0022-538X/82/040020-10$02.00/0
Isolation and Preliminary Characterization of
a
Phosphonoacetic Acid-Resistant
and
Temperature-Sensitive
Mutant
of
Herpes Simplex Virus Type 1
JASMINEI. DAKSIS,MARKM.PRIEMER,AND VOON-LOONG CHAN*
Department of MicrobiologyandParasitology, University of Toronto, Toronto, Ontario, Canada, MSS IAI
Received 29 July 1981/Accepted16 November1981
Agroupof 43phosphonoacetic acid (PAA)-resistant mutants of herpes simplex
virustype1wasisolated after the mutagenesis of infectedcells with nitrosoguani-dine. One of these mutants, designated
PAA1r,tsl,
wasfound tobe temperature sensitive(ts), that is, unable toreplicate at39.5°C, the nonpermissivetempera-ture. Recombination analysis of PAAirtsl indicated that the PAA1r mutation and
the tsl mutation are loosely linked and are located on two separate genes.
PAAirtsl showeda defect in viral DNA synthesisat 39.5°C, whichpresumably
can be attributed to the production ofa PAA-resistant and thermolabile DNA
polymerase. PAAirtslwasalso defectivein the shutoff ofhost DNA synthesis at
therestrictive temperature.
The genome of herpes simplex virus type 1 (HSV-1) is composed of double-stranded DNA withamolecular weight of approximately 100 x
106 (10). This is sufficient genetic material to
code for 80to 100 proteins. One of these
pro-teins isaDNA polymerase that is essential for
thereplication of viral DNA(1).
Phosphonoacetic acid (PAA) isan
antiherpes-virus drug which acts by inhibiting viral DNA synthesis (14). It is aneffective inhibitor of the
virus-induced DNA polymerase (13) by virtue of its interaction with theenzymeattheinorganic
PPi-binding
site(12). Numerous mutantsofher-pes simplex virus whosereplication, DNA
syn-thesis, and DNA polymerase activity are resis-tanttoPAAhavebeen isolated (2, 7-9). None of thesemutantsisolated solely for PAA resistance
(PAAD)
has been reported to be significantlytemperature sensitive (ts). However, the DNA
synthesis-deficient ts mutant of HSV-1, tsD9, which codes for the synthesis ofa DNA
poly-merase that is thermolabile in vivo (1), was
subsequently found to exhibit PAA resistance (9, 15). Revertants of this mutant that were
isolated at 39°C in the absence of PAA were
showntohavereverted bothof the phenotypes (9, 15), suggesting that the PAA-resistant and temperature-sensitive properties of tsD9can be
attributedtoasingle mutation.
Thiscommunicationreports theisolation and preliminary characterization of a
temperature-sensitive PAA-resistant HSV-1 mutant that we
designated as PAAirtsl.
PAA1rtsl,
at 39.50C, showed adefectin viral DNA synthesis and in the shutdown of cellular DNA synthesis. We also specifically examined whether thePAA-resistant and temperature-sensitive phenotypic properties were due to a single or a double mutation.
MATERLALSANDMETHODS
Cellsand viruses. BHK-21/C13 cells wereobtained
from R. Sheinin (University of Toronto, Toronto, Ontario, Canada). CV-1 and LMTK- cells were a
generous giftfrom R. G. Hughes, Jr. (Roswell Park
Memorial Institute, Buffalo, N.Y.). Serially
propagat-ed stock cultures were routinely grown in a-MEM
tissue culture medium (19) supplemented with 10%
fetal calfserum(FCS).
HSV-1 strain KOS 1.1 was obtained from R. G.
Hughes, Jr., and was used as the wild-type virus in this
study. Thets mutants ts478and ts833werefrom the
same source.Thepermissive and nonpermissive
tem-peratureswere34WC and 39.5°C, respectively.
Howev-er,38.5°C is also nonpermissivetoall thets mutants
used in this study. Bu-El, Bu-B4, Bu-E2, and
Bu-C,
arethymidine kinase-deficient (TK-)mutantsisolated
in this laboratory from the HSV-1 KOS strain
(ob-tained from S.Bacchetti,McMasterUniversity,
Ham-ilton, Ontario, Canada) by growth in 30 1.g of
5-bromodeoxyuridine (Sigma Chemical Co., St. Louis,
Mo.) per mlaspreviously described (5). Thets mutant
tsD9was agenerousgift fromP. A. Schaffer (Sidney
FarberCancerInstitute, Boston, Mass.). Virus stocks
were grown onCV-1 cells at a multiplicity ofinfection
(MOI) of0.01PFU per cell.
Mutagenesisand isolation of mutants.Monolayers of
BHK-21/C13 cells in60-mmdishes wereinfected with
wild-type HSV-1 strain KOS 1.1 atamultiplicity of1
PFUpercell. Aftertheadsorptionperiod, the inocula
wereaspirated andtheinfected cells werewashed with
phosphate-buffered saline (PBS).Toeach culturedish,
a-MEMcontaining
10%o
FCSwasadded for aperiod of4h. Infected cells werethen washed with PBS and
givena1-hpulse of
N-methyl-N'-nitro-N-nitrosoguan-20
on November 10, 2019 by guest
http://jvi.asm.org/
PHOSPHONOACETIC
idine (10 rig/ml; Aldrich Chemical Co., Milwaukee,
Wis.) in a-MEMcontaining10oFCS. After the
expo-sure to nitrosoguanidine, the infected cells were
washed twice moreand fed withoa-MEM
supplement-ed with 10%oFCS forafurther incubation of 20 hat
34°C. The resultinglysateswerereplatedonBHK-21/
C13 cells in the presence of 100 ,ug of PAA(disodium
salt, from Abbott Laboratories, North Chicago, Ill.)
perml. Plaques were picked and cloned twice in the
absence of the drug. Each PAA-resistant isolate was
screenedfor temperaturesensitivity by measuring the
plaquing efficiency at 34°C and 39.5°C. Any clone
which exhibited at least a104-foldreduction in
plaqu-ing efficiency when assayed at the nonpermissive
temperature was acceptedas ats mutant.
