JOURNAL OFVIROLOGY, Sept. 1971, p. 275-285 Vol. 8, No. 3 Copyright © 1971 AmericanSociety for Microbiology Printed in U.S.A.
T-Even
Bacteriophage-Tolerant Mutants
of
Escherichia coli B
II.
Nucleic Acid
Metabolism
CHRISTOPHER K. MATHEWS AND MARTINEZ J. HEWLETT
Departmentof Biochemistry, University of Arizona, College of Medicine, Tucson, Arizona 85724
Received for publication 1 June 1971
T-evenbacteriophage-tolerant mutants are strains of Escherichia coli which can
adsorbT-evenphagesbutcannot support thegrowth of infective virus. Under some
conditions,theinfectedcellsarenotkilled.Mutantcells infected by phage T6 are able
tocarryoutseveralmetabolic functions associated with normal virus
development,
including arrest of bacterial nucleic acid and protein synthesis, incorporation of isotopic precursors intoviralnucleic acids and proteins, synthesis of early enzymes of deoxyribonucleic acid (DNA) metabolism, formation of rapidly sedimenting DNA intermediates, and formation of normallevels of early and late messenger ribonucleic
acid species.Phageareunabletomutate toforms capable of growth on these mutants.
Thenatureof the biochemicalalterationleading to tolerance is still unknown.
As anapproachtounderstanding the
contribu-tion of host cell metabolic functions to virus development, ourlaboratory hasisolated T-even
bacteriophage-tolerant (tet) mutants of
Escheri-chia coliB (12). These strains canadsorb T-even phages butare unableto forminfective progeny. Undersome conditions, the cells are not killed. Preliminary investigation showed that someviral genes are expressed after infection of these mutants by phage T6. Early enzymes of
de-oxyribonuwleic acid (DNA) metabolism are
synthesized, albeit in limited amounts (12), and
somephageDNA replication occurs.
The objectives of the work described in the
present paper were, first, to devise
experimental
conditions suitable for extensive analysis of
metabolic events in abortive infections of tet
mutants,and, second,toexploreeventsinnucleic
acid metabolismwhichmight providecluestothe
primary biochemical alteration in tet mutants.
We find that even though tet mutants are
ab-solutely defective in the ability to propagate
infective virus they are able to support a wide range of virus-directed metabolic processes.
MATERIALS AND METHODS
All of the present work was carried out with T6 phage and E. coli B, along with its derivatives E. colitet 1 and tet 2, both described previously (12).
T6 was used because of its rapid adsorption to all threebacterial strains. Culture and assaytechniques and culture media were also as described in earlier
communications from this laboratory, except that
glycerol-Casamino Acids (GCA) medium contains 5 mgofCasamino Acidsperml rather than 15. Except whereotherwise indicated, all experiments were car-ried out in GCA medium.
Because of the tendency of tet mutants to revert to wild type, stocks were reisolated from single col-oniesatfrequent intervals. Moreover, in each experi-mentreported herein, the let phenotypewaschecked as follows. At 6 min after infection, samples were removed to dilution tubes either containing chloro-form for determination of unadsorbed phage or to tubescontaining no chloroform for measurement of
total infectivecenters.At90min, anothersamplewas removed to adilution tube containing chloroform for
determinationoftotal progeny. Suitabledilutions of the abovewere plated, withE. coli Bas theplating
host. The following were taken ascriteria for
satis-factory expression of the tet phenotype: infective
center titer < unadsorbed phage titer and infective
progeny titer < unadsorbed phage titer. Thus, in each experiment reported, there was no detectable formation of infective phage in infection of tet mu-tants.
For studies on
03-galactosidase
induction, 10-ml cultures were induced as described in the legend toFig. 4 and then chilled and centrifuged. Thepellets
wereresuspendedin5ml each of0.02Msodium phos-phate buffer (pH 7.5) and sonically treated for 30
sec. i3-Galactosidase activity was measured in these
sonically treatedextracts asdescribedbyDuckworth and Bessman(4).
Incorporation of 14C-uracil into DNA and ribo-nucleic acid (RNA) and incorporation of 14C-leucine into protein were followed as described previously
(10, 11). Techniques for isolation of RNA and for DNA-RNA hybridizations were also as described 275
on November 11, 2019 by guest
http://jvi.asm.org/
MATHEWS AND HEWLETT
earlier (11), except that in isolating RNA diethyl pyrocarbonate (at 1() was present in the extracting buffer for inhibition of nucleases rather than poly-vinylsulfate.
In studies on DNAmetabolism, the methods used for DNA labeling, cell lysis, sucrose gradient centrif-ugation, and radioisotope countingwere as described by Murray and Mathews (14, 15). The technique used for DNA-DNA hybridization was the membrane filter method of Denhardt (3), as modified in this laboratory by M. Sisson (M. Sisson and C.K. Math-ews, inpreparation).Incorporation of parental phage DNA into"replicating DNA complexes" was meas-ured by the M-band technique of Earhart and his colleagues (5, 6).
Further experimental details are provided in indi-vidual figure legends.
