0022-538X/80/01-0129/11$02.00/0
Virus-Specific
RNA
Synthesis
in
Cells Infected by Infectious
Pancreatic Necrosis Virus
PAULSOMOGYIt ANDPETER DOBOS*
Departmentof Microbiology, College of BiologicalScience, University of Guelph, Guelph, Ontario NIG2W1,Canada
Pulse-labeling experiments with [3H]uridine
revealed that the rate ofinfections
pancreatic necrosis virus-specific RNA synthesis
wasmaximal
at8 to 10h
after
infection and
wascompletely diminished by
12 to 14 h. Threeforms of
RNAintennediates
weredetected: (i)
aputative transcription intermediate (TRI)
which
comigrated in
acrylamide gels
with
virion double-stranded RNA (dsRNA)
after RNase
treatment;(ii)
a24S
genomelength
mRNA which could beresolved
into
twobands
by polyacrylamide gel
electrophoresis,
and(iii)
a14S dsRNA
component
indistinguishable from virion RNA
by gradientcentrifugation
andgel
electrophoresis. The TRI (i)
wasLiCl precipitable; (ii) sedimented slightly faster
and broader (14
to16S)
than the 14S virion dsRNA;
(iii) had a lowerelectropho-retic
mobility
in
acrylamide
gels
than
dsRNA, barely entering acrylamide gels
asa
heterogenous
component;(iv) yielded
genome-sized pieces of dsRNA after
RNase
digestion; and (v)
wasthe
mostabundant
RNAform early
in theinfectiouscycle. The 24S
single-stranded
RNA
wasthought
tobe
the viral
mRNAsince
it:(i) became
labeled during short pulses;
(ii)
wasfound in the
polysomal
fraction ofinfected
cells;
and
(iii) hybridized
todenatured viral RNA, forming
two segmentsof RNase-resistant RNA that
comigrated
with
virion
dsRNA in gels. The 24S
mRNA
componentwasformed before the
synthesis of dsRNA, and radioactivity
could be chased from 24S
single-stranded
RNA
todsRNA,
indicating that 24S
RNA
may serve astemplate for the synthesis of complementary
strands to formdsRNA.
Similar
toreovirus, infectious pancreatic necrosis viral 24S
mRNAcontained
nopolyadenylic
acid
tracts.The
genomeof
infectious
pancreatic
necrosis
virus
(IPNV) consists of
twopieces
of
high-mo-lecular-weight,
double-stranded RNA
(dsRNA)
(2.5
x106 and
2.3 x106)
that sedimented
as anRNase-resistant
14S
componentin
sucrosegra-dients
(5,
12). Upon
denaturation,
the
sedimen-tation
constantincreased
to24S and the RNA
became
sensitive
toRNase
(5).
The
electron
microscopic
diameter
of IPNV
wasfound
tobe
59
nm,
and the
virion
had
asedimentation
coef-ficient
of 435S (7). The
virion contains four
proteins
representing the products of three
genes:one
large (molecular weight,
90,000),
onemedium
(molecular weight,
57,000), and
twosmall
proteins
(molecular weight,
29,000 and
28,000); the latter
two arerelated,
asshown
by
peptide
mapping (8).
Another
protein
(molecu-lar
weight,
27,000) is the product of a fourthgene
and
isonly
present in theinfected
cell (8).The
question
arose:How
can twosegments ofdsRNA mediate
thesynthesis
offour unrelated
proteins?
Fourpossibilities
wereconsidered:
(i)
transcription
of
genomelength
mRNAfollowed
t Present address:Microbiological ResearchGroup, Hun-garian AcademyofSciences,1529Budapest,Hungary.
by
posttranslational cleavage
of
large
precursorprotein(s); (ii) transcription
of
subgenomic,
mon-ocistronic mRNA
pieces; (iii) transcription
of
genome
length
mRNA
with
subsequent cleavage
to
functional
mRNA
pieces,
or(iv) transcription
of
genomelength
mRNA
having
multiple
initi-ation and termininiti-ation sites for transliniti-ation. Since
recent
experiments
have shown
noevidence for
large
precursorpolypeptides
in IPNV-infected
cells (6,
8),
weattempted
todistinguish
between
the other
possibilities by
analyzing
the size and
number of
virus-specific
mRNA
species
in
in-fected cells. Our results showed that there
werethree forms of intracellular virus
specific
RNAs:(i)
aputative
transcription
intermediate
(TRI)
which
barely
entered 2%
acrylamide gels
and
was
partially
RNase
sensitive; (ii)
asingle-stranded
RNA(ssRNA)
species
of about 24Sthat
migrated
as twocomponents between the28S and
18S rRNAmarkers
inacrylamide gels
and
wasassociated with
polysomes;
and
(iii)
twopieces of dsRNA that
comigrated
with
thebi-segmented
virusgenome inacrylamide gels.
Ofparticular
interest was the fact thatnosubge-nomic size
mRNA's
werefound
invirus-infected
129
on November 10, 2019 by guest
http://jvi.asm.org/
cells. These results suggest that IPNV synthe-sizes two genome length mRNA transcripts that are translated into four different proteins by a mechanism that may not involve posttransla-tional cleavage of precursor polypeptides.
(Portions of these results were presented at the Annual Meeting of the American Society for Microbiology in Los Angeles [Abstr. Annu. Meet. Am. Soc. Microbiol. 1979, S134, p. 262]).