Viral DNA synthesis. To determine whether
PAA,rtsl
was capable ofsynthesizing viral DNA atthe nonpermissivetemperature, confluent monolayers
of CV-1 cells in 60-mm disheswerewashed with PBS
and then mock infected or infected with wild-type
virus or
PAA,rtsl
at anMOI of 5. Two milliliters ofa-MEMcontaining 2% FCSwasaddedtoeachdishfor 6
h.This mediumwasthenremoved,and thecellswere
washed with PBS. Cells were refed with 5 ml of
a-MEMplus 2% FCS and 2,uCiof
[methyl-3H]thymidine
(64.0Ci/mmol, NewEnglandNuclear, Boston,Mass.)
perml.After 18h,theinfected cellswerescrapedinto
themedium andpelletedby low-speedcentrifugation.
Each batch ofcells was washedwith 10 ml of TNE
buffer (0.01 MTris-chloride,pH 7.4, 0.1 MNaCl,and
0.001 MEDTA)(16) andrepelleted. Thesupernatants
weredecanted; then thetubesweredrained and frozen
at-70°C.Afterthawing, the cellswereresuspendedin
2.0 ml of TNEbuffer and incubatedat room
tempera-ture for 10 min in the presence of 0.18 ml of 109o
Nonidet P-40 and 0.07 ml of 109o sodium dodecyl
sulfate. Self-digested nuclease-free pronase
(Calbio-chem-Behring, LaJolla, Calif.) wasadded to afinal
concentration of 50 ,ug/mlfora30-minincubationat
37°C.One milliliter of each cellextractwasaddedto4
mlof CsCl dissolved in TNEbuffer,and therefractive
indexwasadjustedto1.4001to2.Samples(5ml)were
loaded into cellulose nitrate tubesforcentrifugationat
25°C inan SW50.1 rotor at25,000rpmfor 60 h ina
Beckman L5-50centrifuge.The tubeswerepunctured
from the bottom, and8-dropfractionswerecollected.
Therefractive indices of selectedfractionswere
mea-sured with a Bausch & Lomb refractometer. The
remaining samples were precipitated with cold 5%
trichloroacetic acid and added onto Whatman GF/A
glassfiber filters. Filterswerewashed threetimes with
cold5% trichloroacetic acid andtwotimes with cold
95% ethanol beforetheywereovendried.
Acid-precip-itable radioactivity was determined by scintillation
counting.
DNApolymerase assay.Theinvitro thermolability
anddrugsensitivity of the HSV-1 DNA polymerase
weretestedbyexaminingthepropertiesofcrude
en-zymeextracts. Approximately 5 x 107 BHK-21/C13
cells grown in roller bottleswerewashed with PBS and
mockinfectedorinfected with wild-type virusorthe
putativeDNApolymerasemutantatamultiplicity of
10 PFUper cell. Infectedcellswere incubated at 34°C for9 h.Cellswerewashed twice with coldPBS; they
were then suspended in 0.75-ml volumes of 10 mM
Tris-chloride (pH 7.5)-150 mM KCl-0.5 mM
dithio-threitol andsonicated with aBransonprobeSonifier
(model S125) for 10 s, at which time cell rupture was complete. An equal volume of 3.4 M KCl, 10 mM EDTA, and 1 mg of bovine serum albumin per ml was
added to each extract, and the extracts wereincubated
at0°C for 20 min. MgCl2 was added to a final
concen-tration of 3 mM, and the preparationswereincubated
at room temperature for 30 min. The extracts were
centrifugedat30,000 x gfor 15 min in aSorvall SS/34
rotor, and the supernatants were dialyzed for 14 h
against 1 liter of 10 mM Tris-chloride, pH 7.5, and 1
mM
P-mercaptoethanol.
These crude enzyme prepara-tions were frozen and thawed once prior to use. Forenzymethermolability assays, samples of 25 ,ul were
incubated at 45°C for various periods of time and then added to reaction mixtures (total volume, 100 ,ul) for determination of the viral DNA polymerase activity. These contained 100 mM Tris-chloride (pH 8.0), 0.5
mMdithiothreitol, 100 mM (NH4)2SO4, 2mM MgCl2,
0.45mgof bovine serum albumin per ml, 10.25 ,ug of
pancreatic DNase-activated calf thymus DNA, 0.1 mMeach ofdATP, dCTP, dGTP, and TTP, and 0.5
,uCi of [methyl-3H]thymidine-5'-triphosphate (44 Ci/
mmol, Amersham Corp.). Mixtures were incubated for
30 min at 38.5°C. In the case of a drug resistance
assay,different concentrations of PAA were included
andtheincubation was at 34WC for 30 min. Samples of
50 ,ul were spotted onto Whatman 3MM filter paper
disks, washed once with cold 5% trichloroacetic acid plus 1% sodium pyrophosphate, twice with cold 5%
trichloroacetic acid, twice with cold95% ethanol, and
oncewithdiethyl ether, and then dried. Radioactivity
was measured by scintillation counting. The counts
from the mock-infected system for each assay were subtracted from those of the wild-type or the mutant virus system.