RESULTS
Effects of host cell survival. The basis for isolation of tet mutants is theirabilityto grow on
agarplatesin the presenceofalargeexcessoftwo
different T-even phages. Under these conditions, infection is not lethal to the hostcell. However, in our early studies wefound that considerable
cell killing occurs in infection of tet mutants in
liquid media, even though infection is always abortive. It is now clear that the ability of a tet
cell tosurvive infection dependsuponits environ-ment; cell survivalishigher in solid media than
in liquid media, and in liquid survival is higher
in nutritionally rich media than in minimal or
other defined media. Figure 1 presents data on
the
ability
oftetcellstoformcolonieswhenplated
in the presence of various amounts of T6. The mutants did lose the ability to form colonies
ultimately but only at an input of phage some
100-fold greater than that necessary to abolish
the colony-forming
ability
of E. coli B platedunder the same conditions. Similarly, in liquid
media tet cells could lose the ability to form colonies when plated after infection at high
multiplicities, but the difference between E. coli
B and the mutants was considerably less under
these conditions. Figures 2 and 3 show bacterial
survival curves at various multiplicities of
infec-tionby T6in nutrient broth and GCAmedium,
respectively.
Note that the slopes of the tetsurvival curves are considerably steeper inGCA
medium than in nutrient broth. Similar results
are seenininfection by T2 (data notshown). In all cases, the survival curves are approximately linear, indicating that killing of the tet mutants is a one-hit process; infection by a single phage
particle is a lethal event, although this occurs
with lower probability in infection of tet mutants than ininfectionofwild-type bacteria.
Because the environment on an agar plate is considerably more anaerobic than that of a
104
105
1O6
107
108
T6 Particles per plate
FIG. 1. Expression ofthe tet phenotype on agar
plates. Foreachstrain, a constant number of
colony-formers was plated on each of a series ofnutrientagar
plates in thepresence of the indicatednumbers of T6 particles.
MOI
FIG. 2. Survival curves bor bacteria injected in
nutrient broth. Bacterial cultures at about 5 X 108
per ml were infected with T6 at the indicated
multi-plicities. At 6 minl, samples were removed, diluted, and platedfbr determination ofcolony-forming
abil-ity.MOI, multiplicity of infection.
2)7
6 J. VIROL.on November 11, 2019 by guest
http://jvi.asm.org/
[image:2.480.255.445.60.295.2] [image:2.480.254.445.359.578.2]T-EVEN-TOLERANT MUTANTS OF E. COLI. 11
.i_
L
Ul)
. c
4-f,
CID
ic
IA
ABN 0 tet1
0 * tet2
0
.-0
0.
A~~~
6.
0 6 12
M01
FIG. 3. Survivalcurvesforbacteria infectedin
glyc-erol-Casamino Acids (GCA) medium. Except for the
substitution of GCA medium for broth, the protocol
wasidenticalto thatdescribedinthelegendtoFig. 1. MOI,multiplicity of infection.
vigorously bubbled liquid culture, it seemed
possible that there is a relationship between
anaerobiosis and cellkilling:themore anaerobic the culture, the lower the probability that a
particularinfection will be lethal. This wastested
by comparing cell survival in GCA medium
either aeratedorbubbledwithnitrogen (datanot
shown). Cell survival was no greater in the anaerobic thanintheaerobic culture and indeed was somewhat less, indicating the absence of a direct relationship between anaerobiosis and cell survival.
Inability of phage to overcome tolerance. We wished to know whether T-even phages could
undergo mutations which would allow them to overcome tolerance, i.e., to plate oneither tet 1
or tet2. Themappositionsof such "host-range" mutations, at least in the genetically
well-char-acterizedT4, might providecluestothebacterial functions altered intheoriginaltetmutations. In
ourpreviousstudy,wefoundthatT2, T4,and T6 wereallunabletoplateoneithertetmutantwhen
as many as 106 viable phage were added to a
single plate. Wehavenowextended thiswork by subjectingboth T4andT6tochemical mutagene-sis by nitrous acid, hydroxylamine,
2-amino-purine, 5-bromodeoxyuridine, and N-methyl-N'-nitro-N-nitrosoguanidine. Even when asmanyas
108 plaque-forming units from mutagenized
stocks were plated on tet 1 or tet 2, no plaques were seen. Thus, phage seem quite unable to
circumvent the effects of the tet mutations by
extending their own host range. In this context, it isof interest that phage T7 plates on tet 1 with an
efficiency equal to that seen on E. coli B (W. C. Summers, personal communication).
By analogy with temperate phages, we consi-dered whether tolerance in the tet systems is at all akin toprophage immunity. In other words, could the tet character result, not from mutation, but from a stable association of phage DNA with the bacterial genome either as an episode or a
plasmid. Although this seemed improbable, we
tested it by attempted hybridization between bacterial DNA, fixed on membrane filters, and
"4C-labeled T6 DNA. Fewer than
0.2%
of theinput counts were bound to bacterial DNAfrom either E. coli
B,
tet 1 or tet 2, and no differences were seen among the three strains. Weconclude, therefore, that the tet genomes do not containsignificantamounts ofphage-specific DNA.
