Copyright © 1977 AmericanSociety for Microbiology Printed inU.S.A.
Visna Virus RNA
Synthesis
M. BRAHIC,I* P. FILIPPI,2 R. VIGNE,2 AND A. T. HAASE'
University of California, San Francisco, Veterans Administration Hospital, San Francisco, California 94121'; and Laboratoire de Virologie,Faculti de Medecine, Secteur Nord, 13326 Marseille Cedex 3, France2
Received forpublication 20 May 1977
Visna is a
classical slow infection in which virus characteristically persists in
the face of the
host immune response. The agent of this disease belongs to the
retravirus group. The persistence
of infection and the slow spread of virus are
at
least
inpart a
consequence of restriction
of the expression of
virusgenetic
information
intissues
of aninfected animal
(A. T. Haase et al., Science195:175-177,
1977), but the point at which the virus life cycle is
interrupted
invivo
and
the mechanism of restriction are unknown. We have embarked on a
molecular
analysis of restriction,
focusing first
ontranscription. In this paper
wehave
established the levels of viral RNA synthesis under permissive conditions, as a
base line for subsequent studies
in vivo.We
show that (i) uninfected cells do
notcontain
RNA sequences
related to the visna virus genome, (ii) parental RNA is
rapidly transported to the nucleus of the infected cell, (iii) virus RNA
issynthe-sized
inthe nucleus and then
transported
tothe
cytoplasm, (iv) synthesis of
RNA
proceeds mostly exponentially
toreach levels of about
4,000 copies percell
at
the end of the
growth cycle, (v) nuclear and cytoplasmic RNA sediment
intwosize
classes, 35S
and 10-20S, (vi) viral mRNA has the
samepolarity
asgenome
RNA
and also sediments
in twosizeclasses of
35Sand
10-20S.Visna is a
classical
model of slow virus
infec-tion
of the central nervous system of
sheep.
The
cauaat6e
agent is
closely related
toRNA
tumorviruses!in
structureand
inthe
characteristic
transfer of genetic information from RNA
toDNA
during replication (9).
There
are twoparticularly
interesting
as-pects
of the
infection
caused
by visna
virus.In
the
infected
animal,
virus
persist
despite
th6
immune
response that it
evokes;
and the
dis-ease
process
evolves
slowly, usually
over ape-riod of
months
oryears.
Both of these features
of
invivo
infection
may
be
a
consequence of
alow
level of
expression of viral genetic
informa-tion:
wehave
recently shown that
proviral
DNA is
synthesized
in
cells
intissues
of
experi-mentally infected
animal.,
whereas
viral
pro-teins are
almost totally absent
in'the
same
cells. This conversion to
latency
in mostin-fected cells
invivoallows
the
virus
topersist;
the occasional
breakdown
of repression
in some
cells
leads to
edntinued&<
production
of
Small
amounts
of
virusand continued but slow
spread
of
infection.
The level
atwhich this host
restric-tion
is
effected has
notyet been
identified.
Itcould
bethat ofproviral DNA transcription
or alater
step inthe
process ofprotein
synthesis.
This
restriction
invivo is in marked contrastto
the situation
intissue
culture cells
(in
vitro).
In
lytic infection of permissive cells
invitro,
theviral life cycle is completed by 72
h/in
a
one-step
growth cycle, and about
100 to
500
infec-tious progeny
virusarereleased per cell (9).
In the present series
of investigations, we
have embarked
ona molecular
analysis of host
restriction
invivo. To
obtain base line
informa-tion
for
ourstudy of proviral DNA transcription
in vivo, we
decided to investigate the RNA
metabolism of visna
virus in in vitroacutely
infected
permissive cells inwhich the
viruspro-duction is
maximal.
Inthis
study
wehave
fol-lowed the fate of parental RNA,
determined the
time
courseof progeny RNA
synthesis, and
examined the
sizesof the
different intracellular
RNAs.