Genetic crosses and isolation of recombinants. A
number of TK-PAAS mutants were crossed with PAAlrts1. BHK-21/C13 cells were infected with 5 x
10'
PFUof each parental virus (total MOI, 10 PFU percell) in a-MEM with 5% FCS. Newly titrated virus
wasused for each cross to ensure an equal MOI for
each parentalstrain. After a 2-h adsorption period, the
unadsorbed virus was removed, the infected cells were
washed with PBS, and a-MEM containing 5% FCS
and a 1/5 dilution of anti-HSV serum (supplied by
FlowLaboratories,Inc.,Rockville, Md.)wasaddedto
each culture dish. After 1 h, the infected cells were
washed once more, refed with a-MEMplus5%FCS,
and allowedtoincubateat34°Cfor 20 h
postadsorp-tion. At thistime,theresulting lysateswereharvested
andreplatedonBHK-21/C13cellsunder nonselective
conditions (i.e., in the absence of PAA and
5-bromodeoxyuridine). Plaques were picked and
re-cloned twice under these nonselective conditions.
Individualrecombinantsweresubsequentlyidentified
by their ability to grow on BHK-21/C13 cells in the
presence andabsenceof 100jigof PAA per mlat34°C,
on LMTK- cells in the presence of 30 ,ug of
5-bromodeoxyuridinepermlat34°C,andonCV-1 cells at34°Cand39.5°C.
Complementationtests. Complementation tests
be-tweenpairsofts mutants wereperformed bya
modifi-cationof theprocedure of Schafferetal.(16).
Approxi-mately 2 x 105 CV-1 cells were infected at the
nonpermissive temperature with thets mutantseither
singly at anMOIof2.5PFU per cell orinpairsat a
total MOI of 5 PFU per cell. The inocula were
21
VOL.42,1982
on November 10, 2019 by guest
http://jvi.asm.org/
aspirated after adsorption,and the cellswerewashed
withPBS. Each infected culturewasfed with 1.0 mlof
a-MEMplus 2%FCS, and thetestplateswere
incu-bated for 20 h at 39.5°C. Cells were collected and
subjected to three cycles of freezing and thawing.
LysateswereassayedonCV-1 cellsat34°C.
Comple-mentation indiceswerecalculatedby dividingthetiter
ofvirus obtained from the mixedinfectionbythesum
of the titers fromthesingleinfections. If this valuefor
any cross wasgreaterthanorequalto2,then
comple-mentationwasdeemedtohave occurred.
RESULTS
Isolation of mutants. The mutagenesis and selection for PAA-resistant clones resulted in the isolation of 43 PAAr mutants of HSV-1. When tested for temperature sensitivity, one
mutant, bearing the designation
PAA'rtsl,
wasfoundtohaveaplaquingefficiencyof 2.9 x
10'
at 39.5°C and 3.0 x
10-5
at 38.5°C relative to 34°C inBHK-21/C13
cells. In comparison, thewild-type
virus had aplaquing efficiency of 2.3 x 10- at 39.5°C and 7.9 x 10-1 at 38.50C relative to 34°C. The plaquing efficiency ofPAA,rtsl
in CV-1 cells is the same as that inBHK cells. The PAA resistance of PAAirtsl
wastestedby plaqueassayand is shown inFig. 1.
Replication efficiency of
PAA,'tsl.
Theisola--j
z
0
I~-U.
20 40 60 80 100
pg PAA/ml
FIG. 1. Inhibition of HSV-1 by PAA. The virus stocksweregrownat34°Cintheabsenceof PAA and
wereplaque assayed with the drug included in theagar
overlayer. The viruses werewildtype(0), PAA1rtsl
(0),and thets+ revertantof PAAlrtsl, R7-2(A).
tionof a PAA-resistant mutant that was foundto betemperaturesensitive presentedthe possibili-ty that this mutant codes for a thermolabile DNA polymerase. To begin thecharacterization of this mutant, PAAirtsl, experiments were performeddealing withtheefficiency of replica-tion ofthe mutant either in the presence of 100 ,ug of PAA per mlor at39.5°C. Theseproperties of the mutant werecompared with thoseofthe wild-type strain from which the mutant was derived.
Triplicate monolayers of 105 CV-1 cells were infected with wild-type HSV-1 KOS 1.1 or
PAA,rtsl
at a multiplicity of 1 PFU per cell. After an adsorption period of 1 h, the infected cellswere washed with PBS and incubated for 20 h at 34°C in a-MEM plus 5% FCS that contained or lacked 100 ,ug of PAA per ml. Progenyvirionswereharvested, and titers were determined on CV-1 cells by plaque assay at 34°C in the absence ofPAA. The yields from eachsetoftriplicatelysates wereaveraged. As shown in Table 1,thewild-type andthe mutant viruses yielded approximatelythe same titer of progenyin the absence ofPAA. However, in the presenceofthe drug, whereas the production of wild-type virus during growth was severely in-hibited (0.051% of the control), the titer of mutant viruswasreducedonly threefold.A similar experimentwas performedto com-pare the growth of wild-type KOS 1.1 and
PAA,rtsl
at34°C and 39.5°C in the absence of PAA. At 34°C, both viruses had similar yields (Table 2); however,at39.5°C,the production of wild-type virus was only slightly inhibited, whereas the yield ofPAAirtsl
at 39.50C was only 0.048% of thatat340C.Viral and celular DNA synthesis. To deter-mine whether the mutation(s) in
PAAirtsl
af-fected the ability of the virustosynthesize viral and cellular DNA in vivo at 34°C and 39.5°C, newly madeDNAin infected cells waslabeled with tritiated thymidine and analyzed by CsCl gradient equilibrium centrifugation. Extracts of mock-infected CV-1 cells (Fig. 2A and B) exhib-ited only one peak at both340Cand 39.5°C. The gradient density at thatpointwas 1.690 g/cm3, corresponding tocellularDNA. Wild-type- andPAAlitsl-infected
cell extracts showed two peaks at both temperatures (Fig. 2C-F), the secondpeak occurringwherethe gradient densi-ty was 1.715g/cm3,
representingviral DNA.As shown in Fig. 2C and 2D, the amount of viralDNA synthesized at39.5°Cwas 2.1 times thatproducedat340C inthewild-typeKOS 1.1-infectedcells. The meanincreaseobtainedinsix separate experiments was 1.2. In comparison, the amount of viral DNA synthesized in the
PAA,'tsl-infected
cellsat39.5°Cwasonly5.6% ofthelevelsynthesizedat340C (Fig.2Eand 2F).on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.492.44.240.374.628.2]PHOSPHONOACETIC ACID-RESISTANT HSV-1 23
TABLE 1. Yield ofwild-type virus and PAA' tsl after growth in the presenceorabsence of 100 ,ug ofPAA/
Mla
Viruls Virus titerb±SD(PFU/ml) +PAAtiter/
-PAA +PAA -PAA titer
Wild type 8.4 ±5.1 x 105 4.3 ±2.8 x 102 5.1 x 10-4
pAArtsl 8.7± 1.4 x 105 2.7± 0.9x 105 3.1 x 10-1
aExperiments wereperformed at 34°C.
