JOURNAL OFVIROLOGY, July 2011, p. 7284–7295 Vol. 85, No. 14 0022-538X/11/$12.00 doi:10.1128/JVI.02472-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
The Diversity of Torque Teno Viruses:
In Vitro
Replication Leads to
the Formation of Additional Replication-Competent Subviral Molecules
䌤
†
Ethel-Michele de Villiers,* Silvia S. Borkosky, Romana Kimmel, Karin Gunst, and Jian-Wei Fei
Division for the Characterization of Tumorviruses, Deutsches Krebsforschungszentrum, Heidelberg, Germany
Received 26 November 2010/Accepted 5 May 2011
The familyAnelloviridaecomprises torque teno viruses (TTVs) diverse in genome structure and organization.
The isolation of a large number of TTV genomes (TTV Heidelberg [TTV-HD]) of 26 TTV types is reported. Several isolates from the same type indicate sequence variation within open reading frame 1 (ORF1), resulting
in considerably modified open reading frames. We demonstratein vitroreplication of 12 full-length genomes
of TTV-HD in 293TT cells. Propagation of virus was achieved by several rounds of infections using supernatant and frozen whole cells of initially infected cells. Replication of virus was measured by PCR amplification and
transcription analyses. Subgenomic molecules (TTV), arising early during propagation and ranging in size
from 401 to 913 bases, were cloned and characterized. Propagation of these TTV inin vitro cultures was
demonstrated in the absence of full-length genomes.
The family Anelloviridae includes torque teno viruses (TTVs), TT-midiviruses (TTMDV), and TT-miniviruses (TTMV), the majority originating from samples of human or-igin (2, 37, 39, 41, 56). The plurality of this family of single-stranded DNA (ssDNA) viruses is reflected not only in DNA sequence, but also in genome size and organization.
Infections occur within the first days of life, with close to 100% of infants being infected at 1 year of age. The primary route of infection, however, remains unclear (23, 38, 48). The ubiquitous nature of TTV infections has hampered efforts to associate it with the pathogenesis of disease (9, 15, 25, 41). A possible etiological association with diseases of the liver (re-viewed in reference 36) and respiratory tract (3, 30, 31, 49), hematopoietic malignancies (9, 10, 13, 15, 25, 53, 59), and autoimmune diseases (9, 26, 28, 54) have been reported.
The presence of a variety of intragenomic rearranged TTV subviral molecules (TTV) in serum samples and thein vitro transcription of a subviral molecule constituting only 10% of the complete genome initiated the discussion whether TTVs may share similarities to the plantvirus familyGeminiviridae(8, 23). Both mono- and bipartite geminiviruses associate with single-stranded DNA satellites to form disease-inducing com-plexes (16, 36, 46, 47, 52, 55).
Multiple attempts have been made to find a suitablein vitro system for the replication and propagation of TTVs. Replica-tive forms of its DNA have been demonstrated in bone marrow cells and in the liver (22, 42, 44, 45). Peripheral blood acts as reservoir for TTVs (43), and replicationin vivoseems to occur preferably in activated mononuclear cells (27, 29, 33). Al-thoughin vitrotranscription has been investigated in a variety
of cell lines (18–21, 35, 50), long-term replication leading to virus production has been difficult to achieve (25).
More than 200 genomes of TTVs have been isolated. The isolates grouping in the genusAlphatorquevirus(ca. 3.8 kb in size) share very low DNA sequence homology and differ in their genome organizations. A short stretch (71 bp) of the intergenic region is highly conserved among all human TTV isolates (48) and is widely used to demonstrate TTV infection. We have analyzed samples from a broad spectrum of diseases for the presence of TTV DNA by applying PCR amplification of this conserved region (9, 15, 25, 54; E.-M. de Villiers and K. Gunst, unpublished results). Identification of individual TTV types, however, requires the amplification of full-length ge-nomes. We have thus far isolated 93 full-length genomes of TTVs (ca 3.8 kb) from human samples (9, 15, 25; present study). These included samples obtained from healthy individ-uals and patients with leukemia and lymphoma, rheumatoid arthritis, multiple sclerosis, and kidney disease. The present study describes thein vitroreplication and transcription of 12 isolates after initial transfection of the genomic DNA and followed by propagation using frozen infected cells or culture supernatant. Intragenomic rearranged subviral molecules (TTV; i.e., microTTV) appearing in early passages were cloned and characterized. These also propagated independent of the mother genome in the 293TT cell line. We propose a putative origin of replication for TTVs based on comparative sequence analyses.
MATERIALS AND METHODS
TTV isolation and characterization. The isolation of TTV isolates TTV Heidelberg 3a (TTV-HD3a) (tth8; accession no AJ620231) and TTV-HD1a (tth25; accession no AJ620222) was previously described (15). Full-length genomic sequences of both TTV-HD3a and TTV-HD1a were cloned into vector pUC18 using restriction enzymes SalI (25) and EcoRI, respectively. Additional TTV sequences were identified in human samples by DNA nested amplification using primers NG472/NG352 and NG473/NG351 as previously described (25, 48). The limited availability of DNA for a number of biopsy and serum samples required prior amplification by rolling-circle amplification with a TempliPhi kit (GE Healthcare). All amplified products were cloned and sequenced (25). Sam-ples harboring TTV DNA were subsequently subjected to long-distance PCR amplification with TaKaRa LATaqenzyme (Takara Bio, Inc., Japan) and
re-* Corresponding author. Mailing address: Division for the Charac-terization of Tumorviruses, Deutsches Krebsforschungszentrum, Im Neuenheimer Feld 242, 69120 Heidelberg, Germany. Phone: 11-49-6221-424655. Fax: 11-49-6221-424822. E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi .asm.org/.
䌤Published ahead of print on 18 May 2011.
7284
on November 7, 2019 by guest
http://jvi.asm.org/
spective primers that had been designed based on the initially identified TTV DNA sequences. These back-to-back primers included the following combina-tions: tth25-1s and tth25-2as, jt34f-1s and jt34f-2as, jt34f-7s and jt34f-8as, jt34f-5s and jt34f-6as, tth4-1s and tth4-2as, and t3pb-1s and t3pb-2as, as well as tth8-1s and tth8-2as (Table 1). Long-PCR amplification was performed using a touch-down stepwise reaction as described previously (25), with the exception of primer combinations t3pb-1/2, jt34f-5/6, and tth4-1/2. The PCR conditions for PCR amplification with the t3pb-1/2 and jt34f-5/6 primers were initial denaturation at 94°C for 1 min, followed by 30 cycles of 94°C for 30 s, annealing at 65°C for 1 min, and elongation at 72°C for 4 min, and a final elongation step at 72°C for 10 min. The PCR conditions for amplification with tth4-1/2 primers were similar, except that annealing was performed at 68°C. All obtained amplicons in the range of 3.8 kb were eluted and purified after gel electrophoresis, cloned into vector pCR2.1 (TA cloning kit; Invitrogen), and propagated in NovaBlue Singles competent cells (Merck Chemicals, United Kingdom). Full-length genomes were sequenced through both strands. A total of 53 full-length genomes were obtained in the present study (for the accession numbers, see Table S1 in the supplemental material).
Sequence analyses and phylogeny.DNA sequences were compared to TTV sequences available in all databanks, using the HUSAR software package (15). The International Committee on Taxonomy of Viruses (ICTV) recently classi-fied TTVs into the familyAnelloviridaebased on the DNA sequence of large open reading frame 1 (ORF1) (2). Characterization of the genomes of our isolates revealed rearrangement of sequences in the ORF1 region. We therefore subjected the full-length genomes of the genusAlphatorquevirusand our isolates to phylogenetic analyses, as previously described (15). The phylogenetic tree was displayed with the Treeview program of the University of Glasgow. Translated ORFs were analyzed for homologous proteins and functional domains with ProtSweep (7).
