, and D.Baltimore.1977.Covalentlinkage of a protein to adefined nucleotide sequence at the 5'-terminus of virion and replicative intermediate RNAs of poliovirus. Proc.Natl. Acad. Sci. U.S.A. 74:961-965.
10. Flanegan, J. B.,and T. A.VanDyke. 1979. Isolation of a soluble andtemplate-dependentpoliovirusRNA polymer-asethatcopiesvirion RNA in vitro. J. Virol. 32:155-161. 11. Flores-Otero, G., C. Fernandez-Tomas, andP. Gariglio-Vidal.1982. DNA-bound RNApolymerases during polio-virus infection: reduction in the number of form II enzyme molecules. Virology116:619-628.
12. Hambling, M. H.,and P. M.Davis.1965.Susceptibilityof the LLC-MK2 line of monkey kidney cells to human enteroviruses.J.Hyg. 63:169-174.
13. Kitamura, N., B. L.Semler,P.G.Rothberg, G. R. Lar-seu, C.J. Adler,A.J.Dorner,E. A.Emini,R.Hanecak, J. J. Lee, S. van der Werf, C.W. Anderson, and E. Wimmer. 1981. Primarystructure, geneorganizationand polypeptideexpressionofpoliovirusRNA.Nature (Lon-don) 291:547-553.
14.Kono, R.,A.Sasagawa,K.Ishii,S.Sugiura,M.Ochi,H. Matsumiya,Y.Uchida,K.Kameyama,M.Kaneko,and N. Sakurai. 1972. Pandemic ofnewtype ofconjunctivitis. Lancet i:1191-1194.
15.Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head ofbacteriophage T4. Nature(London)227:680-685.
16. Lee,Y.F.,A. Nomoto, B. M. Detjen, and E. Wimmer. 1977. A proteincovalentlylinkedtopoliovirusgenome RNA. Proc. Natl. Acad. Sci. U.S.A. 74:59-63. 17. Levintow, L. 1974. Theproductionofpicornaviruses,p.
109-169. In H.Fraenkel-Conratand R. R.Wagner(ed.), Comprehensive virology, vol. 2. Plenum Publishing Corp.,NewYork.
18. Lowry,0.H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951.Protein measurement with the Folin phenol reagent.J.Biol. Chem. 193:265-275.
19. Lucas-Lenard, J. M. 1979. Inhibition of cellular protein synthesis after virus-infection, p. 73-99. In R. Perz-Bercoff (ed.), The molecularbiology of picornaviruses. Plenum PublishingCorp., New York.
20. Lundquist, R. E., and J. V. Maizel, Jr. 1978. In vitro regulation of the poliovirus RNApolymerase. Virology 89:484-493.
21. Miyamura, K., A. Sasagawa, E. Tajiri, and R. Kono. 1976. Growth characteristics of acutehemorrhagic conjunctivi-tis(AHC) virus in monkey kidney cells.II. Temperature sensitivity of the isolates obtained at various epidemic areas.Intervirology 7:192-200.
22. Miyamura, K., S. Yamazaki, E. Tajirl, and R. Kono. 1974. Growth characteristics ofacutehemorrhagic conjunctivi-tis (AHC) virus in monkey kidney cells. I. Effect of temperature onviralgrowth.Intervirology 4:279-286. 23. Miyamura, K., S. Yamazaki, E. Tajiri, and R. Kono. 1978.
Growthcharacteristics of acute hemorrhagic conjunctivi-tis(AHC) virus in monkey kidney cells. III. ViralRNA synthesisatpermissive and nonpermissivetemperatures. Intervirology 9:206-213.
24. Nomoto, A., B. Detjen, R. Pozzatti, and E.Wimner.1977. Thelocation of thepolio genome protein in viral RNAs andits implication forRNAsynthesis. Nature(London) 268:208-213.
25. Penman, S., Y. Becker, and J.E. Darnell. 1964. A cyto-plasmic structure involved in the synthesis and assembly ofpoliovirus components. J. Mol. Biol. 8:541-555. 26. Penman, S., H. Greenberg, and M. Willems. 1969.
Prepa-ration ofpolyribosomes from cells grown in tissue culture, p.49-58. In K. Habel and N.P. Salzman(ed.), Funda-mentaltechniques invirology. Academic Press, Inc., New York.
27. Pettersson, R. F., V. Ambros, and D. Baltimore. 1978. Identification ofa protein linkedto nascent poliovirus RNAand to thepolyuridylic acid ofnegative-strandRNA. J.Virol. 27:357-365.
28. Rueckert, R. R., T. J. Matthews, 0.M.Kew, M.Pollansh, C. McLean, andD. Omilianowski. 1979. Synthesis and processing of picornaviral polyprotein, p. 113-125.InR. Perz-Bercoff (ed.), The molecularbiologyof picomavirus-es, PlenumPublishing Corp., NewYork.
29. Sasagawa, A.,R.Kono,andK. Konno.1976. Laboratory-acquired infection of the eye with AHC virus. Jpn. J. Med.Sci. Biol. 29:95-97.
30. Tershak,D.R.,W.Mitchell,andB. D.Gerfinkle. 1972. Effect ofguanidineonthegrowthof LScpoliovirus.Can. J.Microbiol. 18:747-755.
31. VanDyke,T.A.,andJ.B.Flanegan. 1980.Identification ofpolioviruspolypeptide p63as asolubleRNA-dependent RNApolymerase.J. Virol. 35:732-740.
32. Yamazaki, S.,K.Natori,andR. Kono.1974. Purification and biophysical properties of acute haemorrhagic con-junctivitisvirus. J. Virol. 14:1357-1360.
33. Yin,F.H.,and E.Knight, Jr.1972. Invivoand in vitro synthesisofhumanrhinovirustype2ribonucleicacid. J. Virol. 10:93-98.
J.VIROL.