MATERIALS AND METHODS
Cells andvirus.Visnavirus strain 1514 wasused throughout this work.
Sheep choroid plexus cells were grown in vitro and usedatpassage 5 or 6.Thegrowth mediumwas Leibowitz medium15(L-15)supplementedwith 15% fetal calfserum.
Confluent cellmonolayersweresynchronously in-fectedasfollows. Thegrowthmediumwasremoved,
and the cell monolayer was washed with ice-cold phosphate-buffer saline(PBS) buffer. The cellswere then infected with 3 PFU ofvisna virus per cell. Adsorptionwascarriedoutfor
2.h
at40C.
At theend of thisperiod,the viral inoculum wasremoved and replaced by warm(370C)
L-15 medium containing 74on November 10, 2019 by guest
http://jvi.asm.org/
2% lamb serum.The infectedcellswerethen
incu-bated at 370C. The time at which the cells were
shiftedto37rC was takenastime zero of the infec-tion.
Inpreliminary experiments weshowed, by infec-tious-center assay, that under these conditions 90% or more of the cells areinfected and that the time course ofcytopathiceffect and virusproductionwas notaffected bytheadsorptionat40C.This tempera-turedid blockreplication, however. We foundthat,
at40C, virus particleswere adsorbed but that the early events in the lifecycle,inparticularsynthesis of viral DNA, did not begin until the cells were
shiftedto370C(B. Traynor, P. Ventura, A. Haase,
and M. Brahic, in preparation).
RNA extraction. For all the experiments de-scribed in thisarticle,theglasswarewasheatedat
1800Cfor at least 2 h,and the buffers weretreated with 0.2% diethylpyrocarbonate (DEP) for 1 h and
thenautoclaved.
Total cytoplasmicRNA was extracted as follows. The cells were trypsinized and washed once in PBS buffer containing 3mMMgCl2. The cellpellet was
suspended
(o2
x 107cells/ml)inice-cold reticulocytestandard buffer (10 mM NaCl-10 mM
Tris-hydro-chloride [pH7.41-1.5 mM MgCl2)containing 1% Non-idet P-40, transferred to a Dounce homogenizer, and
subjectedto 10strokes of a tight-fitting pestle. Nu-clei weresedimentedat 600xgfor 4 min at
40C.
The salt concentration of the supernatant wasadjustedto 200 mM NaCl, 10 mM EDTA, and 1% sodium
dodecyl sulfate (SDS). RNA was extracted by the phenol-chloroform technique described by Penman (15) with the following modification: chloroform was not used alone but wasmixed in a 1:1 ratio with redistilled phenol.
Nuclear RNA was extracted according to the pro-cedure of Penman (15), includingthe double deter-gentwashingof thenucleipellet.As for the
purifica-tion of cytoplasmic RNA, chloroform was used mixed ina 1:1 ratio withredistilledphenol.
For the experiments shown in Fig. 1 to 3, cyto-plasmic and nuclear RNAs prepared according to the Penman procedure were subjected to a DNase treat-mentbeforebeinghybridized: the RNA pellets were dissolved in 1 ml of a mixture of 10mMNaCl,5mM Tris-hydrochloride, pH 7.5, and 5 mM
MgCl2
and incubated for 30 min at370C
with 201Lg
of RNase-free pancreatic DNaseper ml. EDTA and SDS were thenadded to a final concentration of 10mM
and 0.5%, respectively, and the incubation mixture was extracted twice with phenol-chloroform.After twoethanol precipitations, the RNA pellets weredried under vacuum and dissolved in a small volume of 20 mM Tris-hydrochloride, pH 7.5-0.1%
DEP. The UVabsorbance (A) was measured at 260 and 280 nm. The
A26I/A2
8 ratio was alwaysgreater than 1.9.Contamination of nuclear RNA by cytoplasmic RNA was monitored in the following way. Sheep choroid plexus cellswere labeled with
[3H]uridine
for12h, and nuclear RNA was extracted. Analysis ofthis RNA(2 x 105cpm) on a sucrose gradient did not showa peak of 18S ribosomal RNA taken as a markerofcytoplasmicRNA (15). We concluded that thenuclear RNApreparations were essentially free
ofcytoplasmiccontaminants.