Cell culture and transfection.The human embryonic kidney cell line 293TT (4, 5) was maintained in Dulbecco’s modified Eagle’s medium (DMEM) supple-mented with 10% fetal calf serum, 1% Glutamax, 1% nonessential amino acids (Invitrogen, Karlsruhe, Germany), and 400g/ml hygromycin B (Roche Diag-nostics, Mannheim, Germany). Cloned virus DNA was released from the vector DNA by restriction enzyme digestion, after which virus and vector DNAs were cotransfected into cells (2g per 1.6⫻106cells on six-well plates) grown without
hygromycin B by using Lipofectamine reagent (Invitrogen) according to the manufacturer’s instructions (12). Vector DNA served as an internal control. Culture medium (2 ml) was supplemented with 800l Opti-MEM prior to incubation for 4 h at 37°C. Transfected cultures were subsequently incubated with fresh medium and propagated when confluence was reached. Hygromycin B was then added to the medium. Full-length genomes of 12 TTV isolates were transfected, maintained, and harvested in parallel at all times. The TTV genomes included TTV-HD14a, TTV-HD14b, TTV-HD14c, TTV-HD14e, TTV-HD15a, HD16a, HD20a, HD3a, HD1a, HD23a, TTV-HD23b, and TTV-HD23d (for accession numbers, see Table S1 in the supple-mental material). Controls included transfection with vector alone and cells
transfected with 1⫻Tris-EDTA (TE). Transfected cells and culture medium were frozen at⫺80°C, and samples for DNA and RNA extraction were taken at each time point during propagation. DNA was extracted with phenol-chloro-form-isoamyl alcohol and RNA by using the RNeasy minikit (Qiagen, Hilden, Germany). All transfection experiments were performed three times with 6-week intervals between primary transfections. Frozen cells and culture medium were passaged between four and seven times. Cloned subviralTTV genomes were transfected into 293TT cells in the same way as the full-length genomes.
Replication of virus DNA as monitored and demonstrated by LD-PCR ampli-fication and real-time qPCR. (i) LD-PCR ampliampli-fication.Long-distance PCR (LD-PCR) on total cellular DNA was performed for each transfected culture with the respective primer combinations and conditions described above. Trans-mission of cotransfected plasmid DNA was controlled for by PCR amplification. M13 forward (5⬘-GCCGTCGTTTTACAACGTC-3⬘) and reverse (5⬘-ATCATG GTCATAGCTGTTTCCTG-3⬘) primers were used under the following amplifi-cation conditions: 94°C for 1 min, followed by 30 cycles of denaturation for 30 s at 94°C, annealing at 63°C for 1 min, and extension at 72°C for 4 min, and a final elongation step of 10 min at 72°C (D. Vester, personal communication).
(ii) Real-time qPCR.TTV-HD14e and TTV-HD1a titers from infected cells were assessed by real-time quantitative PCR (qPCR). Genomic DNA samples (50 ng) in triplicates were amplified with two sets of primers and hydrolysis probes designed for the conserved noncoding region for each TTV-HD tested. For TTV-HD14e, the primers and probes were 33.35q-31-F (5⬘-ACAGACCAA TCAGGACCTTCTAC-3⬘), 33.35q-115-R (5⬘-CGGACGGGCGAAGAAAAA C-3⬘), 33.35q-89-Pr (antisense) (5⬘-6-carboxyfluorescein [FAM]-CTACCATTC GTCCACCGCTGTTGCTT-3⬘–6-carboxytetramethylrhodamine [TAMRA]), 33.35q-326-F (5⬘-GTGCCAGGTAGAGGGAATCAATG-3⬘), 33.35q-411-R (5⬘-GCGAGGAGCAATGCCGTTAAG-3⬘), and 33.35q-373-Pr (antisense) (5⬘-FAM-TCACCACACCCGCAGAAAGCAGCAT-3⬘-TAMRA). For TTV-HD1a, the primers and probes were tth25q-439-F (5⬘-GTTGGCCATCTTAAT CATCTG-3⬘), tth25q-536-R (5⬘-GCTGGCAGGTTTCTTATTTG-3⬘), tth25q-461-Pr (sense) (5⬘-FAM-CAACTACTCTGGGTCGTCCTCCGC-3⬘-TAMRA), tth25q-376-F (5⬘-GAGAGAAACTGGTACGAGAG-3⬘), tth25q-471-R (5⬘-CA GAGTAGTTGCCAGATGATTA-3⬘), and tth25q-437-Pr (antisense) (5⬘ -FAM-AAATCGCCACAGCCACAAACAGCAG-3⬘-TAMRA). The TaqMan assay for TTV-HD14e was obtained from Biomers.net. Beacon designer 7.9 software was used for TaqMan assay design for TTV-HD1a. This included a BLAST search and a predictor of template folding structure tool in order to achieve assay specificity and optimal binding of primers and probes. Additionally, all primers and probes were validated by determining the optimal annealing temperatures, concentration, and absence of dimers. Assay optimization was determined as recommended by MIQE (minimum information for publication of quantitative real-time PCR experiments) guidelines (6), obtaining PCR efficiency andR2
values within the accepted parameters for an optimized qPCR assay (40). Am-plification reactions for TTV-HD14e were performed in 20-l volumes contain-ing 12.5l TaqMan Universal master mix (Applied Biosystems), 0.5l forward primers (10M), 0.75l reverse primers (20M), 0.63l 5⬘-FAM–3⬘
-TAMRA-TABLE 1. Primers used to generate complete TTV-HD genomes andTTV-HD subviral genomes by long-distance PCR amplification
TTV (accession no.) Primer Nucleotide no. Sequence
TTV-jt34f (AB064607) jt34f-1s 223–247 5⬘-GGCCGGGCCATGGGCAAGGCTCTTA-3⬘
jt34f-2as 195–222 5⬘-AGTCAAGGGGCAATTCGGGCTCGGGACT-3⬘
jt34f-5s 205–222 5⬘-CAATTCGGGCTCGGGACT-3⬘
jt34f-6as 186–204 5⬘-ACACACCGCAGTCAAGGGG-3⬘
jt34f-7s 205–223 5⬘-CAATTCGGGCTCGGGACTG-3⬘
jt34f-8as 181–204 5⬘-AGTTTACACACCGCAGTCAAGGGG-3⬘
TTV-HD1 (AJ620222) th25-1s 126–156 5⬘-CCGCAGCGAGAACGCCACGGAGGGAGATCCT-3⬘
tth25-2as 95–125 5⬘-ACTTCCGAATGGCTGAGTTTTCCACGCCCGT-3⬘
TTV-HD3 (AJ620231) tth8-1s 133–164 5⬘-AGAGGAGCCACGGCAGGGGATCCGAACGTCCT-3⬘
tth8-2as 102–132 5⬘-CTTACCGACTCAAAAACGACGGGCAGGCGCC-3⬘
TTV-HD4 (AJ620226) tth4-1s 129–156 5⬘-CAGCGAGAACGCCACGGAGGGAGATCCT-3⬘
tth4-2as 101–128 5⬘-GAATGGCTGAGTTTTCCACGCCCGTCCG-3⬘
TTV-t3pb (AF247138) t3pb-1s 209–226 5⬘-CAATTCGGGCACGGGACT-3⬘a
t3pb-2as 185–208 5⬘-AGTTTACACACCGAAGTCAAGGGG-3⬘
a
The underlined letter indicates that the TTV-t3pb sequence (AF247138) has been modified to A at this position in the primer.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.44.542.82.265.2]labeled probe (10M), 0.62l water, and 5l of DNA, and those for TTV-HD1a were performed in 25-l volumes containing 12,5l TaqMan Universal master mix (Applied Biosystems), 0.75 l each of the forward and reverse primers (10M each), 0.75l 5⬘-FAM–3⬘-TAMRA-labeled probe (10M), 5.25
l water, and 5l DNA. After activation of the AmpliTaq Gold DNA polymer-ase (10 min at 95°C), the reaction mixtures were amplified for 40 cycles (15 s at 95°C and 1 min at 60°C), and the fluorescent signals were detected with an ABI 7300 sequence detection system (Applied Biosystems). Quantification of TTV-HD1a and TTV-HD14e was calculated against a standard curve generated by 10-fold dilutions of either the TTV-HD1a or TTV-HD14e genome, respectively. For dilution of the standards, 50 ng of 293TT untreated cellular DNA in 1⫻TE (Tris-EDTA) buffer was used. The standards were aliquoted and stored at⫺20°C for no longer than a week, and each standard aliquot was used per single reaction. DNAs from untransfected 293TT cells and nontemplate controls were included in each reaction.