bValues represent the averageof triplicate samples.
In three other experiments this value ranged from 0to 10%o, with a mean of4.9% (datanot
shown). This indicates that PAAirtsl is partially deficient in viral DNA synthesis atthe
nonper-missivetemperature.
Withregardtocellular DNAsynthesis,allthe viruseswere very efficient in inhibiting cellular
DNA synthesisat34°C(Fig. 2), asobservedby
other workers(1). At 39.5°C,however, the shut-down of cellular DNA synthesiswas much less pronounced in the
PAA,rtsl-infected
cells thanithad been in these cellsat34°C. Theamountof cellular DNAproduced in the
PAA,rtsl-infected
cellsat39.5°Cwas4.5 times thatsynthesizedat 34°C, adifference significantly greater than the1.2-fold increase observed in wild-type KOS 1.1-infected cells(Fig. 2C-F). The ratio of cellu-lar DNA synthesized at 39.5°C relative to that synthesized at 34°C in the mock-infected cells
was 1.8(Fig. 2A and 2B). Themeanscalculated from several experiments for the cellularDNA synthesized at 39.5°C relative to that synthe-sizedat 34°C in the
PAA,rtsl-,
KOS 1.1-, and mock-infected cellswere3.4(four experiments),1.1 (six experiments), and 1.9 (four experi-ments), respectively. The difference in the meansof the cellularDNAratios between wild-type KOS 1.1 and
PAAi'tsl
was found to be significantataprobability of 0.01 (as judged bya t test). These results suggest that PAAirtsl istemperaturesensitive for the shutdown of
cellu-lar DNAsynthesis. Therefore, PAAirtsl is de-fective in viral DNAsynthesis andinthe shutoff of host DNA synthesis at the nonpermissive temperature.
DNA polymerase assays. The finding that PAAirtsl-infected CV-1 cellswere temperature
sensitive for the synthesis of viral DNAat the nonpermissive temperature, coupled with the fact that
PAA,rtsl
is resistant to PAA, a drugthat bindstoHSV-1-induced DNApolymerase, providedastrongindication that the DNA
poly-merase was indeed affected in the mutant. To examine this possibility, crudeenzyme extracts prepared from wildtype-andPAArtsl-infected cells were heated at45°C for various times and then incubated at 38.5°C for 30 min in the
presence of the DNA polymerization reaction
mixture. The thermal inactivation of the virus-induced DNA polymerase is shown in Fig. 3. After20minat45°C,morethan 70% of the wild-type enzyme's polymerizing activity remained. On the other hand, the DNA polymerase
en-coded by
PAA,rtsl
was rapidly inactivated at45°C. Approximately 65% of this crude
en-zyme'sactivitywasdestroyed after 4minatthe elevatedtemperature,and only 16% of the poly-merizingactivity remained after 20minat45°C, thus showing that the viral DNA polymerase activity of
PAAIrtsl-infected
cells was moresensitivetothermalinactivation than that of the wild-type-infectedcells.
As shown in Fig. 4, similar DNApolymerase
assaysinthepresenceofPAA,at34°C, revealed
that
PAA,rtsl
codes foraDNApolymerase thatis resistant to PAA when compared with the wild-type enzymeinvitro.
*+
revertants of PAA4rtsl. The replicationefficiency, DNA synthesis, and DNA
polymer-ase experiments indicated that the mutant
PAA1rtsl
wasindeedsignificantly PAA resistantand temperature sensitive. In apreliminary
at-tempt to determine whether the two mutant
phenotypes of PAAirtsl were due to a single
TABLE 2. Yield ofwild-type virus and PAAr tsl after growthateither 34 or38.5oCa
Virustiter't SD(PFU/ml) 38.5°Ctiter/
Virus 34°C 38.50C 340C titer
Wild type 1.4 +0.1 x 106 1.1 ± 0.1 x 106 7.9x 10-1
PAAr tsl 1.2 +0.2 x 106 5.8 t 1.2 x 102 4.8x10-4
aExperimentswereperformed in the absence of PAA.
bValues represent the averages of triplicate samples.