Thepossibility that viral RNA might leak during the purification of the nuclei was alsoinvestigated.
Nuclei were prepared from 48 h-infected cells, and the amount of viral RNA was measured, as de-scribed in thelegendofFig. 2,afterone, two,orfour washes with double detergent mixture. Since no differences in the amount of viral RNA were ob-served, we concluded that washing nuclei did not
resultintheleakageof nuclear viral RNA into the cytoplasmic fraction.
Polysome preparation. Themedium of the cells waschanged to growth medium 1 h before trypsini-zation, andcycloheximidewasadded atafinal con-centration of 1
;tg/ml
30min beforetrypsinizationto increase the size ofthepolysomes and to prevent"runoff'of ribosomes. Aftertrypsinization,the cells werewashedonce inPBScontaining 3 mMMgCl2.
The cell pellet was suspended (=108 cells/ml) in buffer A (300 mM NaCl-50 mMTris-hydrochloride
[pH7.51-8mMMgCl2-1 mM dithiothreitol-1 mg of heparin perml) containing 2%NonidetP-40,
trans-ferred to a Dounce homogenizer, and subjected to 10strokesof atight-fittingpestle. The nuclei were
sedimentedat 600 xgfor4min. The supernatant wasdilutedto 6mlwith buffer Aandlayeredontop of a discontinuousgradient consistingof 3 ml of 2 M sucroseand 3 ml of 0.5 M sucrose in buffer A. The
gradientwasspun for1.5h at40,000 rpm at40Cin anSW41rotor.
In some experiments, the polysome pellet was
resuspendedin0.5ml of buffer Aandlayeredontop of alinear 0.5to 1.5M sucrosegradientinbuffer A. Thegradientwascentrifuged at40Cfor 90 min at 30,000 rpm in an SW41 rotor. The sedimentation
profileof thepolysomeswasdeterminedby measur-ing the
A26
ofeach fraction collected from the gra-dient.Inother experiments, RNA was extracted from thepolysome pellet. Thepelletwassuspendedin1 mlof 100 mM NaCl-10 mMTris-hydrochloride (pH 7.5)-i mM EDTA-0.5% SDS and extracted with
phenol-chloroformasdescribed above.
From the hybridization kinetics
(Crtl,2)
oftotalcytoplasmic RNA and polysomal RNA prepared
from the same infectedcells, itwascalculated that
approximately 5% of the total cytoplasmic
virus-specificRNAwasrecovered in thepolysomalRNA. RNA-DNA hybridization. Viral RNA sequences weredetectedby hybridizationinsolution with
3H-labeledcomplementary DNA([3H]cDNA).The [3H]-cDNA was prepared by an endogenous reaction in whichpurified visna virus wasusedas a source of bothRNA-dependentDNApolymerase andviral
60-70SRNA. The reaction wascarriedoutinthe pres-ence of 100
;tg
ofactinomycin Dper ml. The exact conditions for the reaction have been described(8). Thespecificactivity of [3H]cDNA was 4.3x104cpm/ ng. Each [3H]cDNApreparation was characterized for both theextentofhybridizationwithpurified 60-70S viral RNA and theamount of[3H]cDNA neces-sary to protect100% of 32P-labeled 60-70S viral RNA againstdigestionby pancreatic RNase. In all cases,ntore
than90% of the [3H]cDNA hybridized to viral60-70S RNA with a
CAt112
of 1 x 10-2 to 2 x 10-2 mol-s/liter, and the [3H]cDNA was able to protecton November 10, 2019 by guest
http://jvi.asm.org/
76
BRAHIC ET AL.100% of the32P-labeled 60-70S viral RNA at a DNA/ RNA ratio of less than10.