Transcription analyses.Transcripts of TTV-HD full-length genomes were analyzed by two different approaches. 5⬘- and 3⬘-rapid amplification of cDNA ends (RACE) products were generated from single- as well as double-stranded cDNAs. Single-stranded 5⬘-RACE-ready and 3⬘-RACE-ready cDNAs were re-spectively synthesized from 1g purified total RNA in a 10-l reaction mixture using the SMARTerRACE cDNA amplification kit (Clontech catalog no. 634923), in which RNA is reverse transcribed by SMARTScribe reverse trans-criptase at 42°C for 90 min. 3⬘-RACE-coding sequence (CDS) primer A was used for the synthesis of 3⬘-RACE-ready cDNA, whereas the 5⬘-RACE-CDS primer A and SMARTer IIA oligonucleotide were used for the synthesis of 5⬘ -RACE-ready cDNA. Double-stranded cDNA was concomitantly synthesized. Here, full-length single-stranded cDNA was initially synthesized with the SMARTerPCR cDNA synthesis kit (Clontech catalog no. 634925) according to the manufactur-er’s protocol. Purified total RNA (1g) was transcribed with SMARTScribe reverse transcriptase and primers 3⬘SMART CDS primer IIA and SMARTer IIA oligonucleotide. These primers both contain a nontemplate nucleotide stretch, thereby creating an extended template. Second-strand cDNA amplifica-tion was obtained by long-distance PCR (LD-PCR) amplificaamplifica-tion with 5⬘-PCR primer IIA and the Advantage 2 polymerase mix (Clontech catalog no. 639201). PCR amplification was performed for 15 s at 95°C, 30 s at 65°C, and 3 min at 68°C per cycle and with different ranges of cycles in order to determine optimal conditions.
5⬘- and 3⬘-RACE PCR amplification was performed using 5⬘-RACE-ready or 3⬘-RACE-ready cDNA, respectively, or double-stranded cDNA template in both cases. RACE-PCR was performed using Advantage 2 polymerase mix, a univer-sal primer A mix (UPM) from the SMARTerRACE cDNA amplification kit, and forward and reverse primers fitting to the respective TTV types (see Table S2 in the supplemental material). The conditions for amplification were 29 cycles of 30 s at 94°C, annealing for 30 s at 68°C, and elongation for 3 min at 72°C, with a final extension for 15 min at 72°C. All products were analyzed by gel electro-phoresis, purified after gel elution, cloned into vector pCR2.1 (Invitrogen catalog no. K2020-40), and sequenced. Two additional controls were performed in order to control for nonspecific amplification. In one control, amplification was per-formed using only one TTV-specific primer, and in the second, amplification was performed using the UPM primer alone. No products were detected in either of these.
RESULTS
Identification and characterization of TTV genomes.Initial
amplification of the short conserved GC-rich region of TTVs in serum and biopsy samples led to the identification of TTV DNA in the majority of cases. Subsequent amplification of the complete genome is necessary to identify specific TTV types as many share exact DNA homology in the amplified 71 bases lying in the control region, but differ as much as 60 to 80% in sequence identity in the rest of their genomes. We have de-signed a number of back-to-back primer combinations on se-quences obtained during the course of our investigations (Ta-ble 1). Long-distance PCR amplification was performed on TTV DNA-positive samples. Amplicons ranging between 3 and 4 kb were cloned and sequenced. TTV DNA-positive sam-ples originated from healthy subjects as well as patients with leukemia, multiple sclerosis, rheumatoid arthritis, and kidney
disease. Part of these data have been described previously (9, 25, 54).
A total of 53 full-length DNA genomes were characterized in the present study. As many as 12 distinct full-length isolates were identified after sequencing 19 genomes from a single biopsy. The genome organizations of different isolates of one TTV type varied, despite low diversity of nucleotides (ranging from 1 to 4%). Although the large open reading frame ORF1 was mainly involved, differences within the noncoding region and other genes were also noted. These data confirmed our earlier observations (9, 15, 25). Modifications in ORF1 in-cluded premature stop codons leading to separate smaller ORFs in this region, considerable sequence diversity in the hypervariable region (15), or the absence of a stop codon resulting in a larger ORF1 than present in the prototype (see Fig. S1 in the supplemental material). The official classification of the family Anelloviridae is based on comparisons of the ORF1 DNA sequences (2). Due to the ORF1 modifications in our isolates, we chose to include the full-length genomic se-quences in the phylogenetic analyses presented here. The aim of this analysis was to gain an overview of our isolates of TTV-HD in relation to established TTV species (Fig. 1). All of our previous isolates are included in this tree as well (9, 15, 25).
In vitroreplication of TTV-HD.Attempts to associate torque teno virus infection with the pathogenesis of a specific disease have repeatedly been reported in the past. Samples from a large range of diseases have been analyzed.In vitro investiga-tions were hampered by negative attempts by us and others to identify a cell culture system in which these viruses can readily be propagated over longer time periods. TTV particles were initially characterized with the help of density gradients and immunoglobulin aggregates (reviewed in reference 41) and later visualized from sera and feces (14). Torque teno viruses occur predominantly in cells of the hematopoietic system (41). Our first isolates were obtained from the spleen of a patient with Hodgkin’s lymphoma (15). We therefore used the L428 cell line of Hodgkin’s lymphoma origin in our initial attempts to demonstratein vitroreplication and transcription of TTV-HD3a. We achieved replication of the full-length genome for up to 7 days after transfection of the linearized virus DNA (25). Similar attempts including other isolates were less suc-cessful. In order to find a more reproducible cell culture sys-tem, as well as to extend this period of replication, we chose to transfect full-length TTV genomes into the human embryonic kidney cell line 293TT, which was engineered to express high levels of simian virus 40 (SV40) large-T antigen (4, 5). Second, we decided to include 12 full-length isolates in this study in order to determine (i) whether variations in ORF1 would in-fluence replication and (ii) whether divergent TTV types vary in their mode of replication. We took great care in propagating all 12 isolates in parallel in order to exclude, as far as possible, variation which may occur during handling. We chose the fol-lowing isolates for transfection and propagation: TTV-HD1a (15), the closest relative of which is species TTV3 (hel32); and TTV-HD3a (25), the closest relative of which is species TTV12 (ct44f) (Fig. 1). In addition, TTV-HD16a (related to species TTV22); HD15a (related to species TTV12); and TTV-HD14a, TTV-HD14b, TTV-HD14c, and TTV-HD14e (related to species TTV29), all of which were isolated from brain biopsies from patients with multiple sclerosis; TTV-HD20a