VOL.42,1982
on November 10, 2019 by guest
http://jvi.asm.org/
3
a
a
004
00
z
z
*5
a.I
a
0 a,
0
z
2
a z4
10 20 30 40 50
FRACTION NUMBER
FRACTION NUMBER
mutation ortwo separate mutations, ts+
rever-tantswereisolatedby plating individual
plaque-purified lysates ofPAAirtsl at the
nonpermis-sive temperature. The two independently
derivedts+revertantsweresubsequently tested
for PAA sensitivity and plaquing efficiency at
39.5°C. Each of the revertantclones (R7-2 and Ri-i) was capable of forming plaques at the A
o~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
z3~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
J.VIROL.
A
E
1.80<
_-1.700
z
a
I _
1.60Z !
9
°N
2 I
a.
U
I-4
0. 0M
mz
z
z
i O i
O)
0z
4 0<
-I0
o0z
oC-E D-.
60
on November 10, 2019 by guest
http://jvi.asm.org/
PHOSPHONOACETIC
0
a.
a
K
£
zW
0a
z
2
a
i
0 z
a
E D,.
I.
D4A
a a 4
a.
a
6-4
0
U
0
IL
z £ z z FS
E
-1
[image:6.492.56.447.60.342.2]FRACTION NUMBER FRACTION NUMBER
FIG. 2. Separation of viral and cellular DNA synthesized at 34°C and 39.5°C by CsCl equilibrium
centrifugation.Theexperimentswere(A) mockinfected,34°C; (B)mockinfected,39.5°C; (C)wild typeinfected, 34°C;(D) wild typeinfected,39.5°C; (E) PAAlrtsl infected,34°C; (F)PAAlrtsl infected, 39.5°C.
nonpermissive temperature with an efficiency thatwas similar to that ofwild-type virus (the
plaquing
efficiencies ofwild-type KOS 1.1, R7-2,andRi-i were 2.3 x10-1, 6.4 x 10-2, and 4.6 x 10-2, respectively). However, these ts+ re-vertants werestill almost as resistant to PAA asPAA,rtsl
(Fig. 1). These results seemedtoindi-cate that the two mutant phenotypes of PAAirtsl were caused by two separate muta-tions. However, the single-mutation model couldnotbe excludedbythese data, since true reversion mutation (return of the primary base alterationtothe base sequence ofthe wildtype) is
probably
rarecompared
with second-site re-version(suppression)
mutations. Changes of the latter kind neednot apriori
result inreversionof bothcharacteristics.
Segregation of the
PAA,
and the tsl muta-tions. The ts+ revertant studies ofPAAirtsl
suggested the presence of two separate muta-tions. To examine this question further, we crossed
PAA,rtsl
with variousPAASts+TK-strains.PAArTK-and
PAASTK+
recombinants wereidentified, and the degreeoflinkage ofthePAAIrtsl
putative mutations was examined by determining the frequency ofPAAirtsl+
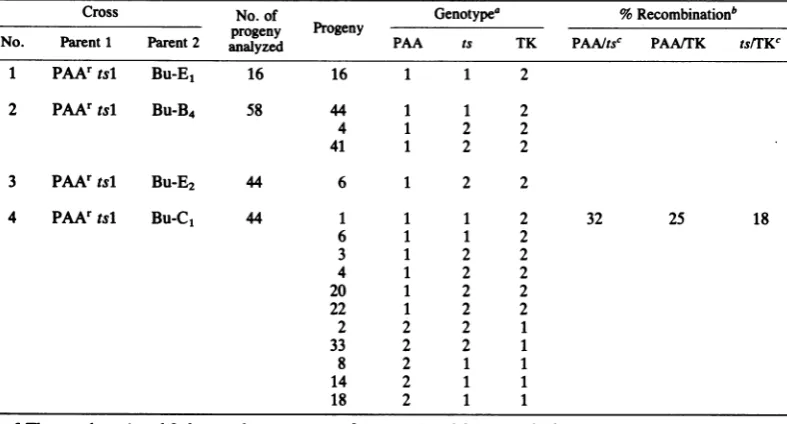
and PAAistsl recombinants among thePAA/TK re-combinants. As shown in Table3,infoursepa-ratecrosses,atotalof16PAA/TK recombinants were isolated, and 10 of these isolates had a crossoverbetween thePAA1r and the tsl muta-tions. Thus, the mutant
PAAirtsl
clearly con-tains twodistinct mutations.In cross 4
(PAAIrtsl
xBu-Cj),
where we obtained 11PAA/TK
recombinants out of 44 plaques analyzed, giving a recombination fre-quency of25%,
the PAAr/tsl andtsl/Bu-C,
respective
recombination frequencies were 32 and 18. The PAA1r and tsl mutations were clearly loosely linked and most likely to be located intwo separate genes.Similarly, the tsl andBu-C,
mutations were located in two sepa-rate genes. The maximum recombination fre-quencybetweentwomutations within the HSV-1 TK gene was reported to be about 2% (18). The aboverecombination data suggesta proba-blelinkagemapofthe tsl,PAA1r
andBu-C,
loci as shownin the diagrambelow.tsl 18%
Bu-C,
25%PAA,r
1( 32%
Complementation studies. Valuable informa-tion on the tsl mutainforma-tion present in PAAirtsl maybe obtainedfrom
complementation
studiesVOL.42,1982 25
on November 10, 2019 by guest
http://jvi.asm.org/
>_ ~~0
<60 X
'060
0
0L 40
z
30-
20-10
2 4 6 10 12 14 16 13 20
HEAT INACTIVATION (min)
FIG. 3. Effect of temperature on the
HSV-1-in-duced DNA polymerase activity inextractsof infected
BHK-21/C13 cells. Extracts of cells infected with
wild-type virus (0) orPAAi'tsl (0) orofmock-infected
cellswereheatedat450Cfor the indicated times and
were assayed for viral DNA polymerase activity as
described in Materials and Methods. 3H counts
ob-tainedfrom the mock-infectedsystem(50to120 cpm)
were subtracted from those of the wild-type- and
PAAIrtsl-infectedsystems.Resultsareexpressed
rel-ativetothepolymeraseactivitiesfoundinthe
unheat-edextracts, whichgave2,500to3,200cpm.
with otherknowntsmutants. Studieshave been performed with tsD9, ts833,andts478,and with other ts mutants isolated in our laboratory. Complementationindices obtained from partof these studiesareshown in Table 4. Indexvalues listedfor eachcross weretheaveragesof dupli-catesamples.