MATERIALS
AND METHODSCells. Chinook salmon embryo cells (CHSE-214)
wereakindgift from R. D.Macdonald, University of Calgary, Calgary, Canada. The cells were grown at 220C asmonolayers inplastic culture flasks (Corning Plastics,Inc.) with Eagle minimum essential medium (MEM) with Earlessalts, supplementedwith 10% fetal calfserum(FCS)andpenicillin (100
IU/IL)
and strep-tomycin(100lg/ml).Virus.TheJasperstrainof IPNVwasused in this investigation since it gave high titers and did not
generate defective interfering particles even during multiple, undiluted passages(13). To grow stock virus, cellmonolayerswereinfectedatamultiplicity of0.1 andincubatedat20°C,and viruswasharvestedwhen grosscytopathogenic effectwasevident, usually2to 3 days later. The infectious titerwasdetermined by
plaque assayasdescribedpreviously (6),and the virus wasdispensedinsmallportionsand storedat-700C. Growth andpurificationof[3Hluridine-labeled
virus. CHSE cellmonolayerswereinfected with
un-diluted virus for1hat20°C.The virusinoculumwas
replacedwithfresh MEMcontaining1%FCS and0.5
jig
ofactinomycin
D(Act-D)
perml. The volume ofthismediumwasreducedsothat it wouldjustcover
themonolayer. Aftera2-h incubation at20°C,
[3H]-uridinewasadded(10,uCi/ml),and thecultureswere
furtherincubateduntil extensivecytopathiceffectwas
evident.The viruswasconcentratedbypolyethylene
glycol precipitationand purified byfreon extraction andalternatecyclesofsucrosegradientcentrifugation
andCsClisopynic density gradient
centrifugation
as described before(8).[3Hluridine
labeling and extraction of intra-cellular RNA. Multiple cultures ofCHSE cells (in75-cm2 Corningflasks) were infected with undiluted stock virus for1hat200C. The virus inoculumwas
replacedwith MEMcontaining0.5,ugof Act-D per
ml,
and incubation continuedat 20°C. At various times after infection, cultures were withdrawn, and the MEM was replaced with 7 ml ofMEM whichcon-tained 1%FCS,0.5
1g
of Act-D perml,
and15,Ci
of[3H]uridineper ml. The cultures were further incu-bated at 20°C for various times, depending on the length of the radioactivepulse (usually2h),atwhich timethe mediumwasremoved,themonolayerswere
rinsed, and the cells were lysed in 1.5 ml oflysing
buffer (0.05 M Tris [pH 7.5], 0.1 M NaCl, 4 mM EDTA, 1%EDTA,1%sodium
dodecyl
sulfate[SDS],
1%,1,5-napthalene-disulfonic acid, disodiumsalt,and 1 mg ofproteinase K per ml). Thepreparation was incubated at370Covernighttoallow theproteinaseK todigest thecellular proteins andrelease the RNA. This protease treatment wasespecially necessarytorelease dsRNA from progeny virions, since it has been shownbefore (14) that it is difficult and very inefficient to extract IPNV dsRNAbyphenol alone. The prepa-ration was passed through a Bio-Gel P30 column, prepared ina5-ml pipette, to removesmallmolecules such as proteinase K, tRNA, andsmall labeled
oligo-nucleotides. The RNA was extracted three times with freshly distilled phenol mixed with chloroform (1:1). The residual phenol was removed from the aqueous phase by ether extraction, and the ether was inturn
removed by bubbling N2 through the preparation. One-tenth volume of 2.5 M ammonium acetate was added, and the RNA was precipitated at -20°C after the addition of two volumes of cold absolute ethanol. Lithium chlorideprecipitation.The alcohol-pre-cipitated RNA was washed three times with 70% ethanol and then resuspended in autoclaved TNE buffer (0.01 M Tris, 0.1 M NaCl, 1 mM EDTA, pH 7.5). Anequal volume of cold, sterile4MLiCl solution was added, and the RNA was precipitated at 4°C overnight. The preparation was centrifuged in the cold at 10,000 rpmfor 30 min, and the supernatant, con-taining LiCl-soluble dsRNA, was removed. The pre-cipitate againwasdissolved in TNE buffer, made2M with respect to LiCl, and reprecipitated in the cold
overnight as described above. This procedure was repeated three times. TheLiCl-solublefractions, after two morecycles of salt precipitation, werepooled;the LiCl-insoluble RNA was dissolved in TNE buffer; and both preparations wereprecipitated with ethanol at -20°C. For further studies, RNA was removed from alcoholbycentrifugation andwasresuspended in the appropriate buffer.
Sucrose gradient centrifugation. Both RNA
preparations (LiClsoluble and insoluble)were
resus-pendedin0.2 ml of TNE buffercontaining 1% SDS
(TNES) layeredonto 5 to20%linearsucrosegradients preparedinthe same buffer. Rate zonal centrifugation was performed at 40,000 rpm at 22°C for 3 h in an
SW50.1rotorbyusingaBeckmanL2-65B preparative ultracentrifuge. The gradients were fractionated by piercing the bottom of the centrifuge tubes. Portions (100pl)from each fractionwereapplied to glass fiber filters (WhatmanGF1A), andafterdrying they were placed in cold 10% trichloracetic acid at 4°Cfor 30 min.Thefilterdisks were washed in ice-coldmethanol and dried. The acid-precipitable radioactivity was counted inaliquid scintillationcounterwitha toluene-based scintillation cocktail [0.4% (wt/vol) PPO (2,5-diphenyloxazole) and 0.01% (wt/vol) POPOP
[1,4-bis-(5-phenoloxazolyl)-benzene].
Unlabeled 28S and 18S rRNA was centrifugedin
parallel tubes and was located in the gradients by measuring the optical densityat 260 nmof gradient fractions.
RNAse digestion. RNA samplesweretested for RNasesensitivityin 2xSSC(SSC:0.15MNaCl,0.015 M sodium citrate, pH 7.4) with 2 yg ofpancreatic RNaseAper mlat roomtemperature for30min.
Preparation ofpolysomes from pulse-labeled cells.Polysomeswerepreparedasdescribedby Wie-gersandHilz(20). The finalpreparationwasdissolved inlysing buffer and incubated for2h at37°Ctoallow theproteinaseK todigest all proteins. TheRNA was then subjectedto sucrose gradient centrifugationas described above.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
Radioactivelabeling of infected cells for poly-acrylamide gel electrophoresis. CHSE cellswere grown in flat-bottomed, multiwell Linbro trays (24
wells per tray). Confluent monolayers were infected with undiluted stock IPNV at 20°C for 1 h. The inoculum was removed, and the cells were overlaid with MEM (0.4ml/well) containing 1%FCS and0.5 Lgof Act-D perml. At various times afterinfection, 0.1 ml of [3H]uridine in balanced salt solution was addedtoeach wellto afinal concentration of10lOCi/
ml. At the endof thelabelingperiod, the mediumwas removed and themonolayers weredissolved in 0.1 ml ofelectrophoreticsample buffer,lysing buffer contain-ing 5% sucroseand a traceof bromophenol blue. The celllysateswereforcedthroughanarrow-gaugeneedle toshear the DNA,dispensed into0.5-ml-capacity mi-crotubes (Beckman, polyethylene),andwereallowed tostandat roomtemperature overnight for the pro-teinase Kdigestion.Samples(10
Il)
werespottedonto glass fiber filters to determine the trichloroacetic acid-precipitable radioactivity. The preparation was then storedat-70°Cuntilgel electrophoresis.Forpulse-chaseexperiments, the medium contain-ing [3H]uridine wasremoved from the cells and re-placed with MEM (2ml/well) containinganexcessof unlabeled uridine. At various times after the chase period, the medium from each well was centrifuged
(40,000 rpm,1h, 4°C, with the SW50.1 rotor)topellet extracellular virus. The monolayer was dissolved in electrophoreticsample buffer, and thiswasaddedto dissolve thepelletbefore incubation and storing.
Polyacrylamide gel electrophoresis.The RNA samples were analyzed on composite
agarose-acryl-amide slabgels by the method of Sinclair and Mindish (17),followed by fluorographyasdescribed by Bonner andLaskey (1).