7286 DEVILLIERS ET AL. J. VIROL.
on November 7, 2019 by guest
http://jvi.asm.org/
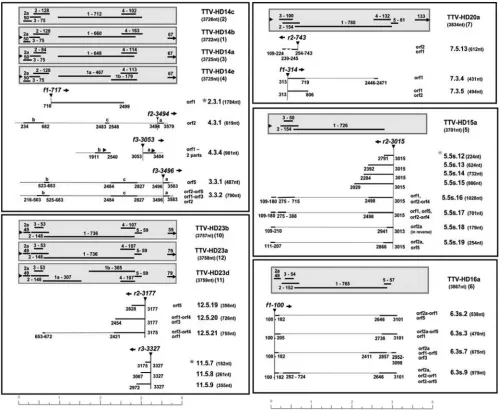
(related to species TTV13), originating from kidney tissue; and TTV-HD23a, TTV-HD23b, and TTV-HD23d (related to spe-cies TTV3) were all three amplified from serum taken from patients with rheumatoid arthritis. The sequences of TTV-HD14a, TTV-HD14b, TTV-HD14c, and TTV-HD14e vary be-tween 1 and 2% in their full-length genomes. The prototype is TTV-HD14a, with an intact ORF1 of 648 amino acids (aa) in size. The ORF1 of TTV-HD14b is 660 aa in size, with only 554 aa sharing identity to TTV-HD14a ORF1, whereas the rest of the ORF indicates fusion to ORF4 (as shown in reference 8). Similarly, TTV-HD14c ORF1 is 712 aa and constitutes ORF1 (first 645 aa) fused to ORF5. TTV-HD14e ORF1 is inter-rupted, resulting in two ORFs of 467 aa and 179 aa in size. The TTV-HD23b, TTV-HD23d, and TTV-HD23a genomes vary only between 1 and 3% in sequence identity, but their ORF1 genes differ as follows: TTV-HD23a ORF1 as the prototype is 736 aa in size, and the TTV-HD23b ORF1 DNA sequence varies from that of TTV-HD23a in the hypervariable region by 18.4% (34.2% in amino acids). The HD23b and TTV-HD23d DNA sequences differ only 1% in overall identity, but the TTV-HD23d ORF1 is interrupted, resulting in two ORFs that are 307 aa and 365 aa in size (see Fig. S1 in the supple-mental material).
The virus genome was released from the vector construct by restriction enzyme digestion, after which the virus genome and vector were cotransfected into semiconfluent 293TT cells. The nature of this cell line with its many rounded cells attached to the monolayer does not permit a clear-cut identification of cytopathic effects. We passaged cells when confluent or when cells started to detach from the surface. Harvesting was done
[image:4.585.138.452.70.346.2]by shaking flasks to loosen all cells, or cells were partially scraped off. In the latter case, the rest of the cells were trypsinized to passage, and the culture medium of the previous culture was added to fresh medium. Aliquots of cells or cells and culture medium were frozen at⫺80°C, as well as used for DNA and RNA extraction. Frozen infected cells were initially used to reinfect new 293TT cultures as reinfection failed if cells used for the inoculum had previously been trypsinized at the time of harvest. Virus replication was monitored by long-dis-tance PCR with virus-specific primers on DNA extracted from infected cells. Long-distance PCR was also performed using primers to amplify vector sequences. This served as a control to determine whether viral DNA was actually replicating or whether the amplification resulted from DNA carried over after the transfection. Periods between reinfection and cell harvest varied between 3 and 7 days, depending on culture density. No obvious morphological differences were noted be-tween cultures of different TTV isolates. Reinfection during the course of one experiment was performed several consecu-tive times using frozen cell aliquots. Long-termin vitro prop-agation of TTVs has not been described before. The novel nature of these experiments evidently required variations in the procedure between the three transfection experiments. DpnI restriction enzyme digestion was performed on cellular DNA obtained from the initially transfected samples to re-move any residual bacterium-generated virus DNA. Long-dis-tance PCR amplification results indicatedde novoreplication of virus DNA. Examples of these TTV DNA amplicons with infected cellular DNA as the template are presented in Fig. 2. Long-distance PCR amplification of the full-length DNA FIG. 1. Phylogenetic tree showing TTV species (isolate names in parentheses) and additional isolates of genusAlphatorquevirus, as well as all TTV-HD (Heidelberg) types isolated by us. TTV-HD types propagated inin vitrocell cultures are circled.
on November 7, 2019 by guest
http://jvi.asm.org/
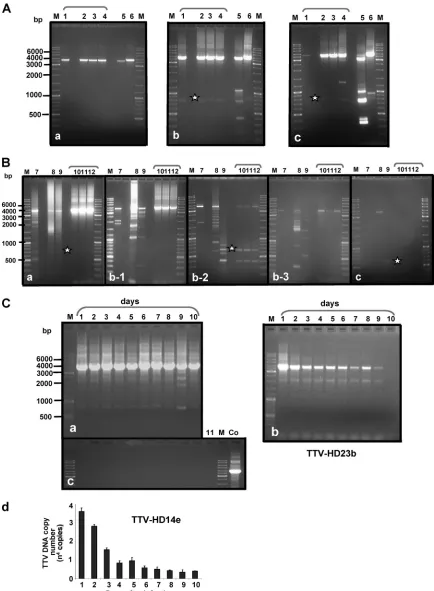
FIG. 2. Propagation of full-length TTV-HD genomes in 293TT cells. Shown are examples of propagation of TTV-HD14b, TTV-HD14c, TTV-HD14a, and TTV-HD14e (lanes 1 to 4), TTV-HD15a (lane 5), and TTV-HD16a (lane 6) after nested PCR amplification (A) and TTV-HD20a (lane 7), TTV-HD3a (lane 8), TTV-HD1a (lane 9), and TTV-HD23b, TTV-HD23d, and TTV-HD23a (lanes 10 to 12) after single PCR amplification (B). (A and B) Panels a, b, and c show examples of propagations harvested approximately 7 days after infection. Panels b-1, b-2, and b-3 indicate variability observed when propagating during the same passage. (C) Daily sampling of TTV-HD14e (nested PCR) (a) and TTV-HD23b (b) cultures. (c) PCR amplification of same DNA samples as in panel a, but using primers to amplify vector DNA as a control. Lane 11, 293TT cellular DNA; Co, vector DNA as a control. (d) Real-time quantitative PCR performed on the same samples as those in panels a and c. M, DNA size marker; *, subviral molecules from different cultures.
7288
on November 7, 2019 by guest
http://jvi.asm.org/
molecules indicated considerable differences between cultures. Second-round amplifications (using the same primers as in the first round) were necessary on all cultures infected with isolates from brain biopsies (i.e., TTV-HD16a, TTV-HD15a, and the four individual TTV-HD14 isolates) (Fig. 2A), despite the phylogenetic divergence (Fig. 1) between these three TTV types (45 to 50% nucleotide homology). Modifications in ORF1 did not seem to influence amplification or propagation, as visualized in the amplification of the full-length DNA (Fig. 2A, panels a to c). Additional DNA amplicons of various sizes were observed in TTV-HD15a-infected cultures. The occur-rence of these molecules increased during subsequent propa-gation (Fig. 2A, panels a to c, lane 5). We previously reported subviral molecules of a similar nature in human serum samples (25). Similar “off-size” amplicons were also occasionally noted in TTV-HD16a-infected cultures (Fig. 2A, panels a to c, lane 6) and rarely in TTV-HD14 cultures (Fig. 2A, panels a to c, lanes 1 to 4).
Large differences were noted in the behavior of the other six isolates. This variation was also evident between experiments and passages (Fig. 2B, panels b1, b2, and b3), reflecting an apparent high sensitivity to very minor modifications in culture conditions. The initially replicating full-length genome (3.8 kb) diminished during propagation (Fig. 2B, panels a to c), in concurrence with prominent subgenomic amplicons of differ-ent size ranges in HD20a-, HD3a-, and TTV-HD1a-infected cells (Fig. 2B, lanes 7 to 9). The amounts of input DNA used for long-distance PCR amplification, as well as the amounts of amplicons loaded onto gels, were the same for all cultures.
Differences observed between the two groups of isolates prompted us to investigate whether variations could be ob-served during serial sampling. We propagated equivalent pas-sages of TTV-HD14e and TTV-HD23b in parallel and took samples daily. Long-distance amplification indicated a con-stant replication of TTV-HD14e (visible after two rounds of DNA amplification) (Fig. 2C, panel b), in contrast to TTV-HD23b (visible already after a single round of DNA amplifi-cation), which was lost after 10 days in culture (Fig. 2C, panel a). These cultures were not passaged, and morphological dif-ferences between cultures were not noticeable. To control for genome replication in contrast to possible carryover of input DNA, we amplified the same cellular DNA using primers for vector amplification, of which an example is presented in Fig. 2C, panel c, for TTV-HD14e. No amplified vector DNA was visible throughout. In addition, we performed quantitative real-time PCR on the TTV-HD14e-infected cellular DNA (Fig. 2C, panel d). Copy numbers decreased gradually until day 4 postinfection, with a slight increase at day 5 when the culture medium was renewed. From day 6 postinfection, the copy number remained stable (approximately 3⫻ 103 to 6⫻ 103
copies) (Fig. 2C, panel d). These findings, together with the absence of vector amplification (Fig. 2C, panel c), confirmed the observation of a replication of TTV-HD14e as described above.