To measure the amount of viral sequences present in agiven RNA preparation, the rate of hybridiza-tionof[3H]cDNA to this RNA preparation was
com-paredwiththe rate ofhybridizationof[3H]cDNAto
purified60-70S viral RNA (12). Hybridization reac-tions wereperformed in solution in a final volume of 20
j.l
per time point. The hybridization mixture contained 600 mM NaCl, 10 mM Tris-hydrochloride (pH 7.5), 3 mM EDTA, 0.1% SDS, 1 mg of carrier RNA per ml, 600 to 800 cpm per 20 'ulof [3H]cDNA, and varying amounts of RNA. Hybridization was performed at680C.The extent ofhybridizationwasassayed bythe resistance of[3H]cDNAtodigestion
with S nuclease (12). Hybridization kinetics were performed by varying either the time of hybridiza-tion ortheRNA concentrationandwereplottedasa function of the
COrt
parameter(RNAconcentration-time of hybridizationexpressed in mole-secondper liter). TheCrt values werenormalized to the
stan-dardsalt concentration (2). Since the value of
COrt
at which 50% of[3H]cDNA hybridizestoanRNAsam-ple
(Crtl/2)
isinverselyproportionaltotheconcentra-tionofviral RNAinthis sample, itwaspossibleto expressthe amount ofviral RNAasthenumber of 60-70S RNAequivalentspresent per cell using the following equation: number of 60-70S RNA
equiva-lent=
(Ctl2
of60-70SRNA)/(Crtl,2
of RNAsample)x (amount of RNA per cell)/(amountof RNA per virion).The amount of RNA percell was taken as 8 x 10-6 lug, and the amount of 60-70S RNA per virion wasassumed to be 1.7 x 10-11
Ag.
Sucrose gradient centrifugation. All gradients used for sizing viral RNA were 5 to 20% linear sucrosegradients inamixture of100mMNaCl, 10 mMTris-hydrochloride (pH 7.5), 1 mMEDTA,and 0.5% SDS. Centrifugations were performed in an SW41 rotor at18'C and 40,000 rpm. One hundred micrograms of RNA was used per gradient. The RNAsamplewasdisaggregated by heatingto100'C for30 sinasolution of100mMNaCl,10 mM
Tris-hydrochloride (pH 7.4), and 1 mM EDTA before
layeringontopof thegradient.
The sedimentation profiles of viral RNA were determined by hybridization with [3H]cDNA. Two hundred micrograms of carrier RNA was addedto each fraction of thegradient, and the RNA of each fractionwas
precipitated
by 2volumes of ethanol. After centrifugation, the RNA pellets were dried under vacuumand dissolvedin 30pul
ofasolution of 20 mMTris-hydrochloride (pH 7.5) and0.1% DEP. Tenmicroliters ofeach fractionwashybridizedwith [3H]cDNAin afinal volume of501.l
using the salt and temperature conditions described above. The extentofhybridizationwasdetermined afterdiges-tionwith
SI
nuclease. Hybridizationwasperformedinalarge RNAexcessand fora timeperiod
suffi-ciently short such thatthepercentageof[3H]cDNA hybridizedwasproportionaltotheconcentration of viral RNApresent.
RESULTS
Absence of
RNAsequences related
tovisna
virus genome in
uninfected sheep
cells. Before
studying visna virus
RNA metabolism, we
de-cided
to investigate the possibility thatunin-fected sheep cells might contain RNA
se-quences that can hybridize to viral
[3H]cDNA.
Such
sequenceshave been described
inchicken
cells and mouse cells
inthe cases of,
respec-tively, avian and murine RNA tumor
viruses
(18, 5).