Purified, labeled virion dsRNAwassometimes run forcomparison. In thiscasethepurified virus pellet wastreated withelectrophoretic sample buffer over-night atroomtemperaturetoallow the proteinase K torelease the RNA. Thereafter thesamplewaskept frozen until used.Heatingto700Cbefore electropho-resiswasomitted for virion RNA.
Unlabeled virion dsRNAwasalso analyzed in 5% acrylamide gels, followed by staining with ethidium bromide,asdescribedpreviously (5).
Viralproteinswereanalyzed in 10% discontinuous SDS-slabgels,asdescribed before (8).
RNA-RNAhybridization. Samplescontaining ra-dioactive 24S ssRNA,0.2 to 0.3absorbance unitsat 260 nm of unlabeled virion dsRNA, and 1 ,umol of EDTA(pH 7.2) in a total volume of 100
pl
were heated for90 s at1000C and then incubatedat50°C for 2 h toallowannealing. Afterincubation,0.1 ml of0.6M NaCl and60 mM sodium citrate containing 2,tg of RNase wereadded, and the samples wereincubated at room temperature for 30 min. Carrier tRNA was added, and theRNA wasprecipitated with 2 volumes ofcold ethanol. The alcohol precipitates were resus-pended in electrophoretic sample and analyzed on composite agarose-acrylamide gels.Oligo(dT)-cellulose chromatography. Oligode-oxythymidylic acid [oligo(dT)]-cellulose (0.1 g) was suspendedinautoclaved binding buffer (0.5MNaCl, 0.5% SDS, 0.01 M Tris-hydrochloride, pH 7.4) and layered over a 5-mm glass wool column and 5-mm
cellulose powder column in a sterile 1-ml plastic sy-ringe.The column wasextensively washed with bind-ing buffer, after which the labeled RNA in binding bufferwaslayeredonthe top of the column. Fractions (0.2 ml) were collected until background levels of radioactivity were attained. Bound RNA was then recovered by washing the column with elution buffer (0.05%SDS,0.01 MTris-hydrochloride, pH 7.4). 3H-labeled 18S rRNA and 3H-labeled encephalomyocar-ditis(EMC) virus RNA were used as controls.
Materials. FCS, MEM, and antibioticswere pur-chased fromGibco Laboratories. Recrystallized acryl-amide, bisacrylamide, and N,N,N',N"-tetramethyl-enediamine and Bio-Gel P30wereobtained from Bio-Rad Laboratories.SDS(specially pure) and proteinase K were obtained from British Drug Houses. [5,6-3H]uridine (specific activity, 40 to 50Ci/mol), PPO, andPOPOPwerepurchased from New England Nu-clearCorp.Oligo(dT)-cellulosewasobtained from Col-laborative Research Inc., and RNase A was from Sigma Chemical Co.
RESULTS
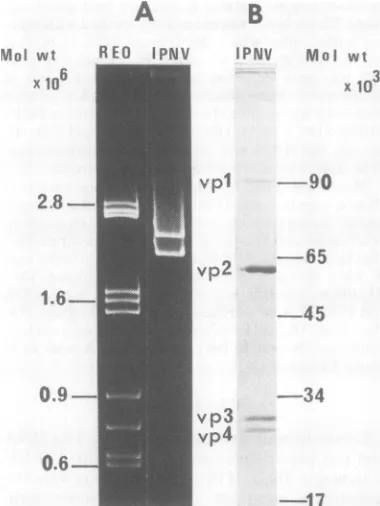
Genome and
proteins
of IPNV. The RNA
and
polypeptide
composition of
purified
IPNVis shown
inFig.
1.Ifthe
virion
proteins
werethe
translation
products of monocistronic viral
mRNA's,
onewould
expecttofind
intracellular
mRNA's with molecular
weights
of
approxi-mately
0.9x106,
0.6 x106,
and 0.3 x106
which
could code for the three size classes of virion
polypeptides. Based
uponthe
molecular
weights
of the
genomedsRNA's,
genomelength
mRNA's
should be
approximately
1.25 x106
and
1.15 x106
daltons.
RNA
synthesis
in
Act-D-treated,
infected,
and uninfected cells.
Act-Dwasused
toinhibit
host
cell RNA
synthesis. Preliminary
experi-ments
showed that the
presenceof Act-D
at aconcentration of
0.5tig/ml
reduced the
yield of
progeny
virus
by
0.5log
atmost, yet itinhibited
host
cell RNA synthesis by
80 to90% within
3to 4 h after
addition.
Therefore,
virus-specific
RNA
could be labeled
preferentially
by addition
of
[3H]uridine
toinfected
cultures
3to 4hafter
the addition of
Act-D.The data
inFig.
2show
the
effect of
Act-D onthe
synthesis
of total
RNA in
uninfected and IPNV-infected
cells.
When infected
cells
werepulse-labeled
for
2hat2-h
intervals from
2 to 12h
after
infection, the
amount
of
RNAsynthesized increased
up to 8to 10 h, followed bya rapid decrease, reaching
negligible
levels
by
12to 14h.
Pattern
of
RNAsynthesis
invirus-in-fected
cells.
Itwasof interesttodetermine thetypes of RNA
synthesized
invirus-infected
cells
under
conditions
in which hostcell
RNAsyn-thesis
wasdepressed by
Act-D. To thisend,
Act-D (0.5
yg/ml)
wasadded to infected andmock-infected
cultures after virusadsorption
(zerotime),
and 2 hlater
the cultures werepulse-33,
on November 10, 2019 by guest
http://jvi.asm.org/
132
A
B
h pulse; however, its presence wasunambiguous
after
4h.REO
IPNV
IPNV
Mol
wt
A second form of virus-specific RNAwhich
3 was
discermible
in thefluorogram
was anRNA x 10 thatcomigrated
with the double-stranded viralgenomic RNA
segments.This RNA
appeared
later than the 24S RNA and could
belabeled
ininfected
cells between
6and
12h
after infection.
A
third form of virus-specific RNA
was aV:p1
-
90
heterogenous
RNA
species
of low
electropho-retic
mobility that barely entered the 2%
acryl-amide
gel. We named this RNA
form TRI
in--65
stead of
replicative
intermediate
(RI)
because
v
2
this RNA
waspresent
at the
sametime
as24S
RNA but
before the
synthesis of
progeny45
dsRNA. This RNA
wasoften obscured
(as in
Fig. 3) because:
(i) there
was asubstantial
amount
of
radioactivity
atthe
origin of the
gel;
(ii) there
was anonspecific band
(marked
X)
-34
present evenin
the
uninfected
celllysates.