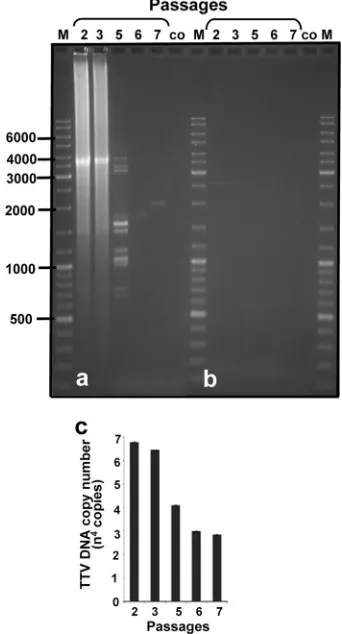
Further confirmation for replication of the virus DNA in 293TT cells by real-time quantitative PCR is presented in Fig. 3. The complete genome of TTV-HD1 was transfected into 293TT cells, as described previously. Transfected cells were passaged 3 days after transfection (passage 1) and harvested
and passaged (passage 2) 4 days later. Passage 2 was allowed to grow for 3 days before harvesting. At the same time, an aliquot of the supernatant from passage 2 was transferred onto unin-fected 293TT cells (passage 3), after which cells were harvested 6 days later. Passaging was repeated every 3 days, and the respective cell cultures were harvested 3 (passage 5), 5 (pas-sage 6), and 3 (pas(pas-sage 7) days after the respective passaging. The higher rate of viral DNA replication as measured by real-time quantitative PCR (Fig. 3c) seemed to decrease during subsequent passages, to level off after five passages. These results may be explained by the formation of smaller subviral molecules during passaging, as visualized after long-distance PCR amplification of the virus genome in these samples (Fig. 3a). The full-length genome and the shorter fragments share the conserved region of the TTV genome in which the inverse primers, used for long-distance PCR amplification, are located. PCR amplification using primers to amplify vector DNA was used as a control (Fig. 3b).
In vitroreplication and characterization ofTTV subviral
molecules.The appearance of smaller DNA amplicons of a
[image:6.585.334.505.72.389.2]constant size in cultures from isolates HD14b, TTV-HD14c, TTV-HD14d, and TTV-HD14e, as well as TTV-HD1a FIG. 3.In vitro replication of full-length genome of TTV-HD1. Each lane represents one passage of infected cells onto uninfected 293TT cells. (a) Long-distance PCR amplification of TTV-HD1 ge-nome in each sample. (b) Control. The same samples were amplifed as in panel a, but using primers to amplify vector DNA as a control. (c) Quantitative real-time PCR measuring the amplification of TTV-HD1 DNA. M, DNA size marker; co, uninfected 293TT cellular DNA.
on November 7, 2019 by guest
http://jvi.asm.org/
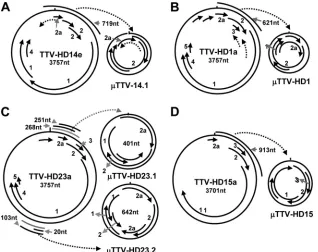
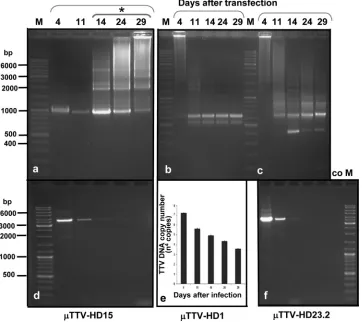
and the three TTV-HD23 isolates, was already noted early after transfection and was maintained during the passages (Fig. 2A and B). They were cloned and characterized. These subvi-ral DNA molecules (TTV-HD14; 719 bases in size [accession no. FR751514]) from TTV-HD14b and the three TTV-HD14 isolates were all identical in DNA sequence and represented circular subgenomic rearranged molecules originating from the parental TTV-HD14 genome (Fig. 4A). Similarly, a rear-ranged subviral DNA molecule (TTV-HD1; 621 bases [accession no. FR751513]) originated from the parental TTV-HD1a genome (Fig. 4B). Interestingly, replication of TTV-HD1 was maintained during the passages, despite the disap-pearance of the full-length TTV-HD1a genome. On the other hand, the presence or absence of the subviral molecules in TTV-HD23-infected cultures may indicate a possible influence of variation in culturing conditions. Here, these molecules ranged from 400 to 900 bases in size, with an increased level of the 642- and 401-base molecules. Characterization of the cloned molecules indicated an apparent evolutionary preferred maturation process as a segment of the 401-base subviral mol-ecule (TTV-HD23.1; accession no. FR751516) was dupli-cated in the 642-base subviral DNA (TTV-HD23.2; accession no. FR751517) (Fig. 4C). Multiple versions of this segment were present in larger molecules. Subviral genomes originating from TTV-HD23b and TTV-HD23d, as well as TTV-HD23a, cultures were all identical in DNA sequence. Transfection of these subviral rearranged molecules in the 293TT cell line resulted in replication of their genomes (Fig. 5a to c), as visualized after PCR amplification and confirmed by quantita-tive real-time PCR forTTV-HD1 (Fig. 5e), even after input vector DNA was not measurable after approximately 14 days in the same cultures (Fig. 5d and f). Interestingly the respective
TTV reacted exactly in the same way as the parental ge-nomes: i.e., genomicTTV-HD15 DNA was visualized after one round of PCR amplification during the first days after transfection, but subsequently only after nested PCR amplifi-cation (Fig. 5a).
In vitro transcription. Detailed transcription patterns of TTV have been reported for the isolates TTV-P1C1 (35), TTV-HEL32 (18, 50), and TTV-HD3a (25). Three main mRNA species (1.0, 1.2 and 3.0 kb) had earlier been reported in bone marrow cells (42) and in COS1 cells (21). Predictions for use of initiation codons according to Kozak rules (15) in combination with use of alternative splice acceptor and donor sites (25) indicated the involvement of nonconserved mecha-nisms during transcription of torque teno viruses. We chose to investigate the transcription of our isolates by using single- as well as double-stranded cDNAs as templates for 3⬘- and 5⬘ -RACE mapping. Double-stranded cDNA reduces the possibil-ity of the formation of nonspecific hybrids. In addition, we selected primers (forward and reverse) that were located within the intergenic regions instead of commonly used gene-specific primers. This was done with the aim of covering the expression of any unpredicted genes in the TTV genome. RNA from all cultures was extracted on day 7 after transfection. RNA from control transfections with vector alone was in-cluded to control for false-positive amplification. We repeated the transcription analyses to control for a suitable time point for harvesting mRNA by extracting RNA 48 h after transfec-tion in the case of isolate TTV-HD14e. The transcriptransfec-tion pat-terns observed did not differ between day 2 and day 7. All results obtained in the transcription analyses are presented in Fig. S2 in the supplemental material.
Abundant transcripts were isolated from TTV-HD23-in-FIG. 4. Schematic presentation of full-length TTV-HD genomes with their respectiveTTV-HD molecules. Numbers indicate ORFs in the DNA genome.