Cytoplasmic
RNA was
extracted
ftom
unin-fected
SCP
cells
and
hybridized
wi'thIcA
as
described
in
Material
and Methods,
up to a
Crt
of
10'
mol
-s/liter. Figure 1 shows that no
hybridization was
observed
even for the
highest
Crt
value
attained. Since
a
CAt,2
of
5
x 104
mol
s/liter
corresponds
to
the presence
of 0.1
viral genome equivalent per cell,
we
concluded
that RNA
sequences
related
to visna virus are
virtually absent from
uninfected
sheep
cells.
The
study of visna virus RNA metabolism was
therefore
possible without the problems created
by "background" hybridization.
Time course of viral RNA synthesis. The
major purpose of this study was to determine
the time
course and the extent of transcription
of
visna
proviral DNA during the lytic
replica-tion
cycle of the virus.
Therefore, permissive
cells
derived from SCP
weresynchronously
infected with 3
PFUof visna virus per
cell and
harvested at different times
after infection.
Cytoplasmic and
nuclear RNAs
wereextracted,
and
the amount of viral RNA
wasdetermined
in
both fractions 0.5, 1.5, 3, 6, 9, 12, 24,
48and 72 h after infection. The results of the
hybridization reactions are presented in Fig. 2.
1001
a
N
a 50
I
*1 ~~Li-. *
103 10' 10'
Crt(Mole sec/1)
FIG. 1. Quantitation of viral RNA in the cyto-plasm ofuninfectedsheep choroidplexuscells. Cyto-plasmic RNA waspreparedfrom100uninfected 75-cm2tissuecultureflasks. TheRNA was hybridized
withvirus-specific[3H]cDNA prepared byan
endoge-nous reverse transcriptasereaction. Thefinal RNA concentration inthehybridizationmixturewas6mg/
ml.The variousCrt valueswereattainedbyvarying the time of incubation. Crt values are in mole-seconds per liter. Theextent ofhybridization
wasassayed byresistancetotheS, nuclease. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.504.296.420.445.557.2]D~~
/a ~~I I
10-3 10-1 10-1 10OU 10l 101 l03 104 10, 10, 10 Crt(Mole sec/I)
FIG. 2. Quantitationofvirus-specific RNAinthecytoplasmicand nuclearfractionsofsheepchoroidplexus
cellsatdifferenttimesofasynchronousinfection.Foreachtimepoint,45 tissuecultureflasks (75 cm2) were
synchronously infectedwith3 PFUof visnaviruspercell. At each timepoint,the cellswereharvested and fractionated into cytoplasmicand nuclear fractions, and RNA wasextractedfrom both fractions. All RNA
samplesweredissolvedin 50
pi
of2OmM Tris(pH7.5)-0.1 % DEP. ThedifferentRNAswerehybridizedwithvirus-specific[3H]cDNA in afinal volumeof20p. perCrtvalue examined. Quantitation ofviral RNA in nucleican be unreliable because nuclear RNA cannotbeprepared entirely free of DNA, which introduces uncertainty intoRNA concentrationdeterminedsolely fromthe
A2(.
Thisproblemwasobviatedby preparingnuclearand cytoplasmic RNAfromthesamecells, dissolvingboth in thesamevolume,andusingthe RNA concentrationsfrom cytoplasmicRNAtoconstructthe Ctcurvesofbothcytoplasmicand nuclear RNA. The
num1~er
of viral genomeequivalents inthe nucleiwascomputed fromtheCrt112
ofnuclear andcytoplasmicRNA,sincethe
Crtii2
isinverselyproportionaltothe concentrationofviral RNA sequences,and both nuclear andcytoplasmic RNA wereprepared fromthesamenumberofcells and dissolved in thesamevolume. The RNA concentrationsfor cytoplasmicRNAswere:0.5h,3,480pg/ml;
1.5h, 4,000Mg/ml;
3h, 2,560 pg/ml;6 h, 3,920ug/ml;9h, 3,920pg/ml;12h, 3,760pg/ml;24h,180Mg/ml;
48h,196Pg/ml;72h,59Mg/ml.
Forpurified60-70S viralRNA, the RNA concentrationwas02
pg/ml.