Effects on bacterial polymer synthesis. One of the most powerful biochemical research tech-niques is the use of radioisotopes for labeling
metabolic intermediates, whereas one of the most attractive features of T-even phages as objects for metabolic studies is the fact that infection com-pletely arrests the synthesis of macromolecules coded by the bacterial genome. Thus, one can label T-even phage-infected cells with isotopic protein or nucleic acid precursors secure in the knowledge that only virus-specific polymers will belabeled. In ourearlier work intetmutants,we
had no such security; since bacterial survival was high, the level ofbacterial nucleic acid and protein synthesis almost certainly was also high, and a number of informative biochemical experiments could notbe performed. However, once we had
learned that infection at high multiplicity in
chemically defined media leads to extensive cell
killing,
itseemedthat theseapproaches mightbe available after all. First, of course, we had toestablish that host cell gene expression is
ar-rested in the tet mutants in a fashion similar to
that seen inwild-type cells. As a prerequisite to
theseexperiments, weneededtoknow thatphage adsorb to tetcellsasrapidlyas toE.coli B. This
wasindeedthe case,atleast underourconditions of infection(5 X 108to8 X101cells/ml,in GCA
medium,andmultiplicities of6to10). With each
bacterialstrain, morethan 60% of added phage
was adsorbed by 2 min after infection andover
85%7,
by5min.As afirstapproachtohost cell geneexpression,
we examinedthe ability of infected cells to
syn-thesize 3-galactosidase after induction by
iso-VOL.8, 1971 277
on November 11, 2019 by guest
http://jvi.asm.org/
[image:3.480.39.231.61.281.2]MATHEWS AND HEWLETT
propylthiogalactoside (IPTG). The specific
ques-tionaskedisthefollowing:does everycell which
is killed (i.e., loses colony-forming ability) also
lose the capacity to synthesize
#3-galactosidase?
This was asked by infectingcultures at differentmultiplicities. Six minutes after infection, samples
wereremoved for determination of viablecount. IPTG was added tothe remainder of eachculture
for a 12-min period, after which the cells were
harvested and assayed for
3-galactosidase
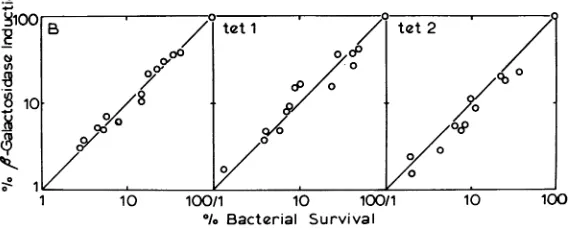
activity. Now, if every cell which is killed also loses thecapacityfor induced enzymesynthesis,we expect
a plot ofresidual enzyme induction versus
bac-terial survivaltobeastraight linewitha
450
slope.However,ifsomekilled cells do notloseenzyme inducibility with the same kinetics, then we expect
significant upward deviation from a
450
line. Asseen inFig.4,thedatamostclosely approximated
450
linesforallthreestrains.Inotherexperiments(Fig. 5), nodifferenceswere observedamong the
three strains in enzyme inducibility when IPTG
wasadded earlier ininfection, i.e.,at 3 min.Thus,
it appears that the kinetics of loss of induced
enzyme
synthesis
arethesamein themutants asin the wild-type strain and that every killed cell
losesthecapacityfor bacterialenzymeinduction.
We have investigated the kinetics ofarrest of
host-specific RNA synthesis
indirectly by
fol-lowing the rate ofphage-specificRNA
synthesis
early in infection. RNA was
pulse-labeled
withuracil-5-3H, either in uninfected cells or at
early
times after T6 infection. After
purification,
theRNA
samples
werehybridized toT6DNA. TheRNA toDNA
input
ratio wasadjusted
1:20, acondition
favorable
forcomplete hybridization
of
all
RNAspecies
which arehomologous
tophage DNA (9). Thus, the percentage of
input
counts bound to phage DNA represents the
percentage of the total labeled RNA which is
phage-specific.Ourresults for T6-infectedE.coli
B(Fig. 6)aresimialrto those reportedbyKennell
(9) for T4-infected E. coli; a relatively
large
fraction of theRNAbeing synthesized aslateas
5 min after infection is not phage-specific and
hence is host-specific. The important point from Fig. 6 isthat the data for E.coil
B,
tet 1, and tet 2 areidentical. In otherwords,the rate ofreplace-ment of host transcription by virus-directed
transcriptionis the same inall threestrains.
The kinetics of arrest of host cell DNA
syn-thesis were studied in similar fashion (Fig. 7).
Thymidine-methyl-3Hwas used tolabelDNA for
2-min periods either before or at the indicated
periodsafterinfection. Figure7A showsthe total
radioactivity incorporated into acid-insoluble
material during each labeling period. The
dif-ferences among the three strains in counts
in-corporated before infection may well represent
not truedifferences in rates of DNAsynthesisbut
rather differences in thymine nucleotide pool
sizesor someotherfactor. At any rate,thedata
on total counts incorporated during each
post-infection labeling period are quite similar,
sug-gesting that the rates ofhost DNA shutoff are
similar, ifnotidentical, inthe threestrains. The
upturn in rate oftotalthymidine incorporation
seen with each strain between 5 and 7 min
representsthe initiationof phage DNAsynthesis,
since DNAlabeled during thisperiodhybridizes
efficiently withT6 DNA(data not shown).