7290 DEVILLIERS ET AL. J. VIROL.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.134.452.69.321.2]fected cultures. Their transcription patterns, as well as those for TTV-HD20a, TTV-HD15a, and TTV-HD16a, were in gen-eral similar to previously described transcription patterns (re-viewed in reference 18). An exception is the absence of a full-length ORF1 transcript from all of our isolates. Tran-scripts covering sections of the ORF1 gene (either the 5⬘or the 3⬘ends) and which could code for smaller proteins were pres-ent (Fig. 6). In silicoanalyses for putative proteins revealed additional information to previous reports. Examples include splicing (fusions) between either ORF2 or ORF2a with ORF1 or with ORF5 in TTV-HD16a (6.3s.2, 6.3s.3, and 6.3s.9). Splic-ing between ORF1 and ORF5 is another possibility (6.3.7). Short transcripts covering the region of ORF2 in TTV-HD20a may also be expressed as a smaller ORF1 protein (7.3.5, 7.3.4, and 7.5.13) (Fig. 6). Transcripts were also obtained by using primers (forward or reverse) located in the control region. Two observations were made. Reverse primers resulted in spliced or nonspliced transcripts covering extended regions of the ge-nome (12.5.19, 12.5.20, 12.5.21, 5.5s.16, 5.5s.17, 5.5s.18, and 5.5s.19) or transcripts of various lengths which did not have any coding capacity (5.5s.12, 5.5s.13, 5.5s.14, 5.5s.15, 11.5.7, 11.5.8, and 11.5.9). Amplification with forward primers in this region resulted in other short noncoding transcripts or spliced tran-scripts with coding capacity even as distant as ORF5 (4.3.4,
3.3.1, and 3.3.2) (Fig. 6). Functional analyses of these tran-scripts are presently conducted throughin vitroexpression.
DISCUSSION
The ubiquity of torque teno viruses, together with the ab-sence of suitablein vitroculture systems, has hampered prog-ress in investigating this group of viruses. The multitude and heterogeneity of types (2, 41), as well as their ubiquitous pres-ence in hematopoietic cells (22, 57, 58), have added to the delay in gaining information on whether these viruses are in-volved in the pathogenesis of any disease. We have isolated a spectrum of TTV types during the course of our investigations (9, 15, 25; present study). Full-length genomes of a number of TTV types were often isolated from an individual sample, depending on the composition of primers used for long-dis-tance PCR amplification. The scattered distribution of our new isolates on a phylogenetic tree of genusAlphatorquevirus(Fig. 1) indicates their heterogeneity, irrespective of origin. The variation in genome organization resulting from minor differ-ences in sequence identity across the genome was often ob-served between isolates of the same type and has prompted questions as to the functionality of these modified genes.
[image:8.585.112.474.72.394.2]We have in the past attempted to propagate TTV genomes FIG. 5. Independent propagation ofTTV-HD. (a) The higher replication ofTTV-HD15 after initial transfection decreased over time.ⴱ, nested PCR amplification. (b and c) Replication of transfectedTTV-HD1 (b) andTTV-HD23.2 (c) DNA.TTV-HD23.1 molecules formed during replication ofTTV-HD23.2. (d and f) Amplification of the same samples as in panels a and c, respectively, but using primers for amplification of vector DNA as a control. (e) Real-time quantitative PCR indicating replication ofTTV-HD1. M, size marker; co, control (293TT cellular DNA).
on November 7, 2019 by guest
http://jvi.asm.org/
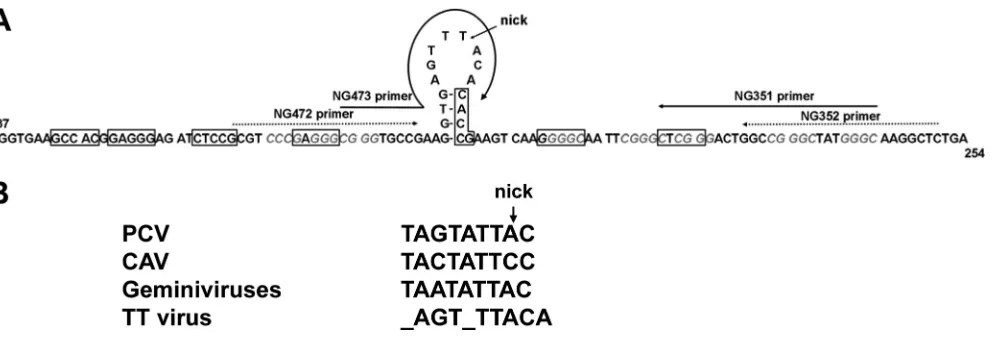
in a number of cell lines and in peripheral blood monocytes under variousin vitroculturing conditions. We achieved mod-erate success with single isolates in Hodgkin⬘s lymphoma cell lines and in the 293T cell line. Replication occurred at low levels (23; L. Leppik and E.-M. de Villiers, unpublished data). The human embryonic kidney cell line 293TT was engineered to express high levels of SV40 large-T antigen (5). Transfection of TTV genomes into these cells resulted in virus DNA repli-cation. We propose an origin of replication for the TTV group in the conserved region used for the identification of anellovi-ruses (25, 48) and used in the present study (Fig. 7). A con-served nanonucleotide required for rolling-circle replication is present in other single-stranded DNA viruses, including circo-viruses and geminicirco-viruses (reviewed in references 32 and 34).
[image:9.585.42.546.70.480.2]We identified an octanucleotide (_AGT_TTACA) in the TTV genome that, although only partially conserved to the men-tioned nanonucleotide, forms a stem-loop with a possible nick-ing site between nucleotides (nt) 5 and 6. In addition, a 4-bp motif (CGGG and GGGC) is found five times adjacent to the stem-loop sequence. Large-T (LT) protein of polyomaviruses requires variable numbers of GAGGC-like pentanucleotide motifs in various orientations as recognition sites for the initi-ation of repliciniti-ation (reviewed in reference 11). A number of additional distinct characteristics are required for initiation of replication in polyomaviruses. The fact that we were able to propagate TTV replication in the 293TT cell line in the present study, as opposed to meager replication in the 293T cell line (unpublished results), strongly indicates a helper effect of FIG. 6. Examples of transcripts obtained during propagation of full-length TTV-HD. TTV-HD genomes with the respective transcripts are grouped together in boxes. Shaded boxes show a schematic presentation of TTV-HD genomes indicating ORFs with amino acid sizes within the genome. Numbers in parentheses indicate the nucleotide size of the full-length genome (TTV-HD), as well as a serial number used for labeling TTV-HD transcripts. ORF1 of TTV-HD23b, as well as that of TTV-HD23a, is 736 aa in size, but the two ORFs differ in the hypervariable region. The transcripts’ nucleotide lengths (nt) and coding capacities (ORFs) are indicated. Primers (f, forward; r, reverse) and locations (nucleotide positions) are presented above each transcript. The range (nucleotide positions) of each transcript sequence is indicated. Gray stars indicate transcripts that were more frequently isolated.
7292 DEVILLIERS ET AL. J. VIROL.
on November 7, 2019 by guest
http://jvi.asm.org/
SV40 large T-antigen in TTV DNA replication. The conserved region of TTVs harbors modified pentanucleotides that are suspected of acting as recognition sites for the SV40 LT pro-tein expressed in the 293TT cell culture system and under the conditions used in our experiments (Fig. 7). These modified pentanucleotides often overlap with the 4-bp motif of TTV adjacent to the stem-loop. Additional investigation is needed to confirm both the proposed origin of replication for the TTVs, as well as the basis for the helper effect of SV40 LT protein for the TTV replication in the 293TT cell line.
The differences in the levels of DNA replication observed between TTV isolates cannot presently be explained. Phyloge-netic information does not provide an answer. It is noticeable that the six isolates (types HD14, HD15, and TTV-HD16) that originated from brain biopsies of patients with multiple sclerosis all replicated to a lesser degree in our sys-tem. Various levels of DNA replication or modifications in the genome organization, which included modified ORF1 genes, did not influence serial passaging (Fig. 2). Transcription levels seem to depend on TTV type. Fewer of the common tran-scripts described for other TTV types were detected in the four TTV-14 isolates than in TTV-HD15a and TTV-HD16a cul-tures. Transcripts previously reported (17, 25) were also isolated from all infected cultures in the present study. Inter-estingly, no transcript was identified which would code for full-length ORF1 protein of any of the TTV-HD types studied. In silicoanalyses of putative proteins resulting from the tran-scripts stress the need for future in-depth analyses of TTV transcripts in general. A number of putative protein sequences were identified which may have resulted from fusion products of any two or three genes. Translation strategies known to be used by viruses, such as leaky scanning, reinitiation, and ribo-somal shunting (51), might be involved here. Dual coding in alternative reading frames is an additional mechanism that may be involved (24). Interestingly, transcripts of the control region were also isolated. Here two groups of transcripts were identified. One group involved transcripts spanning at least
part of the intergenic region and extending into the rest of the genome covering the known genes. The second group con-sisted of transcripts of various lengths and without recogniz-able coding capacity. It is tempting to speculate that the nature of the torque teno virus intergenic region with its high GC content may play a role in transcription-dependent replication blockage (1).