Theextentofhybridizationwasassayed byresistance tothe
SI
nuclease.Symbols (---) 60-70SRNA, (E)0.5-hcytoplasmic,(3) 0.5-hnuclear, (0)1.5-h cytoplasmic, (0)1.5-h nuclear, (A) 3-hcytoplasmic, (A) 3-hnuclear, (E)6-hcytoplasmic, (a) 6-hnuclear,(V)9-hcytoplasmic, (v)9-hnuclear, (0) 12-hcytoplasmic, (*)12-hnuclear,(0) 24-hcytoplasmic, (0) 24-h nuclear, (A) 48-hcytoplasmic, (A)48-hnuclear, (e) 72-h cytoplasmic, (*) 72-h nuclear. For the sake of clarity, Crtcurveshavebeenrepresentedintwopartsofthesamefigure.(A) Cotcurvesfor60-70S RNA and 12-, 24-, 48-,and72-h RNAs. (B) Curvesfor0.5-, 1.5-,3-,6-, and 9-h RNAs.
The
data obtained
from
these
hybridization
studies
aresummarized
in
Table
1and
Fig.
3,
which
also
presents the time course of
produc-tion
of infectious virions.
During
the
first 3 h of the infection, we
de-tected
atotal of
eight
tonine
viral
genome
equivalents
per
cell.
This value
is in
reasonably
good
agreement with
the
multiplicity
of
infec-tion
used
(3
PFU/cell), taking
into
account a
particle-to-PFU
ratio
of
5in
the
caseof visna
virus (9).
'wo
copies of viral genome were
pres-ent in
the
nucleus
even at
the
earliest time
studied
(30
min). The
e,
afraction of the
infecting
RNA
molecules
must
be rapidly
transported to the
nucleus
(seeDiscussion).Be-tween 6
and 12 h
postinfction,
there was a
marked
decrease in
vir-alRNA
* ie
cytoplasm,
whereas
the
amount of viral
RNA
remained
icoistant
in
the nucleus. These
phe-nomena were
observed in three independent
experiments. The accumulation of viral RNA
was
first
detected
in the nucleus 10 to
12.
h
postinfection and
inthe cytoplasm
approxi-mately
2h
later.
By 24
h
after infection, the
amountof
viral
RNA was
still
four times
greater
inthe
nucleus
than
in
the
cytoplasm;
at
the same time,
the
first
progeny
viral
particles
were
detected
inthe
culture medium.
Between
24h
postinfection and
the
end
of
the
lytic
cycle
(72
h),
the amount of
viral RNA
increased
al-most
exponentially in
both the nucleus and the
cytoplasm
and
was
always higher in
the
cyto-plasm. At 72 h postinfection, the cells contained
atotal
of
about
4,000
viral genome equivalents.
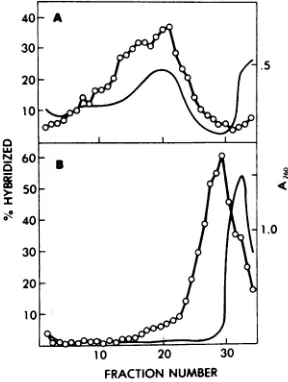
Size of
cytoplasmic and
nuclear viral RNA.
The visna virus genome is a 60-70S RNA
mole-cule
made
of two or
three 35S RNA subunits
(21).
Rapidly
harvested
virions contain
free
35S
RNA
subunits that assemble to form the 60-70S
RNA
complex during aging of the
particle (1).