This
v3
nonspecific
bandwasalsopresent
whensucrose vp4
gradient-purified
28Sor 18S labeled rRNAwasanalyzed in these
gels
(Fig. 4). Since
weused
[5-3H]uridine, it
washighly
unlikely
that the
non-specific band would
representDNA,
such
as-17
mitochondrial DNA. The amount of RNAnrion
RNA andproteins
of IPNV. (A) trapped in this region also depended on the ide gel electrophoresis of IPNV RNAamount
ofmaterial
loaded
ontothe
gel.
InsomehreovirusRNA
(type
3Dearing
in5% casesthere
waslittle
radioactivity
left
atthe
gels
followed bystaining
withethidium
c
t
Polvacrvlamide gel electroDhoresis of
orgin,
and thenonspecific
band was absent, yet1v.0vvv 8,v> ,rvv >g5VVW, V%RIWVO proteinsofpurified IPNV in10%discontinuous SDS-gel. Thegelswerestained with Coomassie brilliant blue. Thepositionand the molecularweightx103of
the markerproteinsareshown on theright,andthat of the virionproteinsis shownontheleft.Themarker proteins used werephosphorylaseA(90,000), bovine serumalbumin(65,000),ovalbumin(45,000),aspartate
transcarbamylase (34,000),and tobacco mosaicvirus protein (17,000).
labeled with
[3H]uridine for
2h
at2-h
intervals
from
2 to 12hafter infection. The
cell
lysates
were
analyzed for RNA by
acrylamide
gel
elec-trophoresis and fluorography. The results
in Fig.3
show
that
IPNV-specific
RNA
species
weresynthesized
in
infected cells
by
2 to 4h
after
infection.
The
earliest form of viral RNA detected
inthese
gels
was anRNAspecies
whichcould
beresolved
into two components andmigrated in
the
gels between the 28S and
18S
rRNAmarkers.
ThisRNA
wascalled 24S RNA (see
Fig.
4below) and
wasthe
predominant fonn of
RNA.
The
amountof24S RNA
synthesized
atvarious times
after infection
(as judged from
theintensity
ofbands)
paralleled
theaccumulation
of
virus-specific
RNAshown inFig.
2.The
syn-thesis
of24S RNA
wasdetectable
in the 2-to4-5
4
3
x
E2
Q
m 1
2 4 6 8 10 12 14
HRS pi.
FIG. 2. RNAsynthesisinAct-D-treateduninfected andinfectedcells.IPNV-infectedandmock-infected CHSE cells werepulse-labeled for2 h at 120-min intervalsfrom 2to 12 h after infection with [3H1-uridine. The monolayers were dissolved in
electro-phoretic sample buffer, and the trichloroacetic acid-precipitable radioactivity was determined as de-scribed in thetest. Symbols:0, Uninfected cells;0,
infectedcells. Mol wt
x
106
2.8
-
1.6-0.9
-FIG. 1. Vi,
Polyacrylam
together wit)
acrylamide j bromide. (B)
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.504.65.255.72.325.2] [image:4.504.292.427.385.562.2]UN. U N.
Act-o 2 4 6 8 10 12
28S _
18So
FIG. 3. Time course ofRNA synthesis in
IPNV-infected,Act-D-treated CHSE cells. Mock- and virus-infectedcellmonolayersweretreated with Act-D(0.5
pg/ml) from0to2hafterinfection followed by pulse-labeling with [3H]uridine for 2h at 2-h intervals from2 to12hafterinfection. Themonolayerswere dissolved in electrophoretic sample buffer, and heatedto70°C, and the RNAwasanalyzed by
elec-trophoresis in 2%acrylamide-agarose slabgels
fol-lowedbyfluorography. Labeled virion dsRNA was also includedforcomparison.Thenumbers above the wells indicate the time (after infection) when the isotopewasadded. Thepositionof28Sand188rRNA is shown on the left. Theposition of TRI, virion
dsRNA,and 24Svirus-specificRNA is shown on the right. The locationof the nonspecific band described in thetextismarked with X.
the TRI could
clearly be observed (Fig.
4D).Further
evidence for
aTRI. dsRNA is
sol-uble in
2M
LiCl,
whereas ssRNA and
transcrip-tion
complexes
containing single-stranded
nas-cent
chains
(such
asthe
TRI)
are precipitatedby LiCl and
canbe
subsequently
separated fromeach other
by
sucrosegradient centrifugation
oracrylamide gel
electrophoresis.
To investigatethe
natureof 24S RNA
andthe TRI,
phenol-extracted RNA from
labeled, infected
cells
wasseparated into LiCl-soluble and
LiCl-precipita-ble fractions by repeated
cycles of precipitationand
analyzed
onsucrosegradients as describedin
Materials and Methods. The
majority ofLiCl-precipitable
RNA
sedimented between
the tworRNA markers
as a24S
component (Fig. 5A). Aminor
peak
sedimented
behind the18S rRNA as a 14 to16S
component.Samples
fromfractions 11 to 29were subjected to acrylamide slab gelelectrophoresis and fluorography
as shown inFig.
5B. The RNA in the 24S peak had a highelectrophoretic mobility
andmigrated in the gelbetween
the
RNA markers. The 14 to16S RNA showed lowelectrophoretic
mobility and washeterogenous in
nature, twocharacteristics
of
V
transcription
intermediates.
Note
that the
14 toTRI.
16S
RNA(TRI)
sedimentedonsucroseclose tothe
genome14S dsRNA
butmigrated slower
indsRNA
acrylamide
gels than
thedsRNA. Sucrose
gra-dient fractions
representing
the
24Sand
14 to16S
peaks
werepooled
separately,
and
the
amount
of
acid-precipitable
radioactivity
was24S
determined
before and after RNase
treatment.The data in Table
1show
that the
24S RNA
wascompletely sensitive
toRNase, whereas about
14%
of the
14 to16S RNA
wasresistant
toRNase, and
this resistant RNA
comigrated
with
[image:5.504.47.240.71.244.2]virion
dsRNA in the
acrylamide
gel
shown inFig. 5B
(lane
A).
24S RNA is viral mRNA. The
size andA
B
C
D
E
--- --TR.I.
x
--'3Mf.