Hybridization
of DNA isolated from eachlabeled cultureagainst unlabeled E. coilB DNA
100/1 10 10C
1/o Bacterial Survival
FIG. 4. Comparisonofcellkillingwithf-galactosidase inducibiliiy.Log-phase bacterial cultures at about 8 X 108 per mlinglycerol-CasaminoAcidsmedium were subdivided into 10-ml amounts, each of which was infected with
T6 over arangeof multiplicities between 0 and 5. At6min, samples were removed for determination of viable
count,andisopropylthiogalactosidewasadded to the remainder of each culture to a level of S X 10-4M. Twelve
minutesafter addition of inducer, each culture was chilledandcentrifuged. The pellets were resuspended, and
fl-galactosidase
wasassayedasdescribedinMaterials and Methods. Thefigure presents combined datafrom three separateexperimentsoneach bacterialstrain.278 J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:4.480.112.397.445.560.2]T-EVEN-TOLERANT MUTANTS OF E. COLI. II
0
x
-I._
oracy act---, 0h- 0-0-.--O
t0-0
O.-U'.
-0 15 30 0 15 300 15 30
Minutes After Infect ion
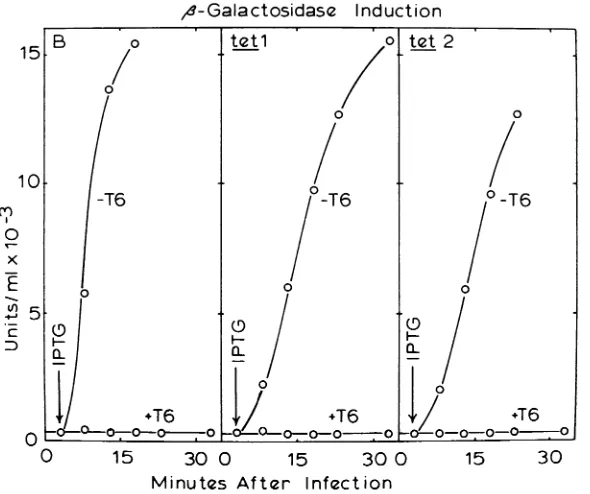
FIG. 5. fi-Galactosidase induction after T6 infection. Log-phase bacterial cultures inglycerol-Casamino Acids medium were infected with about 6 T6particles per bacterium. Isopropyllhiogalactoside (IPTG) was added to 5 X 1O4 Xat3min,andsampleswereremovedatthe indicated times andassayedfor
f3-galactosidase
asdescribed inthelegendtoFig.4.Results are compared with control values from uninfected cultures.B
tet1
tet
280
260-
r 00
40- F--0
0
20-0
2 40
2 40
2 4 6 [image:5.480.94.389.71.318.2]Minutes Af
terInfection
FIG. 6. T6-specificRNAsynthesis. Bacterial cultures (100-ml)weregrown in glycerol-Casamino Acids (GCA) mediumplus20figofuracilper ml. At aturbidity corresponding to about8X 108cells/ml, each culture was cen
-trifuged, washed in GCA medium, and resuspended in 100 ml offresh GCA medium containing nouracil.
Pulse-labelingwascarriedoutfor 1.5-minperiods with50MCieach ofuracil-5-3H onuninfected cellsand T6-infected cultures (multiplicity of infection = 6),beginning at 2.5min and at 4 min after infection. RNA was isolatedas describedin MaterialsandMethods.Labeled RNA samples containing 2.5Mgeach were hybridizedagainst 50lsg eachofT6 DNA immobilizedonmembranefilters.Eachpointinthefigure correspondstothemidpointofalabeling
period. The ordinate denotes the percentage of labeled RNA which is phage-specific in each preparation.
279 VOL. 8, 1971
on November 11, 2019 by guest
http://jvi.asm.org/
[image:5.480.96.387.383.555.2]MATHEWS AND HEWLETT J. VIROL.
[image:6.480.99.387.70.251.2]-1 1 3 5 7 -1 1 3 5 7
FIG. 7.Incorporation ofthymidine-methyl-H intoE. coliDNA. Bacterial cultures(20 ml) wereinfectedwith
T6atamultiplicity of about6. At various times, 1-ml sampleswereremovedtosmallincubation tubescontaining
10 MACieachof 3H-thymidiiie. After 2minof labeling,metabolism washaltedbyadditionof
cyanide-ethylenedi-aminetetraaceticacid-lysozyme lysing fluid (14). Lysiswascarriedoutbyahigh-temperature procedure (14).Each
lysate wasextracted twice in succession with water-saturatedphenol, and the solutions weredialyzed overnight
against 0.02Vsodiumphosphate buffer, pH6.8. A 0.5-mlamountofeachpreparation wasmixed with0.5 mlof
I NNaOH, and allpreparationswereheatedinaboiling-waterbathfor7min. Eachpreparation wasneutralized
andmadeupto3.0 mlil 3X SSC (SSC = 0.15 mNaCl plus0.015-fsodiumcitrate). Duplicate0.5-mnlsamples
ofeachwerehybridizedagainstmembranefilters containing20jugeachofE. co/i B DNA.Hybridizedcountswere
corrected forthe small numberofcountsboundtoblankfilters. (A) Totalcountsincorporatedintoacid-insoluble
material. Eachpointon thegraph represents the midpoint ofapulse-labelingperiod. (B) Hybridizable counts.