One
would, therefore, expect at least part of the
viral
intracellular RNA to consist of
35S RNA
molecules. We
investigated this point by
exam-ining the size of viral cytoplasmic and nuclear
RNA 48 hafter
infection, a time that
corre-sponds
to the activeperiod
ofRNA
synthesis
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.504.97.403.72.213.2]TABLz 1. Amount of viral RNA present in the cytoplasm and the nucleus ofsheep choroid plexus
cells atdifferent times after. synchronous infection with 3 PFUofvisna viruspercell
Cytoplasmic RNA Nuclear RNA
Time No.of No.of
after viral viral
infec- genome genome
tion (h) Cti12 equiva- Crtii2i
equiva-lents lents
per per
cellb cell
0.5 7.5x102 6 2.3x103 2
1.5 7.5 x 102 6 2.3 x 103 2
3 6.4 x 102 7 2.3 x 103 2
6 1.0 x 103 5 3.0 x 103 2
9 3.0 x10 2 3.0 x103 2
12 3.0 x 103 2 2.0 x 103 3
24 2.0 x 102 23 6.0 x 101 77 48 1.0 x101 471 1.4 x 101 336 72 1.2 x100 2,350 1.8 x 10° 1,570
aCot valuesareinmoles secondperliter.
CAtM,2
values [image:5.504.292.442.228.453.2]weregraphically determined from thecurvespresentedin
Fig. 2.
&Numbers of viral genomeequivalents percell were
calculatedasindicated under Material and Methods and in
thelegend of Fig. 2. The number given in the table is the nearestintegertothecalculatedvalue. The Ct,,2 for puri-fied viral60-708wasdetermined fromFig. 2. The valuewas
10-2mol*s/liter.
Ui
I--z
u 100 10lo
z
10 10'
I
36912 24 48 72
TIME AFTER INFECTION(hours)
FIG. 3. Timecourseofaccumulationof viralRNA
in the nucleus (a) and the cytoplasm (0) of sheep choroid plexus cells synchronously infected with visna virus.Theamountof viral RNA is expressedin numberof viralgenomeequivalentspercell,and the values were takenfrom Table 1. Symbol: (0) time
course ofappearanceof infectious viralparticles in
theculture medium (PFUpermilliliter).
(Fig. 3).
Cytoplasmic
and-iuclear RNAs wereprepared asdescribed in
ode andsedimentedthrou * sucrose
gradient.To determine the sedimentation
pro-file of
viral RNA, the RNA present in each
fraction was
hybridized to [3HlcDNA.
Ribo-somal and
transfer RNA were
used
as
sedimen-tation
coefficient markers.
Two
classe$
of
viral RNA were found in both
the
nucleus
and the cytoplasm
(Fig. 4).The
first
is anRNA
specieswith
sedimentation
coef-ficient of 35& identical
tothat of the
gnome
subunits. The second class
sedimented between
208 and 10S with
aconstantpeak
at208.
How-ever,
the
sedimentation profile
inthis
partof
the gradient
wasvariable from
one experimentto
another.
60- A 35S
50- 40-30 9U20 I 10
I
I~~~~~~8
IOFRACTION NUMBER
FIG. 4. Sedimentation profile of virus-specific RNA extracted from (A) the cytoplasm and (B) the nucleus of sheep choroid plexus cells 48 h after infec-tion with visna virus. Twenty 75-cm2 tissue culture flasks were used in each case. Cytoplasmic and nu-clear RNA were extracted as described in Materials and Methods and dissolved in the same volume of 100 mMNaCl-10 mM Tris-hydrochloride (pH 7.5)-i mMEDTA-0.5% SDS -0.1 %DEP. One hundred mi-crograms of cytoplasmic RNA and the corresponding volume of nuclear RNA were heated to1000C for30s8
to disaggregate RNA and layered on top ofa 5 to 20% sucrose gradient. Centrifugation was from right to left in an SW41 rotor at 40,000 rpm for 120 min.A mixture of 32P-labeled 60-70S RNA and[3H~rRNA
wascentrifuged in a separate tube ofthe same rotor to serve as a sedimentation coefficient
marker.
After fractionation, the RNA present in each fraction was concentrated by ethanol precipitation in the presence of200jug of carrier RNA per fraction and hybridized withf3HlcDNA. The extent ofhybridization was as-sayed by the resistance toSo nuclease digestion. The locations of60-705, 28S, and 188 marker RNAs are indicated.on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.504.71.263.362.550.2]Identification
of
viral mRNA's.