A
w 24SFIG. 4. Analysis of cellular and virus-specific
RNAs in 2%acrylamide-agarosegel followed by
fluo-rography. Lanes A and B show the electrophoretic
patternof labeled,phenol-extractedrRNAwhich has beenpurified bysucrosegradientcentrifugation
be-fore gelelectrophoresis.LaneA,
288
rRNA;laneB,18SrRNA.Nonspecificband markedwith X. Lane C showsanautoradiogramwhenrRNA,24SRNA,and viraldsRNAare runin thesamewell. LaneD dem-onstratestheTRI without thenonspecificband. Here
infectedAct-D-treated cellswerepulse-labeledwith
[3HJuridine
from 6 to 7h after infection; the cellswere
lysed
inelectrophoreticsamplebuffer,heatedto70°C for2min, andsubjectedtogelelectrophoresis
andfluorography. LaneEshows thefluorogram of
labeled RNA present in thepolysomal fraction of
infected Act-D-treated cells.Samples oflane D and Ewereelectrophoresedon adifferent gel than those
ofA, B,andC; thus, thepositionof24SRNA does notcoincide in thetwosets(see dashedline).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.504.258.440.109.460.2]~~~~~~0
.~~~~~~~~ e
B ABC
l4 dsRNA
FIG. 5. Analysis of the LiCl-precipitable, virus-specificRNAbysucrosegradient centrifugationand polyacrylamide gel electrophoresis. (A) Infected Act-D-treated cellswerepulse-labeledwith[3H]uridine
from5to6 hafter infection. The cellswerelysed and incubated withproteinaseK andpassed througha
Bio-Gel P30column,and the RNAwasextractedby phenol, followed bythreecycles ofprecipitation of2 M LiClasdescribedin the text. Rate zonal
centrifu-gationina30to15% SDS-sucrosegradientinTNES buffer (24,000rpm,22°C, 16 h withanSW27rotor).
The acid-precipitable radioactivity ofa portion of
eachgradient fractionwasdetermined. (B)Portions of gradient fractions from11 to29weresubjectedto
acrylamide-agarose slab-gel electrophoresis, fol-lowed by fluorography. Lane A representspooled gradient fractions from25 to29afterRNase treat-mentasdescribed under Table 1. Lane Brepresents total virusspecificRNAfromcellslabeledfrom6to 8 hafter infection.LaneC shows28Sand 18S rRNA markers.
RNasesensitivityof the24SRNAindicated that
itwas a genome length transcript ofthe viral
RNA andthat it most likely representedviral
mRNA. To furtherinvestigate this possibility, infected cellextractswerepreparedwith
Noni-det P-40 and Dounce homogenization, and the
postmitochondrial fraction was pelleted by
ul-tracentrifugation (140,000x g,2h).The
result-ing polysomal pelletwastreated withproteinase
K andsubjected to acrylamide gel electropho-resis. The predominant, labeled RNA was the
24S RNAasshown inFig. 4E.
Ifthe 24S polysomal RNA wasviral mRNA representing "plus" strands of the
double-stranded viralgenome,then it shouldhybridize
to viral RNA. To test this, labeled 24S RNA was
prepared from infected cell
lysates by sucrosegradient centrifugation.
The 24S RNA wasmixed with unlabeled
purified viriondsRNA,
melted, and reannealed.
Half of the preparationwas treated with RNase in high
salt,
and bothsamples
wereanalyzed
in acrylamide gels (Fig.6).
The
24S
RNA did not self-anneal, but in the presenceof virion
RNA itformed
two dsRNAhybrids which
comigrated with viriondsRNA
inacrylamide gels. This experiment
also indicatedthat the
two close bands that made up the 24SRNA in
gels
represented molecules from each ofthe
two genome dsRNAs and were notsimply
due
to aconformational
modification of oneRNA.
Another attribute of mRNA's
is that they canbe
rapidly
labeled when, during their synthesis,
cells
areincubated with
RNA precursors for ashort time. When virus-infected
cells
werela-beled with [3H]uridine for
15 or 30 min at 6 hafter
infection, only the 24S RNA
becamela-beled,
asshown in
Fig.
9.Taken
together, theseexperiments
indicate that the 24S
RNArepre-sents
viral
mRNA.Kinetics of virus-specific RNA
synthesis.Although acrylamide gel electrophoresis
was auseful method for
qualitative analysis of the
types
of
virus-specific RNA synthesized
inin-fected
cells,
it
wasunreliable for quantitative
studies due
tothe variable
amountof
radioactiv-ity
left
ontopof the
gel and
tothe
presenceof
the
nonspecific band (X) in all preparations.
Denaturing gels
were notused
because they
mayhave denatured the dsRNA.
Forthis
reason, thetime
courseof
synthesis of the three forms of
virus-specific RNAs
(24S, TRI, and dsRNA)
wasTABLE 1.RNasesensitivity of threefornsof virus-specific RNAs
RNA
form
Acid-precipitable
%RNasere-cpm sistance
24Sa 32,000 1
TRI(14-15S) 6,200 14
dsRNA(14S)b 8,000 92
a24S RNA representsgradient fractions from11 to
23inFig. 5, and TRI represents fractions 25 to 29 from thesamegradients. The representative fractionswere pooled,precipitatedwithethanol, and resuspended in 0.2ml of 2xSSC. Half of each preparationwastreated withRNase A(5,ug/ml,30minat roomtemperature), and the acid-precipitable radioactivity of untreated and RNase-treated samples were determined as de-scribedinthetext.
b Labeled virion dsRNAwasreleased from purified virus by proteinase K treatment. It was separated from the proteaseby phenol extraction and alcohol precipitation; RNase treatment was carried out as described for 24SRNAand TRI.
on November 10, 2019 by guest
http://jvi.asm.org/
a
b
CFIG. 6. Hybridization of labeled 24S RNA to
un-labeled viriondsRNA. Purified[3H]uridine-labeled 24SRNA and unlabeled virion RNA in 1
jmol
ofEDTA (pH 7.2) wereheatedfor90sat 100°C and then incubatedat50°C for2htoallow annealing. The salt concentration was adjusted to that of 2x SSC, and half of the samplewastreated with RNase
(2p,g/ml, 30minatroomtemperature). Lanea,24S
RNA self-annealing followed by RNase treatment;
lane b, 3H-labeled 24SRNA annealedtounlabeled virionRNA;lanec,3H-labeled 24SRNAannealedto virionRNAfollowedby RNasetreatment.
analyzed by sucrose
gradient
centrifugation ofLiCl-soluble
andLiCl-precipitable
RNApulse-labeled with [3H]uridine. Cell extracts were
passed through a column of Bio-Gel P30 to
remove small molecules such as peptides,
pro-teinase K, tRNA, and unincorporated [3H]uri-dine. Thesampleswerephenol extracted,
alco-holprecipitated, and separated bythreecycles of LiCl precipitation. Both LiCl-soluble and
LiCl-insoluble
RNAs were precipitated withethanol, and the precipitateswereresuspended
in TNES buffer, followed by sucrose gradient
centrifugation in 5 to 20%sucrose gradients in
TNES buffer (40,000rpm at220C for3 hinan
SW50.1rotor).ThegradientRNAprofilesofthe
three forms of RNA at different times after infectionareshown inFig.7.