Allof the tsmutantstested werefoundtobe complementary to one another, indicating that the mutations in each of these mutants are
located in differentgenesof the HSV-1genome.
DISCUSSION
Previous studies(1, 9, 15) reported the isola-tion of a DNA synthesis-deficient mutant of HSV-1, tsD9,whichwassubsequentlyfoundto
exhibit PAA resistance and code fora
thermola-bile DNApolymeraseinvivo.Thisstudy report-ed the isolation of a novel PAA-resistant and temperature-sensitive mutant of HSV-1 (PAAirtsl), which wasisolated asbeing PAAF.
Reversion studies and recombination analyses
of
PAA,'tsl
indicated that thePAA1F mutationand tsl mutation were two distinct mutations located on separate genes. The tsl mutation mapped at approximately 18% recombination units to the left of the TK- mutation, Bu-C1,
whereas the PAA1r mutationmappedat approxi-mately 25% recombination units to the right of Bu-C1. The probablelocationofthe tslmutation with respect to the TK and PAA markers is tsl-TK-PAA.
ViralDNAsynthesisin the
PAAlrtsl-infected
cells was substantially reduced at 39.5°C, the nonpermissive temperature. The observation that viral DNA synthesis was nottotally blocked at39.5°Cindicates that PAAirtsl is not quite as temperature sensitive for DNA synthesis as tsD9, tsC4, and tsC7 (1). The in vivo DNA synthesisdefect and the increasedthermal sensi-tivity of the PAAirtsl-inducedDNApolymerase demonstratedin vitro are consistent witha no-tion thatthe PAA1r mutation is responsible for the production of a viral PAA-resistant DNA polymerasethat isthermolabile at39.5°C.These results support the studies withtsD9 (1, 9, 15), which showed that a PAA-resistant mutation is located inthestructural genefor the viralDNA polymerase. Thestructurallyaltered viral DNA polymerasewas stillpartly functionalthoughat 39.5°C, sinceminimal levels of viralDNA were synthesizedin
PAA,rtsl-infected
cells.PAAirtsl was also defective in the shutoff of
110 O
100
0
:t
'S
5S.
02 13
10 20 3S sU
Jug PAA/ml
FIG. 4. Effect ofPAAon theHSV-1-inducedDNA
polymerase activity in extracts of infected BHK-21/
C13 cells. Cellswereinfected withwild-type virus(0)
orPAAirtsl (0)or weremock infected for the
prepa-ration of extracts to determine the in vitroactivityof
the viralDNApolymerase in the presence of PAA. 3H
countsobtained from the mock-infected system (50to
120cpm) weresubtractedfrom those of the
wild-type-and
PAA,rtsl-infected
systems. Results are expressedrelative to the polymerase activities found in the
absence of PAA, which gave2,500 to 3,200 cpm.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.492.49.244.56.265.2] [image:7.492.255.446.362.566.2]PHOSPHONOACETIC ACID-RESISTANT HSV-1 MUTANT
TABLE 3. Three-factor crosses between PAAr tsl and TK- PAAS mutants
Cross No. of Genotype' %Recombination'
progeny Progeny
No. Parent1 Parent 2 any PAA ts TK PAA/tsc PAA/TK ts/rKc
1 PAArtsl Bu-E1 16 16 1 1 2
2 PAArtsl Bu-B4 58 44 1 1 2
4 1 2 2
41 1 2 2
3 PAAr tsl Bu-E2 44 6 1 2 2
4 PAAr tsl
Bu-C,
44 1 1 1 2 32 25 186 1 1 2
3 1 2 2
4 1 2 2
20 1 2 2
22 1 2 2
2 2 2 1
33 2 2 1
8 2 1 1
14 2 1 1
18 2 1 1
a Thenumbers 1 and 2 denote the genotypes of parents 1 and 2,respectively.
bRecombinationfrequencieswere calculated from the data obtained from cross 4only.
cOnly one-halfof the recombinants between the
PAAT/tsl
andtsl/TK
loci were detected in ouranalysis. Therefore,inthe calculations of the recombination frequencies between these loci, the numbers of recombinantsdetectedweremultipliedby afactor of 2.
cellular DNA synthesis at the restrictive
tem-perature, as evidenced by the4.5-fold increase
in cellular DNA synthesized in the
PAA,rtsl-infectedcellsat39.5°C compared with thatsyn-thesized at34°C. Wild-type-infectedand mock-infected cells exhibited onlya1.2-fold anda 1.8-foldincrease, respectively. The contributions of
the
PAA,r
mutation and the tsl mutation inaffecting the levels of viral and cellular DNA synthesizedat34°C and 39.5°C have been
exam-ined with the various
PAA,Stsl
and PAAirtsl+ recombinants (see Table 3) andPAA,rtsl+
re-vertants.PAA,rtsl+recombinants
are not significantlydefective in viral DNAsynthesis and growthat
39.50C. ThePAArtsl+ revertants(R7-2 and
Rl-1) exhibit intermediate to wild-type levels of viral DNA production at 39.50C. PAAistsl
re-combinants also exhibit wild-type viral DNA synthesisat39.50C.However, they haveavery
lowplaquingefficiency at39.5°C, ranging from
3.0 x 10-6 to4.1 x 10-6(J. I.Daksis and V.L.