TheseareplottedaspercentofE.coliDNA-hybridizablecountsrelativetothelabeled unionfected cultures.Again,
each pointrepresentsthemidpoint ofalabeling period.
providesmorespecificinformationontherelative
ratesof hostDNA shutoff (Fig. 7B).These data
make it appear that host DNA synthesis is
actually turned off faster in infection of the tet
mutantsthaninnormal infection. Thedifferences
are more apparent than real, since the E. coli
DNA-hybridizablecountsineachlabeling period
areplotted relative to the counts incorporatedin the uninfected cultures. Thus, the apparent
differences derive in part from the differencesin
total counts per minute incorporated amongthe three uninfected cultures. However, it does ap-pearthat host cell DNAsynthesis is arrested at
least as fast in the tet mutants as in the parent strain,ifnotfaster. Ofcourse,quantitativestudies
of DNA metabolism mustawait the isolation of thymine auxotrophsof the tetmutants, andthese
are currently being preparedinourlaboratory.
DNA metabolism. Our earliest work onDNA
metabolism in T6 infection of tet mutants
in-volved studies on the fate of labeled parental
phage DNA. We find (Table 1) that parental DNA doesbecome attached tothe "M band," a
complexdescribed by Earhartand his colleagues (5, 6), which apparently contains replicating
DNA and the membrane material present atthe
TABLE 1. Incorporation of pareiiial T6 DNA ilto
Mbandsa
Bacteria Totalcounts/min Percentoftotal Bacteria Tot~nl
zin
Mband parentalcounts/mininMband E.coli B... 1.48 X 104 25.1
E. colitet 1.... 1.78 X 104 30.2 E. coli tet 2.... 1.47 X 104 24.8
aBacterialcultures(5.0ml) growing in tryptone
broth mediumwereinfectedatamultiplicityclose to 5.0with T6 containing 14C-labeled DNA. At 7 min after infection, the cells wereharvested and M bands were prepared and visualized on
mag-nesium-sarkosylgradientsasdescribed by Earhart (5). EachM bandwas removed from itsgradient
centrifuge tubebylowering the collection tube of a Buchler gradient collector to the level of the band andpumpingoutthewhite, viscous material
by suction.
replicating site. This, of course, isnotunexpected, since wehad earlier found (12) that tetmutants
support T6DNAreplication.
Inourprevious study, wefound that parental
phage DNA is not degraded to acid-soluble
280
Iq0
x
E
0
on November 11, 2019 by guest
http://jvi.asm.org/
[image:6.480.258.447.386.476.2]T-EVEN-TOLERANT MUTANTS OF E. COLI. II
material in infection of tet cells. In the present 4 Minutes
Alkaline
Gradients study, we asked whether any breakdown isde-tectable. Again, parental DNA was labeled by tet 2
growth of phage in the presence of 14C-uracil (10). 10 Cells were harvested at 4minand lysed by a
low-shear, low-temperature
process(7, 15);
thelysates
were subjected to sucrose gradient analysis in the 5
presence of
32P-labeled
mature T6DNA present as a sedimentation marker. At 4min,
no break- 0 down is evident, as shown by the fact that the t tsedimentation profiles of marker and parental et DNA are virtually identical in neutral gradients 10
(Fig. 8). The same results are seen when portions Nc
of the same lysates are sedimented through alka- °
line sucrose gradients (Fig. 9); parental and x 5. I
marker profiles are identical. Thus, within the E o **-.
first 4
min
of infection, parental phage DNA is a 0 -0neither broken nor subjected to significant B
endonucleolytic cleavage. 10
4 Minutes NeutraI Gradients
tet 2
10- 0
-.__.0
b0to0o,
'Q
10 20 3C
Fraction Number
[image:7.480.242.433.60.319.2]5 /
FIG. 9. Alkaline sucrose gradientcentrifugation of Hi\_ *
parental
T6 DNA at 4 minafter
infection.
Samples
0 ofthe lysates preparedfor the experiment described
in Fig. 8 were adjusted to0.3Xy with NaOH, and 0.2-tet 1 mlportions were layered onto 5.0-ml alkaline sucrose
10 gradients (14), which were centrifuged at 40,000 rev/
0/ min for 60 min. Symbols: Q, 32p marker colts per
a minute; *, 14C parental counts per minute.
x5 / \
E / \t Analysis of the fate of parental DNA later in
Q
infection
reveals
differences between
the wild
and
B mutant strains. Asshown by Frankel (7), much of
the parental DNAduringperiods ofactive DNA
10
replication
ininfectionby
T4 ispresentinaformwhich sediments more
rapidly
than does matureDNA and which contains individual DNA
i \ strands longer than those of mature DNA. As
J
v*
**- ;* shown in Fig. 10, much of the parentalDNA at 0>&e~e...*g t 0 13 min afterT6 infection ofE. coli B sediments10 20 30 through neutral gradients heterogeneously but
Fraction Number more rapidly than mature
T6
DNA. Infected tet FIG. 8. Neutral sucrose gradient centrifugation of cells also form rapidly sedimenting intermediates parental T6 DNA at 4 miii after infection. Bacterial containingparentalDNA. However, thismaterialcultures (about 8 X 108 per ml) were infected with does not sediment as rapidly as that seen in
in-T6 phage containing '4C-labeled DNA (15). At 4 fection ofE.
coli
B,
and a smaller proportion ofmin,
cells were lysedby a low-temperature procedure the total parental DNA sedimentsrapidly.