Between2to4h afterinfection, only the 24S
mRNA and the 14 to16S TRI becamelabeled. Between 4to 6h the 24S mRNAwasthe
pre-dominant species, although asmall amount of 14SLiCl-soluble (dsRNA) was also detectable.
The synthesis of 24S mRNA reached its peak
between 8and 10 hafterinfectionanddeclined
thereafter. The kinetics of
synthesis of the TRIroughly
paralleled
thatof
the24S mRNA. Thesynthesis
of
the14S dsRNA
reached itspeak
at8to 10 h
after
infection, and although it slowly
declined thereafter
(dueperhaps
tovirion
re-lease),
its
contribution
tothe
total labeled
RNAincreased
so thatbetween 12 to 14 h, almost halfof the labeled
RNA wasdouble-stranded.
When the
radioactivity
represented by three
peaks
wassummed
ateach time
interval,
acom-posite
graph
wasplotted
toshow the
amountof
each
of the three RNA forms
presentrelative
toeach other and
tothat of the
total RNA (Fig. 8).
A
numerical
presentation of the precentcontri-bution of each RNA
to thetotal
is shown inTable
2.24S mRNA
maybe
the
precursorfor
progeny
dsRNA. The synthesis of 24S mRNA
preceded that of
progenydsRNA,
as shownin
Fig.
3and
7. Inreovirus-infected
cells, the
"mi-nus"
strands
of
viral
RNA aremade
onplus-strand
templates
toyield
progenydsRNA (22).
We
attempted
to seewhether this
wasalso the
casein
IPNV-infected cells
by
pulse-labeling
24S
mRNA with
[3H]uridine
and
thenchasing
itwith
excessunlabeled uridine. The data
inFig.
9
show that when
infected
cells
werepulse-la-beled
for 15,30,and
60minat 7hafter
infection,
the
24S
mRNAbecame labeled but the dsRNA
was not
labeled
(dsRNA became labeled
only
if
2-h
orlonger
pulses
wereused
asshown
inFig.
3).
When
infected cells
werepulse-labeled
with
[3H]uridine
for
30minand chased for
a6-or12-h
period, the
amountof
radioactivity decreased
in the
24S mRNA
componentand
increased in
the dsRNA. These
data
arecompatible
with
(but
do
notprove)
the
hypothesis
that the
com-plementary strands of the dsRNA
aresynthe-sized
asynchronously,
possibly
via
single-stranded
plus
RNA
intermediates.
Furthermore,
the
fact that
intracellular dsRNA
cannotbe
labeled
by
shortpulses
suggeststhat, like
reo-virus, IPNV
RNA
replicates by
aconservative
mechanism.
The
drawback of this
typeof
experiment
is
the
long
period
of chase
required
before
radio-activity
canbe
demonstrated
inputative
product
molecules (in this
case,dsRNA). This
isdue
tothe
presenceof
large
intracellular uridine
pools
which
cannotbe
diluted
out except overlong
periods
of time.Therefore,
the data shown inFig.
9 canalso
beinterpreted
in a manner that does notimply
aprecursor-product
relationship
between mRNA
and
dsRNA(see
Discussion).
However,
the results ofhybridization
experi-mentsmake a "reovirus-like" mechanism a more
likely
possibility.
24S
mRNAcontains
nopoly(A) tracks.
The mRNA's of
all
animal viruses so far studiedon November 10, 2019 by guest
http://jvi.asm.org/
[image:7.504.105.182.65.292.2]'ox
1
AA12
E 2865 185 285 185 285 185
8-1
5 0h2 51 15 20 25h 12h0214
6
4
3
2
5 10 1520 25 5 1015 20 25 5 10 5 20 25
FRACTION NUMBER
FIG. 7. Timecourseof LiCl-precipitableandLiCi-solubleRNA synthesis in IPNV-infectedCHSE cellsas
determined bypulse-labeling with [3HJuridinefollowed bysucrosegradient centrifugation. Cultureswere
labeledfor 2 hat120-minintervals from2to12h after infection,andthe RNAwasextractedasdescribed in
thelegend of Fig.5. TheLiCl-soluble (0 - 0) andLiCl-precipitable (a-_)preparationswereanalyzed byratezonalcentrifugationin5to20%SDS-sucrose gradients.
60~
50f
x
E
a
zr
v~-40[
I I \
%
I
I
I
il P
30[
20~
10
2 4 6 8 10 12 14
HRS p.i.
FIG. 8. Synthesis ofthe three forms of virus-spe-cificRNAininfectedcells.At each timeinterval,the amountof radioactivitypresentin thegradient frac-tions undereachpeak in Fig. 7was summed and
[image:8.504.120.407.67.312.2]plotted with respect to time (A---A) totalRNA; (@-*) 24S mRNA (LiCl-precipitable); (0-0 ) 14SdsRNA (LiCI soluble); (A A) 14to 15S TRI (LiClprecipitable).
TABLE 2. Temporalsynthesis of IPNV RNA intermediatesafter infectiona
Time of TotalRNA % of total RNA pulse(h) (cpm) TRI
mRNA dsRNA
2-4 6,400 55 45 0
4-6 12,100 25 69 6
6-8 43,850 22 60 18
8-10 60,950 18 53 29
10-12 41,700 13 53 35
12-14 17,250 8 51 41
aTheamountofradioactivityrepresented byeach
peakinFig.7 wasexpressedasthe percentage oftotal
radioactivityfound in eachgradient.
[image:8.504.89.239.371.581.2]except reovirus contain a
polyadenylic
acid[poly(A)]
tractof variablelength
atthe 3' end ofthe RNA
(18).
Thelength
of
thepoly(A)
tracts rangesfrom
15nucleotides
as inEMC virus
RNA
(2)
to about 200nucleotides
as in Rous sarcomavirusRNA
(4).
To
determine
whether IPNV 24S mRNAcon-tained
poly(A),
wehave
subjected
[3H]uridine-labeled
purified
24S
mRNAtochromatography
on
oligo(dT)-cellulose.
Labeled 18S
rRNAserved
asnegative control and labeled
purified
EMC
RNA wasused
aspositive
control. Thedata
inFig.
10show
that underconditions in
k
k
4
1
I
I
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.504.269.462.392.481.2]IPNV-SPECIFIC RNA SYNTHESIS 137
3+0'
3Q0'
6h
12h Vam _
X~~~~~~~~~~~~~~~~~~~~~~
X
6w
__o_ _ dsRNA
24S_-FIG. 9. Possible precursor-product relationship between24S mRNAand viraldsRNA.Infected, Act-D-treated cellswerepulse-labeledfor 15, 30,and60
min with
[3H]uridine
7hafterinfection. The radio-activitywas"chased" intwocultures that had been pulse-labeled for 30min, by incubation for6and12 h in thepresence ofexcessunlabeled uridine. Thelysed cellsweresubjectedtoacrylamide-agarose gel electrophoresis followed by fluorography, together withpurified, labeledviral dsRNA. Theposition of the nonspecific band is marked with X.
which 86% of
EMC
RNA was bound to theoligo(dT)-cellulose,
both IPNV 24SmRNA and18S rRNA could be eluted from the column. Since EMC RNA contains a short poly(A)
stretch ofonly14 to18nucleotideresidueslong,
even such a short poly(A) tracton IPNV 24S
mRNA would have been detected.