Chan, unpublished data). The observation that the
PAA,rtsl
mutant is defective in viral DNA synthesisat39.5°C, whereas the PAAirtsl+ andPAA,stsl
recombinants arenotsignificantlyde-fective, suggests the possibility thatthePAAr and tsl gene productsmay be components ofa
DNA polymerase enzyme. Alternatively, they
maybeseparateenzymesexistingas acomplex
thatis involved in DNAreplication. The altered
PAA1r and tsl polypeptides perhaps form a
thermolabile complex.However,when the
com-plex consists of either the PAA1r and tsl+orthe
PAA'S
andtslgeneproducts, it isnotdefective in viral DNAsynthesis at 39.5°C. Presumably, the defective interaction of the altered viral PAA1r DNA polymerase with the altered tslgeneproduct was responsible for the observed thermolability of the PAAirtsl-induced DNA polymerase (see Fig. 3). Determination of the thermolability of the DNA polymerases ofthe
PAA,stsl
andPAArts1 + recombinants, aswellas the
PAA,rtsl+
revertants, should provide valuableinformation.At themoment,wedonothave an explanation for the defect in plaquing
TABLE 4. Complementation betweentsmutants
Complementation index' frommixed tsmutant infection with tsmutant
PAAirtsl tsl-8 tsD9 ts199 ts833
PAA,rts1
1.2 13.3 3.9 24.1tsl-8 4.6 26.2 27.5
tsD9 4.5 4.0
tsl99 8.1
ts833
aComplementation index = (Ax
B)39.5C/A39.5-c
+B39.s-C.
assayed at34°C.Values greater than 2.0 wereconsidered to bepositive.
VOL.42, 1982 27
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.492.255.449.562.652.2]efficiency atthe nonpermissivetemperature for the PAA1'tsl recombinants, which are not de-fective in viral DNA synthesis, butaredefective in the shutdown of host DNA synthesis at 39.50C.
Animportantnewobservationhasbeen made inthe presentstudies.Itis thatthe
PAAirtsl
and PAAistsl mutants werealldefective in the shut-off of cellularDNAsynthesisat39.5°C(only the results ofPAAirtsl
are presented here). These observations imply the involvement of the tsl geneproduct intheshutoff of hostDNA synthe-sis. The intriguing observation thatPAA,rtsl+
recombinants were also defective in the shut-down of cellular DNA synthesis implies the involvement of the PAA1r gene product in the shutdown of host DNA synthesis. We propose that the shutoff of cellular DNA synthesis by HSV-1requiresaninteraction between the viral DNA polymerase and the tsl+ polypeptide. Modification of the structure of either gene productmaygiverisetoimproper interaction in the complex,leadingtoadefect in theshutoff of cellular DNA synthesis. Augmentation of this effectmay occurwhenbothproteins
are mutat-ed. One of thePAAirtsl
+ revertants(R7-2)was normal in the shutoff of host DNA synthesis. Since all thePAAirtsl+
recombinants were de-fective in the shutoff of host DNAsynthesis, the above resultimplies that thetsl+revertantgene product could interact efficiently with thePAA1r geneproducttoallow normalshutoff ofcellular
DNA synthesis.
ts+
revertantsof the PAA1'tsl recombinants will beisolated and tested for theirability
toshutoff cellular DNAsynthesis. Itwill also be interesting to determine whether otherPAAF
HSV-1 mutants aredefective in the shut-down of host DNA synthesis at an elevated temperature.No tsHSV-1 mutantsdefective in the shutoff of cellular DNA
synthesis
havepreviously
been reported. Two ts viralDNAsynthesis-deficient mutantsof HSV-2 (ts9 and tsll) intwodifferentcomplementation
groups have beenreported
to be defective in the shutdown of host DNA synthesisatthe restrictivetemperature (6, 20).It was not clear whether the viral DNAsynthesis defect of thesetwoHSV-2mutantswas aresult ofthedefective shutoff of cellularDNA synthe-sisatthenonpermissive temperature. Our find-ingthat thePAA15tsl
andPAArtsl
+ recombi-nant HSV-1 mutants synthesizednormal levels of viralDNA at39.50C,
while stillbeing defec-tive in shutdown ofhost DNA synthesis, indi-catesthat theshutoffof cellularDNAsynthesis isnotessentialfor viralDNAreplication.Clear-ly,
PAA,rtsl
is invaluable in elucidating themechanismof inhibitionof
cellular
DNA synthe-sisby HSV-1.Previous fine
mapping
studies of two PAArmutantsof HSV-1 indicated that they may pos-sessmutations resident inseparate genes(4,11). More recently, however,
Chartrand
et al.(3)
presented evidence for the existenceof onlyone PAArgenein HSV-1 and HSV-2.Itwill, there-fore, be of interest to
physically
mapboth the PAA1r and the tsl mutations of PAAirtsl, by markerrescueexperiments.
As shownin Table 4,PAA,'tsl
(or thePAA1'tsl recombinant, tsl-8) wasfound to complement the ts DNA poly-merase mutants,tsD9and ts833(which is in the samecomplementation
group astsC4
[17]),
indi-cating'that
thetsmutations in thesemutants are locatedin differentgenesof the HSV-1genome. A more extensivecomplementation
study with otherts mutantsisolated from other laboratories would benecessary todetermine whether thetsl mutation of PAA1rtsl represents anovel com-plementation group.ACKNOWLEDGMENTS
We thank RoseSheininfor the BHK-21/C13 cells and for valuable criticism during the course of this work, L. Simino-vitch for interest andvaluable suggestions on thepreparation of thismanuscript,R.G.Hughes, Jr.,P. A.Schaffer,andS. Bacchettifor theirgiftsof cellsandvirus, ChristinaA.van't Hof fortheisolationof PAA-resistantHSV-1mutants,andS. Guttman for expert technicalassistance.