The(7) in the presence of 32P-labeled T6 DNA added as a
sedimentation marker. Portions (0.2 ml) were layered
rue
anitue
ofthe ifferenebetwendtheswil
onto 5.0-ml neutral 5 to20% sucrose gradients, which strain and the mutants is probablyunderesti-were then centrifuged at 32,000 rev/min for 35 min. mated in this figure. Recovery of input parental Symbols: Q, 32P marker counts per minute;
*,
14C countsin the tet 1 and tet2 gradients was 87 andparental counts per minute.
96%,
respectively, whereas that in the E. coli BVOL.8, 1971
281
on November 11, 2019 by guest
http://jvi.asm.org/
[image:7.480.36.226.265.527.2]MATHEWS AND HEWLETT
2. 0 15
)~~~~~'
LoDoooo0
oo@
03-0-0-0-0 0- 00-0Di0--0I0:~ ie ~0
0 10 20 30
[image:8.480.55.245.59.298.2]Fr2ction Number
FiG. 10. Neutral sucrose gradient centrifugation of parental T6 DNA at 13 min after infection. Except for thetime ofinfection, theprotocol wasidenticalto
thatdescribedinthelegendtoFig. 8.
gradientwasonly78 %. It isprobableinthelatter case thatconsiderable parentalDNA sedimented
to the bottom of the tube and was lost in this
gradient.
When the above lysates, obtained at 13 min,
wereanalyzedinalkaline gradients,an even more
strikingdifferencewas seen.As shown inFig. 11,
parental DNA strands in infection of E. coli B sedimentheterogeneously,butmostof the strands sediment asthough they are longerthanmature
DNA strands. By contrast, virtually no
long-stranded DNA was seen in infection of the tet
mutants. This implies that parental DNA is
subjectto considerable endonucleolytic breakage
or nicking. Whether this is a direct or indirect consequence ofthe tet alteration remains to be
determined.
Oncewehadestablished thatarrestof host cell
gene expression occurs normally in T6 infection
of the tet strains, it became possible to design isotope incorporation experiments for measuring
relative rates of nucleic acid and protein syn-thesis. When cells are grown in the presence of
high levels of uracil, such that they arerepressed
in the pyrimidine biosynthetic pathway, it
be-comes possible to use 14C-uracil for following
accumulation ofboth DNA and RNA (10, 11).
When such an experiment was performed (Fig.
0 0 2C 30
[image:8.480.259.452.59.277.2]Fraction Number
FIG. 11. Alkaline sucrose gradient centrifugation of parental T6 DNA at 13 mill after infection. Except forthe time ofinfection, the protocol was identical to
thatdescribedinthelegend to Fig. 9.
12), we found, somewhat to our surprise, that
the ratesofaccumulationofisotopeinboth DNA
andRNAwere
comparable
ininfectedtetcellstothoseseeninnormal infection. Similarly,ratesof
incorporation of
'4C-leucine
into protein reveal no gross defects inprotein synthesis ininfectionof the mutants (Fig. 13). Similar results for T4
infection of thesemutantshave been observed by
A. Kaji (personal communication). We have
relatively little information on the synthesis of
specificphageproteins. However, itis ofinterest
that infected tet cultures incubated for several
hoursultimately undergo partial
lysis.
Thus, eventhough the synthesis ofphage
lysozyme
issub-normal(12), someexpressionof thislategenedoes
occur.Wehavenot yet
investigated
thesynthesis
ofotherspecific late phage
proteins.
RNA synthesis. When tet mutants were
origi-nally isolated, an attractivehypothesis toexplain
their
inability
tosupportphagereplication
was analteration in the enzymatic
machinery
fortran-scription: perhaps an alteration in RNA
poly-merase which prevented the replacement of the
host cell sigma factor by comparable phage
factors (17,18).Ifthisweretrue,thenonewould
expectto seeafailureofinfectedtetcellstocarry
outthenormalprogram ofphagegene
transcrip-tion (forreview, seereference 13).Wehave used
competitive DNA-RNA hybridization to try to
detect this. To ask whether any abnormalities
282 J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
T-EVEN-TOLERANT MUTANTS OF E. COLL II2
0
6
0
x
LT)
4-,
.E_
z
4-,-C:
j
(13
z
0
x
cE
0.
E
z :3
E
U3
u u
0 20 40 60 0 20 40 60
[image:9.480.90.380.62.291.2]Minutes After Infection
FIG. 12.Accumulation ofphageDNA and RNA. Bacterial cultures (20ml) inglycerol-CasaminoAcids medium were grown inthe presenceof20,ugofuracilperml, centrifuged, resuspendedinfresh medium,andinfectedinthe presenceof14C-uracil, asdescribedpreviously (10, 11). Removalof samples,determination oftheradioactivity incorporatedintoDNA andRNA,and calculationoftheradioactivityin DNAasphage-equivalentunitsojDNA permilliliterofculture were alsoas describedpreviously (11). Note thedifferentordinatesforthe twopanels.
exist in transcription of early phage genes, we
hybridized earlypulse-labeled (2to5minat37C)
RNAfromT6-infected E. coli B toT6 DNA in
the presence of unlabeled early RNA species prepared from cultures of E. coli B, tet 1, and tet2 harvested 5 min after infection by T6. As
showninFig. 14, both the initial slopesand the
final plateau values of all threecompetitioncurves are similar. Hence, we conclude that at 5 min infected tet cells contain the same messenger RNA (mRNA) species, and in thesamerelative abundance, asthose present 5min afterinfection
ofwild-type bacteria.