DISCUSSION
AnimalviruseswithdsRNAgenomessuchas
reovirus replicate via ssRNA
intermediates
which serve as templates for the synthesis of
their
complementary
strands (11). These ssRNA intermediates inreovirus-infected
cells arecalledplus strands because they also functionas
mRNA. Evidence that thetwo segments of24S ssRNA described in thispaper areviral RNAs
and are probably intermediates in the
replica-tion of IPNV dsRNAincludesthefollowing: (i) they are found only in infected cells and are
labeled in the presence of Act-D; (ii) theycan
be "chased into dsRNA"; (iii) they have sedi-mentationproperties thatareidenticalto that
of denatured virion dsRNA (5); and (iv) they hybridize with viriondsRNA. The 24S ssRNA's
mayfunctionasmRNA'ssinceboth speciescan
berapidly labeled by short pulses and bothcan
be isolated from the polysomal fraction of
in-fected
cells. Similar
toreovirus, the IPNV
mRNA contains
nopoly(A)
tracts.The properties of IPNV TRI
aresimilar to
those
reported by Franklin
(9)for
the RI ofphage
R17:(i)
itsedimentsslightly faster
thandsRNA; (ii) it has
alower
electrophoretic
mo-bility in
acrylamide gels than
dsRNA;
in
fact, it
barely
enters2%
gels;
(iii) it
precipitates in
2M
LiCl;
and(iv)
ityields
genome-sized pieces
ofdsRNA
after RNase
digestion. These data
areconsistent with
a structurecomposed of
afull-length dsRNA
molecule and
one or morepar-tially completed ssRNA
transcripts. Although
the RI of the R17
phage exists
as asingle-stranded
complex that anneals
upondeprotein-ization
(19), it is
unlikely
that this is the
casewith IPNV since it has
adsRNA
genome.The
TRI of IPNV
makes
upmostof the intracellular
virus-specific RNAs
during the early
partof the
infectious cycle
(Table
2) when there is
arapid
increase
inthe rate ofsynthesis of 24S
mRNA.Although
noTRI
(or RI) have been
reported
in
reovirus-infected
cells per se,these kinds of
mol-ecules would be found in
parental and
progenysubviral
particles which
synthesize mRNA
ondsRNA
templates using
the
ds
--ssRNA
poly-merase(s)
(27). Since reovirus transcribes
plus
strands in
aconservative
manner(16), rapid
labeling of progeny dsRNA does
notoccurbe-cause
the
complementary
strands
aresynthe-sized
atdifferent times. Our results indicate that
a
similar
mechanism is
operating
in IPNV
be-3.
2
-1I
C)
E
C-F
_o
unbound bound
[image:9.504.44.236.71.262.2]5 10 15 20 fract on no.
FIG. 10. Chromatographyof[3H]uridine-labeled,
IPNV-specific 24SmRNA,EMC virusRNA,and188 rRNAon oligo(dT)-cellulose. The RNAs in binding bufferwereaddedtothe columnandwashedwiththe
same bufferuntil 10fractions werecollected. Then theeluantwaschangedtoelutionbuffertodisplace the boundRNA.
15'
30' 60'
_.
anmwt
_on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.504.279.400.402.589.2]cause: (i) dsRNA becomes labeled only when
pulsesof 2 h (orlonger)areused; (ii)
radioactiv-ity could be chased from 24S mRNA into
dsRNA; and (iii)
24S
mRNA hybridized with denatured viral dsRNA.Themostlikelyreasonwhy dsRNAcannotbe labeled by short pulses is because onlyasmall
minority
of moleculesarereplicatingatanyonetime and are not detected until longer times elapsed. The data in Fig. 9 show thata major
problem in the examinationofRNAsynthesisin
animal cells isthe
difficulty
infollowingthefateof RNA that has been synthesized and labeled during ashortexposureor pulsetoradioactive RNAprecursors.Thisis because thelarge intra-cellularpools of RNAprecursors cannot be
di-lutedoutoremptied exceptoverlong periodsof
time. Forthisreason,24S mRNAwasstillbeing
labeled
during
the 6-h chase, and to a muchsmallerextentevenduring the 12-h chase period.
Nevertheless,
radioactivity couldbechased,al-beitslowly, from 24SmRNA todsRNA.
Altematively,
the data shown in Fig. 9 arealso compatible with the theory that dsRNA became labeled during the 6-h chase (but not
during the 30-min pulse), merely because the
rateofsynthesis ofdsRNAwasslower than that
of
24S
mRNA. Hence, eventhe"slowly
made"dsRNA became labeled during the 6-h chase, which could be regarded as a long (6-h) pulse
because of thelarge intracellular labeled uridine pool. Indeed theamountoflabel in24SmRNA
increased
during this period compared with thesample thatrepresentsthe30-minpulse. Itmay
also be argued that the loss of label from
24S
mRNA after a 12-h chase was due to RNA
breakdown rather thanto movementofmRNA intodsRNA, especially since theamountoflabel increased but little in the dsRNA region after thisadditional 6-h chaseperiod.
It would be highly desirable to be able to
firmly establish the mechanism of IPNV dsRNA synthesis by the elegant method of
Schonberg
et al. (16) who showed unequivocally thatthecomplementary
strandsofreovirusgenomeweresynthesizedasynchronously.
Unfortunately,
this experiment requireslargeamountsofunlabeled plus strands which can be produced readily invitro, by using reoviruscores;however, repeated
attempts tosynthesizeIPNVmRNA in vitro in
ourlaboratory havesofarbeenunsuccessful.
During
the course of thisinvestigation,
wehavenotdetectedanysubgenomic-sized mRNA in IPNV-infected cells; therefore, the relation-ship between the virus-specific proteins and the virus genome still remains to be determined. One possibility is that the 24S mRNA's have multiple initiation and termination signals for translationsimilartothemRNAofRNAphages
J. VIROL.
(3). However, eucaryotic mRNA's
usuallycan-not
display
more than oneinitiation site
at atime for
protein synthesis,
evenif
asecond
ini-tiation site ispresent
onthe mRNA and
canbemade functional in
adifferent
configuration
asin the
caseof
togaviruses (12). This kind of
protein synthesis is
unlikely in IPNV-infected
cells,
for we haveshown
previously that
all threesize classes of
virus-specific polypeptides
arepro-duced
throughout the replicative cycle (6). Of
course, farfetched
asitmay
seem, it ispossible
that in the IPNV mRNA
population each
of
thefour initiation sites for the four
different
proteins
are
represented
ondifferent
mRNA's
by
second-ary
folding right from the
beginning
of the
infec-tion. A
second possibility is that
IPNV mRNA
is
functionally
similar
tothat of Kunjin virus
(member of
flaviviruses)
which contain multiple
internal initiation sites for
translation of
virus-specific proteins
asshown
by Westaway (21).