A very prominent observation in our study is the formation of subviral molecules already early during the replication cycle of the majority of our isolates. Two different groups of subviral molecules were distinguished. The formation of multiple sub-viral DNA molecules ranging in size occurred frequently and extensively in TTV-HD20a-, TTV-HD3a-, and TTV-HD1a-infected cultures. We previously demonstrated similar rear-ranged subviral molecules in serum samples (25). Transfection into the Hodgkin⬘s lymphoma L428 cell line of a small number of these subviral genomes originating from sera resulted in limited replication and transcription for a few days (9). Similar subviral molecules of various sizes were occasionally and in-constantly demonstrated in cultures of the other six isolates, but did not influence the replication of the full-length genome. This difference underlines not only the diversity between TTV types, but also that this phenomenon does not result from PCR artifacts. Similar defective molecules have also been reported in geminiviruses, where they accumulate during improper rep-lication (16).
[image:10.585.48.545.67.236.2]The second group of subviral molecules,TTV (ranging in size between 401 and 913 bases), evolved during replication of TTV isolates HD14b, HD14c, HD14a, TTV-HD14e, TTV-HD15a, TTV-HD16a, TTV-HD1a, TTV-HD23b, TTV-HD23d, and TTV-HD23a and remained con-stant in size and composition during propagation, as evidenced after cloning and sequencing. Their production in the case of the latter four isolates seemed to be influenced by culture conditions. Interestingly, the subviral moleculeTTV-HD1 in the TTV-HD1a-infected culture was detectable in the cell cul-ture even after loss of detectable parental full-length genome FIG. 7. Proposed origin of replication for TTVs. (A) Conserved region of TTV sequence indicating the proposed origin of replication in the stem-loop structure. Nucleotide numbers corresponding to TTV1 (AB008394) are indicated. The 4-bp motif (CGGG or GGGC) is indicated in italic and gray. Boxes indicate conspicuous GAGGC-like pentanucleotides which may serve as putative recognition sites for SV40 large T-antigen. The primers (NG351, NG352, NG472, and NG473) used for PCR amplification are indicated. An arrow indicates a putative nicking site necessary for the initiation of rolling-circle amplification. (B) Conserved nanonucleotide motifs of porcine circovirus (PCV), chicken anemia virus (CAV), and geminiviruses in comparison to the octanucleotide motif in the loop of the stem-loop in TTVs.
on November 7, 2019 by guest
http://jvi.asm.org/
(Fig. 2B, panel c). Two molecules,TTV-HD23.1 (409 bases) andTTV-HD23.2 (642 bases), were isolated from all three TTV-HD23-infected cultures.TTV-HD23.2 is composed of theTTV-HD23.1 molecule plus a duplication of 306 nt of the smaller molecule. The subviral molecules (TTV-HD14) that were isolated from the four TTV-HD14 cultures were all iden-tical in sequence and appeared very early after the initial trans-fection of the parental genome. The production of these smaller molecules did not seem to be influenced by the varia-tion in genome structure between isolates of the same TTV type. All TTV molecules were composed of parts of the parental TTV type, although the genome regions involved dif-fered. They were all amplified by long-distance PCR using the same back-to-back primers used for amplification of the pa-rental genome. These primers are located in the conserved region of the TTV genome harboring our proposed origin of replication and were present in all TTV isolates. We had previously observed the episomal replication of a TTV subviral molecule isolated from a serum sample over a period of 23 days. Multimeric subviral RNA was demonstrated during this process (9). The subviral molecules reported in the present study are able to replicate autonomously in 293TT cells and can be propagatedin vitro, as evidenced by real-time quanti-tative PCR (Fig. 5). It is not known whether they are trans-mitted as part of an infectious TTV or whether they are in-duced only after infection by the parent virus and then transmitted by autonomously infecting other cells. Similar sub-viral DNAs have been associated with the geminivirus disease complex (55) where-satellites enhance symptom phenotypes in plants. They share a network of protein interactions with geminiviruses and are dependent on them for transreplication, encapsidation, and vector transmission. The only sequence shared between-satellites and geminiviruses lies in the short origin of replication (36, 46, 47). TTV subviral molecule (TTV) sequences are almost identical to parts of the parental genome, including the proposed origin of replication for TTVs. The cytopathic effect observed duringin vitropropagation of our TTV subviral molecules allows for speculation on their possible role as the disease-inducing component of some torque teno viruses. Signature motifs of proteins involved in autoimmune disease (9) have been identified byin silico anal-yses of putative proteins expressed by these subviral molecules, as well as from virus transcripts isolated from the TTV-in-fected cultures. Presently, we cannot exclude that a number of these transcripts may have originated from transcription of these subviral molecules. Investigations into this aspect are currently ongoing.
ACKNOWLEDGMENTS
We thank Birgit Hub, Imke Grewe, Helen Rahn, and Sonja Stephan for excellent technical assistance. We also thank Roland Martin, Jochen Kalden, and Markus Hohenfellner for providing samples.
REFERENCES
1.Belotserkovskii, B. P., et al.2010. Mechanisms and implications of transcrip-tion blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. U. S. A.107:12816–12821.
2.Biagini, P., and P. de Micco.2010. La famille desAnelloviridae: virus TTV et genres apparente´s. Virologie14:3–16.
3.Biagini, P., R. N. Charrel, P. de Micco, and X. de Lamballerie.2003. Asso-ciation of TT virus primary infection with rhinitis in a newborn. Clin. Infect. Dis.36:128–129.
4.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller.2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol.78:751–757. 5.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller.2005. Generation
of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med.119:445–462.
6.Bustin, S. E., et al.2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem.55:611– 622.
7.del Val, C., et al.2004. High-throughput protein analysis integrating bioin-formatics and experimental assays. Nucleic Acid Res.32:742–748. 8.de Schmidt, M. H., and M. H. M. Noteborn.2009. Apoptosis-inducing
proteins in chicken anemia virus and TT virus. Curr. Top. Microbiol. Im-munol.331:131–149.
9.de Villiers, E.-M., R. Kimmel, L. Leppik, and K. Gunst.2009. Intragenomic rearrangement in TT viruses: a possible role in the pathogenesis of disease. Curr. Top. Microbiol. Immunol.331:91–107.
10.de Villiers, E.-M., R. Schmidt, H. Delius, and H. zur Hausen.2002. Heter-ogeneity of TT virus related sequences isolated from human tumor biopsy specimens. J. Mol. Med.80:44–50.
11.Fanning, E., and R. Knippers.1992. Structure and function of simian virus 40 large tumor antigen. Annu. Rev. Biochem.61:55–85.
12.Fei, J.-W., Q.-X. Wei, P. Angel, and E.-M. de Villiers.2005. Differential enhancement of a cutaneous HPV promoter by p63, Jun and mutant p53. Cell Cycle4:689–696.
13.Garbuglia, A. R., et al.2003. Detection of TT virus in lymph node biopsies of B-cell lymphoma and Hodgkin’s disease, and its association with EBV infection. Int. J. Immunopathol. Pharmacol.16:109–118.
14.Itoh, Y., et al.2000. Visualization of TT virus particles recovered from the sera and feces of infected humans. Biochem. Biophys. Res. Commun.279:
718–724.
15.Jelcic, I., A. Hotz-Wagenblatt, A. Hunziker, zur H. Hausen, and E.-M. de Villiers.2004. Isolation of multiple TT virus genotypes from spleen biopsy tissue from a Hodgkin⬘s disease patient: genome reorganization and diversity in the hypervariable region. J. Virol.78:7498–7507.