Thisworkwassupported bythe NationalCancerInstitute and theMedical Research Council of Canada. V.L.C. is a Research ScholaroftheNational Cancer Institute ofCanada.
LITERATURE CITED
1. Aron, G. M.,D.J.M.Purifoy,andP. A.Schaffer.1975. DNAsynthesisand DNApolymeraseactivity of herpes simplex virus type 1 temperature-sensitive mutants. J. Virol. 16:498-507.
2.Becker, Y., Y. Asher, Y. Cohen,E.Weinberg-Zahlerlng, andJ.Shlomal.1977.Phosphonoaceticacid-resistant mu-tantsofherpes simplexvirus: effect ofphosphonoacetic acidon virusreplication and in vitrodeoxyribonucleic
acid synthesis in isolated nuclei. Antimicrob. Agents Chemother. 11:919-922.
3. Chartrand, P., C. S.Crumpacker,P. A.Schaffer,and N. M. WDkie. 1980. Physical and genetic analysis ofthe herpes simplex virus DNA polymeraselocus. Virology 103:311-326.
4. Chartrand, P., N. D.Stow, M.C. Timbury,andN. M. Wilde. 1979. Physical mapping ofpaar mutations of herpes simplex virus type 1 and type 2 by intertypic markerrescue.J. Virol.31:265-276.
5.Dubbs,D.R.,andS.Kit.1964.Mutantstrain^sofherpes simplex deficient in thymidine kinase-inducing activity. Virology 22:493-502.
6.Halliburton, I.W.,andM.C.Timbury. 1976. Tempera-ture-sensitive mutants ofherpes simplex virus type 2: description of three new complementation groups and studiesontheinhibition of host cellDNAsynthesis. J. Gen. Virol. 30:207-221.
7. Hay, J.,andJ.H.Subak-Sharpe. 1976. Mutants ofherpes simplexvirus types1and2thatareresistantto phospho-noacetic acid induce alteredDNApolymeraseactivities in infectedcells. J. Gen. Virol. 31:145-148.
8. Hone",R.W.,andD. H. Watson.1977. Herpessimplex virus resistance andsensitivitytophosphonoaceticacid. J. Virol.21:584-600.
9. Jofre, J. T., P. A. Schaffer, and D. S. Parris. 1977. Genetics of resistancetophosphonoaceticacidin strain KOS ofherpessimplexvirus type1.J.Virol. 23:833-836.
on November 10, 2019 by guest
http://jvi.asm.org/
PHOSPHONOACETIC ACID-RESISTANT HSV-1 MUTANT
10. Kieff, E. F., S. L. Bachenhelmer, and B. Roizman. 1971. Size, composition andstructureof thedeoxyribonucleic acid of herpes simplex virus subtypes 1 and 2. J. Virol. 8:125-132.
11. Knipe,D. M., W. T.Ruyechan,and B. Roizman. 1979. Molecular genetics of herpes simplex virus. III. Fine mapping of a genetic locus determining resistance to phosphonoacetate bytwomethods ofmarker transfer. J. Virol.29:698-704.
12. Lelnbach,S.S.,J. M. Reno, L.F. Lee,A.F. Isbell, and J. A. Boezi.1976. Mechanism of phosphonoacetate inhibi-tion of herpesvirus-induced DNA polymerase. Biochem-istry 15:426-430.
13. Mao, J. C.-H.,E. E.Robishaw,andL. R.Overby.1975. Inhibition of DNA polymerase from herpes simplex virus-infected Wi-38 celis byphosphonoacetic acid. J. Virol. 15:1281-1283.
14. Overby, L. R., E. E. Robahaw, J. B. Schlekcher, A. Rueter,N. L.Shipkowitz,andJ. C.-H.Mao.1974. Inhibi-tion of herpes simplex virus replicaInhibi-tion by phosphonoace-tic acid. Antimicrob. Agents Chemother. 6:360-365. 15. Purlfoy,D. J.M.,andK.L.Powell.1977.Herpes simplex
virus DNA polymeraseasthesite ofphosphonoacetate
sensitivity: studies withtemperature-sensitivemutants.J. Virol. 24:470-477.
16. Schaffer,P. A., G. M. Aron, N.Biswal,andM. Benyesh-Melnlck. 1973.Temperature-sensitive mutantsof herpes simplex virus type 1: isolation, complementation and partial characterization. Virology 52:57-71.
17. Schaffer,P. A., V. C. Carter, and M. C.Timbury. 1978. Collaborativecomplementation study of temperature-sen-sitivemutants of herpes simplex virustypes1 and 2. J. Virol. 27:490-504.
18. Smnley,J.R.,M.J. Wagner,W.P.Summers,andW. C.
Summers. 1980. Genetic and physical evidence for the polarity of transcription of the thymidine kinasegeneof herpes simplex virus. Virology 102:83-93.
19. Stanners,C. P., G. L.Eliceiri,andH. Green.1971. Two
typesofribosome in mouse-hamster hybrid cells. Nature (London) New Biol. 230:52-54.
20. Subak-Sharpe,J. H., S. M.Brown, D.A.Ritchie, M. C. Tlmbury,J.C.M.Macnab, H. S. Marsden,andJ. Hay. 1974. Genetic and biochemical studies with herpesvirus. ColdSpring Harbor Symp. Quant. Biol. 39:717-730.
VOL.42, 1982