Poliovirus RNA
mayalso have
twoinitiation
sites for
protein
synthesis (10).
Athird
possibil-ity
isthat
VP1 isthe
primary gene product ofone
of the
genomesegments, and
the
medium-and small-size IPNV
polypeptides (VP2, VP3,
and the nonstructural
polypeptide of
27,000mo-lecular
weight)
areproduced by
rapid
posttrans-lational
cleavage from
apolyprotein, the latter
being the
primary
geneproduct of the other
genome segment.
As
reported previously,
wehave failed
todetect
polyprotein precursor(s)
inIPNV-infected cells with amino
acidanalogs,
protease
inhibitors,
ZnCl2, supraoptimal
temper-atures,
and
pulse-chase experiments (6,
8). Itshould be
noted,
however,
that
sometimes these
conventional
techniques
todemonstrate
precur-sor-product
relationships
are notsufficient.
Forexample,
the existence of
a precursorpoly-peptide for Sindbis virus structural proteins
wasdemonstrated
convincingly
only with the
useof
temperature-sensitive
mutantswhich
failed
toexhibit
proteolytic cleavage
atthe
nonpermis-sive
temperature(15).
Whichever of these possibilities turns
out tobe true,
the
results
sofar show that
the mecha-nism ofvirus-specific
RNAsynthesis
inIPNV-infected cells
issimilar
tothat ofreovirus,
butvirion
proteins
areproduced
from IPNVmRNA's
by
a differentmechanism
than that found in reovirus-infectedcells.
ACKNOWLEDGMENT-S
Wethank R.Macdonaldforhishelpfulcriticismduring the preparationofthismanuscript.
ThisinvestigationwassupportedbytheNational Research Council ofCanada.
LITERATURE CITED
1. Bonner, W.M., and R. A.Laskey.1974. Afilm detection method fortritiumlabeledproteins and nucleicacids in
on November 10, 2019 by guest
http://jvi.asm.org/
polyacrylamide gels. Eur.J. Biochem. 46:83-88. 2. Burness, A. T. H.,I.V. Pardoe, E.M.Duffy, R. B.
Bhalla, and N. 0. Goldstein. 1977. The size and
location of the poly(A) tractin EMC virus RNA. J.
Gen.Virol. 34:331-345.
3. Cancedda, R., L. Villa-Komaroff,H. F. Lodish,and
M. Schlesinger.1975.Initiationsites for translationof
Sindbis virus 42Sand 26S messengerRNAs. Cell 6:
215-222.
4. Coffin, J.M.,andM.A. Billeter.1976.Aphysicalmap
of the Roussarcomavirusgenome.J. Mol. Biol.100:
293-318.
5. Dobos, P. 1976. Size and structure of the genome of
infectiouspancreatic necrosis virus. NucleicAcid Res.
3:1903-1924.
6. Dobos,P. 1977. Virus-specific proteinsynthesis incells
infected by infectiouspancreatic necrosis virus.J. Virol.
21:242-258.
7. Dobos, P.,R.Hallett,D. T. Kells,0.Sorensen,and
D. Rowe. 1977.Biophysical studiesof infectious
pan-creaticnecrosis virus.J. Virol. 22:150-159.
8. Dobos, P.,and D. Rowe.1977.Peptidemapcomparison
of infectiouspancreatic necrosis virus-specificproteins. J. Virol.24:805-820.
9. Franklin,R. M.1966.Purification andproperties of the
replicative intermediateof the RNA bacteriophageR17.
Proc.Natl. Acad. Sci. U.S.A. 55:1504-1511.
10. Jense,H., F.Knauert, andE.Ehrenfeld.1978.Two initiation sites for translation ofpoliovirus RNA in
vitro:comparison of LSc andMahoney strains. J. Virol. 28:387-394.
11.Joklik,W.K.1974.ReproductionofReoviridae,p.
231-334. InH. Fraenkel-Conratand R. R. Wagner (ed.), Comprehensive virology, vol.2.Plenum Publishing Co., NewYork.
12.Kennedy, S. L.T. 1976.Sequencerelationship between
the genomeand theintracellularRNAspeciesof stan-darddefective-interferingSemliki forestvirus.J. Mol. Biol. 108:491-551.
13.Macdonald, R. D. 1978. Ringed plaque formation in infectiouspancreatic necrosisviruscorrelateswith de-fectinginterferingparticleproduction.J.Gen. Virol.41: 623-628.
14.Macdonald, R. D., and T. Yamamoto.1977.The
struc-ture of infectious pancreatic necrosis virus RNA. J. Gen. Virol.34:235-247.
15. Schlesinger, M.J., and S. Schlesinger. 1973. Large-molecular-weightprecursors ofSindbisvirusproteins. J.Virol.11:1013-1016.
16. Schonberg, M., S. C.Silverstein,D.H.Levin,and G.
Acs.1971.Asynchronoussynthesisofthe
complemen-tarystrands of thereovirusgenome.Proc.Natl.Acad. Sci.U.S.A. 68:505-508.
17. Sinclair, J.F.,and L. Mindich. 1976.RNA synthesis
during infection with bacteriophage 4,6. Virology75: 209-217.
18. Stoltzfus,C.M.,A.J.Shatkin,and A. K.Banerjee.
1973. Absence ofpoladenylicacid from reovirus
mes-sengerribonucleic acid. J. Biol. Chem. 248:7993-7998. 19.Tach, S.S.,and R. E.Tach.1973.Mechanism of viral replication. I. Structure ofreplicationcomplexesof R17 bacteriophage. J. Mol. Biol. 81:367-380.
20. Weigers,V., and H. Hilz. 1972.Rapidisolation of
un-dergradedpolysomalRNAwithoutphenol.FEBS Lett. 23:77-82.
21. Westaway,E.G.1977.Strategyoftheflavivirusgenome: evidence formultipleinternal initiation of translation ofproteinsspecified by Kunjinvirus.Virology 80:320-335.
22. Zweerink,H.J.,Y.Ito,and T. Matauhisa. 1972.
Syn-thesisof reovirus doublestranded RNA within virus-likeparticles. Virology 50:349-358.
VOL. 1980