16.Jeske, H.2009. Geminiviruses. Curr. Top. Microbiol. Immunol.331:185–226. 17.Kakkola, L., et al.2008. Expression of all six human torque teno virus (TTV) proteins in bacteria and in insect cells, and analysis of their IgG responses. Virology382:182–189.
18.Kakkola, L., K. Hedman, J. Qiu, D. Pintel, and M. So¨derlund-Venermo.
2009. Replication of and protein synthesis by TT viruses. Curr. Top. Micro-biol. Immunol.331:53–64.
19.Kakkola, L., et al.2007. Construction and biological activity of a full-length molecular clone of human torque teno virus (TTV) genotype 6. FEBS J.
274:4719–4730.
20.Kamada, K., T. Kamahora, P. Kabat, and S. Hino.2004. Transcriptional regulation of TT virus: promoter and enhancer regions in the 1.2-kb non-coding region. Virology321:341–348.
21.Kamahora, T., S. Hino, and H. Miyata.2000. Three spliced mRNAs of TT virus transcribed from a plasmid containing the entire genome in COS1 cells. J. Virol.74:9980–9986.
22.Kanda, Y., et al.1999. TT virus in bone marrow transplant recipients. Blood
93:2485–2490.
23.Kazi, A., et al.2000. High frequency of postnatal transmission of TT virus in infancy. Arch. Virol.145:535–540.
24.Kovacs, E., P. Tompa, K. Liliom, and L. Kalmar.2010. Dual coding in alternative reading frames correlates with intrinsic protein disorder. Proc. Natl. Acad. Sci. U. S. A.107:5429–5434.
25.Leppik, L., et al.2007. In vivo and in vitro intragenomic rearrangement of TT viruses. J. Virol.81:9346–9356.
26.Maggi, F., et al.2007. Torquetenovirus in patients with arthritis. Rheuma-tology46:885–886.
27.Maggi, F., et al.2010. Role of hematopoietic cells in the maintenance of chronic human torquetenovirus plasma viremia. J. Virol.84:6891–6893. 28.Maggi, F., et al.2001. Low prevalence of TT virus in the cerebrospinal fluid
of viremic patients with central nervous system disorders. J. Med. Virol.
65:418–422.
29.Maggi, F., et al.2001. TT virus (TTV) loads associated with different pe-ripheral blood cell types and evidence for TT replication in activated mono-nuclear cells. J. Med. Virol.64:190–194.
30.Maggi, F., et al.2003. TT virus in the nasal secretions of children with acute respiratory disease: relations to viremia and disease severity. J. Virol.77:
2418–2425.
31.Maggi, F., et al.2003. TT virus loads and lymphocyte subpopulations in children with acute respiratory diseases. J. Virol.77:9081–9083.
32.Mankertz, A., F. Persson, J. Mankertz, G. Blaess, and H.-G. Buhk.1997. Mapping and characterization of the origin of DNA replication of porcine circovirus. J. Virol.71:2562–2566.
33.Mariscal, L. F., et al.2002. TT virus replicates in stimulated but not in nonstimulated peripheral blood mononuclear cells. Virology301:121–129. 34.Meehan, B. M., J. L. Creelan, M. S. McNulty, and D. Todd.1997. Sequence
of porcine circovirus DNA: affinities with plant circoviruses. J. Gen. Virol.
78:221–227.
7294 DEVILLIERS ET AL. J. VIROL.
on November 7, 2019 by guest
http://jvi.asm.org/
35.Mu¨ller, B., A. Ma¨rz, K. Doberstein, T. Finsterbusch, and A. Mankertz.2008. Gene expression of the human torque teno virus isolate P/1C1. Virology
381:36–45.
36.Nawaz-ul-Rehman, M. S., and C. M. Fauquet.2009. Evolution of geminivi-ruses and their satellites. FEBS Lett.583:1825–1832.
37.Ninomiya, M., et al.2007. Identification and genomic characterization of a novel human torque teno virus of 3.2kb. J. Gen. Virol.88:1939–1944. 38.Ninomiya, M., M. Takahashi, T. Nishizawa, T. Shimosegawa, and H.
Oka-moto.2008. Development of PCR assays with nested primers specific for differential detection of three human anelloviruses and early acquisition of dual or triple infection during infancy. J. Clin. Microbiol.46:507–514. 39.Nishizawa, T., et al.1997. A novel DNA virus (TTV) associated with
ele-vated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem. Biophys. Res. Commun.241:92–97.
40.Nolan, T., and S. Bustin.2009. Article 3: qPCR assay design. Eur. Pharm. Rev.3:26–32.
41.Okamoto, H.2009. History of discoveries and pathogenicity of TT viruses. Curr. Top. Microbiol. Immunol.331:1–20.
42.Okamoto, H., et al.2000. TT virus mRNAs detected in the bone marrow cells from an infected individual. Biochem. Biophys. Res. Commun.279:700–707. 43.Okamoto, H., et al.2000. Sequestration of TT virus of restricted genotypes
in peripheral blood mononuclear cells. J. Virol.74:10236–10239. 44.Okamoto, H., et al. 2000. Replicative forms of TT virus DNA in bone
marrow cells. Biochem. Biophys. Res. Commun.270:657–662.
45.Okamoto, H., et al.2000. Circular double-stranded forms of TT virus DNA in the liver. J. Virol.74:5161–5167.
46.Paprotka, T., V. Metzler, and H. Jeske.2010. The first DNA 1-like a satellite in association with New World begomovirus in natural infections. Virology
404:148–157.
47.Patil, B. L., and C. M. Fauquet. 2010. Differential interaction between cassava mosaic geminivirus and geminivirus satellites. J. Gen. Virol. 91:
1871–1882.
48.Peng, Y. H., et al.2002. Analysis of the entire genomes of thirteen TT virus variants classifiable into the fourth and fifth genetic groups, isolated from viremic infants. Arch. Virol.147:21–41.
49.Pifferi, M., et al.2005. Associations between nasal torquetenovirus load and spitometric indices in children with asthma. J. Infect. Dis.192:1141–1148. 50.Qiu, J., et al.2005. Circovirus TT virus genotype 6 expresses six proteins
following transfection of a full-length clone. J. Virol.79:6506–6510. 51.Ryabova, L. A., M. Pooggin, and T. Hohn.2006. Translation reinitiation and
leaky scanning in plant viruses. Virus Res.119:52–62.
52.Saunders, K., et al.2000. A unique virus complex causes Ageratum yellow vein disease. Proc. Natl. Acad. Sci. U. S. A.97:6890–6895.
53.Shiramizu, B., Q. Yu, N. Hu, R. Yanagihara, and V. R. Nerurkar.2002. Investigation of TT virus in the etiology of pediatric acute lymphoblastic leukaemia. Pediatr. Hematol. Oncol.19:543–551.
54.Sospedra, M., et al.2005. Recognition of conserved amino acid motifs of common viruses and its role in autoimmunity. PLoS Pathog.1:e41. 55.Stanley, J.2004. Subviral DNAs associated with geminivirus disease
com-plexes. Vet. Microbiol.98:121–129.
56.Takahashi, K., Y. Iwasa, M. Hijikata, and S. Mishiro.2000. Identification of a new human DNA virus (TTV-like mini virus, TLMV) intermediately re-lated to TT virus and chicken anemia virus. Arch. Virol.145:979–993. 57.Takahashi, M., et al.2002. TT virus is distributed in various leukocyte
subpopulations at distinct levels, with the highest viral load in granulocytes. Biochem. Biophys. Res. Commun.290:242–248.
58.Zhong, S., et al.2002. Frequent detection of the replicative form of TT virus DNA in peripheral blood mononuclear cells and in bone marrow cells in cancer patients. J. Med. Virol.66:428–434.
59.zur Hausen, H., and E.-M. de Villiers.2005. Virus target cell conditioning model to explain some epidemiologic characteristics of childhood leukemias and lymphomas. Int. J. Cancer115:1–5.