The ability of infected tet cultures to form
mRNA transcripts normally synthesized late in
infection was tested by using unlabeled 20-min
RNA preparations as competitors against T6-B
RNAlabeled from 15to20min. Theleftpanel of Fig. 15 shows competition curves for unlabeled
5-and20-min RNA.Thismerely confirmsforT6
what has long beenknown for T2 andT4 (1, 8, 11), namely, that there are somemRNA species
whicharesynthesized late butnotearly in infec-tion. If the tetmutantswereunableto carryout part or all of the transitions involved between earlyand latepatternsoftranscription,onemight
expectcompetitioncurves withlate unlabeled tet
RNA to resemble those seen with normal early
RNA. However, as shown inFig. 15, the
com-petitioncurves for late infectedtet RNA species
are
virtually identical
tothecorresponding curvefor late T6-B RNA. Thus, within the limits of
resolution of this technique, we can see no
dif-ference intheability of infectedtet cellstocarry
out the normalprogram ofviral gene
transcrip-tion.Itis
possible
thatone ora small number ofcritical lategenes are not transcribedin infected
tet
cells,
but no gross defects in transcriptionpatternsexist.
DISCUSSION
The genetic alteration in tet mutants which
prevents phage replication seems to affect a
relatively late viral functionin view of the large
number ofphage-directedprocesses which occur
ininfection oftetcells.Theseinclude adsorption,
cell killing,
arrest of host cell nucleic acid andprotein synthesis, early changes in phospholipid
metabolism (L. I. Pizer,personalcommunication),
initiation of DNA replication, formation of
rapidly sedimenting DNA intermediates,
ac-cumulation of newly synthesized nucleic acidand
protein, formation of specific early enzymes,
formation of
early
and late mRNA species innormal amounts, and partial lysis of infected
cultures. Thusfar, thechiefdistinctive properties
of tet cells relative to wild-type include
slightly
slower growthrate in defined media(12),relative
283 VOL. 8,1971
on November 11, 2019 by guest
http://jvi.asm.org/
MATHEWS AND HEWLETT
, resistance to ultraviolet light (12), subnormal
o levels of both early and late enzymes
(12),
AB extensiveendonucleolytic cleavageof intracellular
otet1 0
DNA, high
survival to infection in solidmedia,
3-
e*
tet 2 and, of course, failure of infective progenyphage
to be formed under any conditions yet devised.
An additional difference relates to the ability to
adsorb T4. When the mutants were
originally
a o/ isolated, both tet 1 and tet 2 could adsorb T4,
///-
although
moreslowly
and to a lesser extentthan2 the
adsorption
of either T2 orT6.However,
withincreasing time of cultivation in the laboratory,
/./ both strains gradually lost theabilitytoefficiently
0 adsorbT4,although T2 and T6areboth adsorbed
0 at rates
comparable
tothose seen withwild-type
X 0 bacteria. Whether this is a result of the
primary
E tet mutation or some
secondary mutation
we doE1 // not know. We are presently tryingto
isolate
newtet derivatives capable ofadsorbingT4.
Westill have verylittleidea of thebiochemical difference between
wild-type
and tet bacteria. A* difference in membrane structure and function
seems an attractive
possibility,
first,
because somany phage-directed processes, including tail
o 20 40 60 morphogenesis (16), occur at membrane
attach-Minutes After Infection ment sites and, secondly, because bacterial cells
FIG.
13. Incorporation of14C-leucine
into protein with membranedefects
can lose the ability to after T6 infection. Bacteria were grown in M9 mediumsupport
T-even
phage
growth
(2).
Weplan
to containing leucine, centrifuged, washed, resuspendedanalyze
further intracellularphage
DNAinter-in fresh medium, inter-infected with T6, and labeled with mediates, since the extensive endonucleolytic
'4C-leucine as described previously (10). Determina- cleavageofintracellularparentalphage DNAmay
tion of the amount ofradioactivity incorporated into
protein was also as described previously (10). The po.int t
ards
aefectininA
met
asnthe
higher rate of incorporation seen in tetI relative toB primary basis for explaining the let phenotype.was noted in three separateexperiments. Finally, we planto analyze the process ofphage
Competition vs. 2-5' Labeled T6-B RNA
10 'B teti tet2
d~o
0'\lo
0*280
L
0~~~~~~~~~~~~
C
E40-
C1 °~~~~~~~0
0a.
00
00 0
3'20 0
0 10 20 30 0 10 20 30 0 10 20 30 40
[image:10.480.60.251.63.323.2]Unlabeled/Labeled RNA Ratio
FIG. 14. CompetitiveDNA-RNA hybridization: early RNA. Pulse-labeledRNA waspreparedasdescribedin
the legendtoFig.6, withlabelingcarriedoutfrom 2to5 min after infection ofE.coliBwith T6. UnlabeledRNA
samplesforcompetitioncurveswerepreparedfrom cells harvestedatS min afterinfection of E. coliB,tet1,ortet
2 with T6.Hybridizationmixtures contained5sAgeachofT6 DNAimmobilizedonfilters, either2.5or5,ugeach of labeledearly T6-BRNA,andamountsofunlabeled RNAspecies sufficienttogivethe indicatedratiosof
com-peting(unlabeled) to labeledRNA. Thefigurecontainscombineddata fromtwo separateexperiments.
284
J. VIROL.on November 11, 2019 by guest
http://jvi.asm.org/
[image:10.480.114.401.422.574.2]T-EVEN-TOLERANT MUTANTS OF E. COLI. IL
Competition vs. 15-20' Labeled T6-B RNA
10O B
D 80
>I
ox 95 RNAa*40 O
-0 0
20 \ 2o' RNA
o .
0 20 40 60 0
\ 20' RNA
of~~
20 40 60 0
2'RA
20 40 60
Unlabeled/Labeled RNA Ratio
FIG. 15. Competitive DNA-RNA hybridizations: late RNA. The experimentalprotocol was similar to that
described in thelegendtoFig.14,exceptthatlabeledRNA wasobtainedinaperiodbetween 15and20min after
infectionat37 C,and unlabeled competitorRNAspecies wereisolatedfrom cellsharvestedat20min.Acontrol hybridization withunlabeled5-min T6-B RNA isalsopresentedinthe left panel. Thisfigurepresents combined datafromtwoseparateexperiments.
morphogenesis, since the high rates of phage nucleic acid and protein synthesis seen
through-out, plus the normal operation of the transcrip-tionprogram, suggest that the tet phenotype, at
least in liquid
media,
is expressed at a very latestage. At present, we have no idea why tet cells
survive infection under some conditions but are
efficiently killed under others. Further investiga-tionof thispointwill bedeferreduntilwehavea
better understanding of why these cells are so
totally unabletoform infectivephageprogeny. ACKNOWLEDGMENTS
This investigation was supported by Public Health Service researchgrant AI-08230from theNational Institute of Allergy and Infectious Diseases and research grant 70 1013 from the American HeartAssociation.
We thank MildredCeizyk and Celta Lacambra for capable technical assistance.
LITERATURE CITED
1. Bolle, A., R. H. Epstein, W. Salser, and E.P.Geiduschek. 1968. Transcription during bacteriophage T4 development: synthesisandrelativestabilityofearlyand late RNA. J.
Mol.Biol.31:325-348.
2. Cronan,J.E., Jr., andP.R.Vagelos.1971.Abortive infection by phage T4 under conditions of defective host membrane lipid biosynthesis.Virology43:412-421.
3. Denhardt,D. T. 1966. Amembrane-filter technique for the detection ofcomplementary DNA. Biochem. Biophys. Res. Commun.23:641-646.
4. Duckworth,D.H., and M. J. Bessman. 1965. Assay for the killing properties of T2 bacteriophage and their "ghosts." J. Bacteriol. 90:724-728.
5. Earhart, C.F.1970.Theassociationof host andphageDNA
with the membrane ofEschertchiacoli. Virology42:429-436.
6. Earhart, C. F., G. Y. Tremblay, M. J. Daniels, and M.
Schaechter. 1968. DNA replication studied byanewmethod fortheisolation of cell membrane-DNAcomplexes. Cold Spring Harbor Symp. Quant. Biol.33:707-710.
7. Frankel, F. R. 1968. DNA replication after T4 infection. Cold Spring Harbor Symp. Quant.Biol. 33:485-493.
8. Hall, B. D.,A.P.Nygaard, and M. H. Green. 1964. Control ofT2-specificRNAsynthesis. J. Mol. Biol.9:143-153. 9. Kennell, D. 1968. Inhibition of host protein synthesis during
infection ofEscherichiacolt by bacteriophage T4.I.
Con-tinuedsynthesisof host RNA. J. Virol.2:1262-1271. 10. Mathews, C. K. 1966. DNA metabolism and virus-induced
enzyme synthesis in a thymine-requiring bacterium
in-fectedbyathymine-requiringbacteriophage. Biochemistry 5:2092-2100.
11. Mathews, C. K. 1968. Biochemistry ofDNA-defective amber
mutantsofbacteriophage T4.I.RNAmetabolism. J. Biol.
Chem. 243:5610-5615.
12. Mathews, C. K. 1970. T-Evenbacteriophage-tolerantmutants
ofEscherichta colt B. I. Isolation and preliminary char-acterization.J. Virol. 6:163-168.
13. Mathews, C. K. 1971. Bacteriophage biochemistry. American Chemical Society Monograph No. 166. Van Nostrand-ReinholdBooks, NewYork.
14. Murray, R. E., and C. K.Mathews. 1969. Addition of nucleo-tidestoparentalDNAearlyin infectionby bacteriophage T4.J. Mol. Biol. 44:233-248.
15. Murray, R. E.,and C. K. Mathews. 1969. Biochemistry of DNA-defective amber mutants ofbacteriophage T4. II.
Intracellular DNA formsininfectionbygene44mutants.
J. Mol.Biol. 44:249-262.
16. Simon, L. D. 1969. The infection of Escherichtacoltby T2 and T4bacteriophagesasseeninthe electronmicroscope.III.
Membrane-associated intracellular bacteriophages. Virology 38:285-296.
17. Travers, A.A. 1969.Bacteriophage sigma factor for RNA polymerase.Nature(London)223:1107-1110.
18. Travers, A. A. 1970. Positive controloftranscription bya
bacteriophage sigma factor. Nature (London)
225:1009-1016.
VOL. 8, 1971
285
tet1l tet 2.