Visna virus
is a
member of the family of retraviruses (9).
Genome
RNA
istherefore
expected
tohave the
same
polarity
asviral mRNA. It follows that
[3H]cDNA
should hybridize
toviral RNA
se-quences that
cosediment
with
polyribosomes,
and
further,
these
sequencesshould be
released
when the
polysomes
aredisrupted
with
EDTA
(16). In
the
experimentdescribed below,
wedemonstrate
that these
predictions
wereful-filled.
Purified
polyribosomes
from
infected cells
were
divided into two
frtions, dEDTA,
at
afinal
concentration of
20
mM,
was
added to
one.
The two fractions were then sedinted
sepa-rately in linear 0.5 to 1.5
M sucrose
gradionts.
The sedimentation profile oftotal cellular
poly-ribosomes was
determined from the
Am
of
each
fraction,
and that of
viral sequences
was
determined by
hybridizing the RNA of each
fraction with
[3H]cDNA. Figure
5
shows the
40- A
30
.5 20
00
60-
>_50-40
~~~~~~~~~~~1.0
30-
20-FRACTION NUMBER
FIG. 5. Virus-specific RNA inpurified
polyribo-somes.Purified polyribosomes were prepared from
40 75-cm2 tissue culture flasks 48 hafter infection
with visna virus. Thefinal polyribosome pelletwas dividedinhalf. One-half was directly layered on top
ofa0.5 to1.5Mlinearsucrosegradient; the other half was treated with 20 mM EDTA before being
layeredontopofasimilarsucrosegradient.
Centrif-ugationwas for 90 min at 30,000 rpm and
4'C
in an SW41 rotor.After fractionationofthegradients,theA260of each fraction wasdetermined. Thecontentof eachfraction wasconcentrated by ethanol precipita-tion in the presenceof200pg of carrier RNA and hybridized with
[3HJcDNA.
The extent of hybridiza-tion wasassayed by S, nuclease digestion. Sedimen-tation wasfromrightto left. (A) Polyribosomesnot treated with EDTA; (B) polyribosomes treated with EDTA. Symbols:(-)
Ama,
(0) percentage of [3HlcDNAhybridized.LU
40-
30-20
10
I@
10 20 30
FRACTION NUMBER
FIG. 6. Sedimentation profile of visna virus mRNA.Polyribosomeswerepurified from 40 75-cm2 tissue cultureflasks48 hafter infection with visna virus. The RNAs presentin thepolyribosome pellet were extracted with the phenol-chloroform-SDS method. One hundred microgramsof polyribosomal
RNAwasheated to 100'Cfor30s todisaggregate the RNA and then layeredontop ofa 5to20%linear sucrosegradient. Centrifugationwas,from rightto left, for 3 h and 20 min at 40,000 rpm inanSW41 rotor. The amountof virus-specific RNA presentin eachfractionofthegradientwasdeterminedasfor
the experimentpresentedinFig.4.[3H]rRNA'swere
centrifugedonaseparategradientinthesame rotor and usedassedimentationcoefficientmarkers. Their positionsareindicated.
result of the
experiment.Before
EDTA
treat-ment,
the hybridization profile and the
A2,
were
nearly coincident. Treatment with EDTA
disrupted the cellular polyribosomes,
asshown
by the shift of
all UV-absorbing
material
tothe
top
of the
gradient.
The
same treatmentalso
resulted
in
displacement
of
viral RNA
se-quences tothe
topof the
gradient. From the
hybridization profiles of
Fig. 5, it canbe
esti-mated
that
morethan
85%
of the
viral RNA,
which
cosedimented with cellular
polyribo-somes
before treatment, was
displaced -to the
top
of the
gradientu
From
this
experiment
we
conclude that
more tha 85%
of
the
viral
ENA
sequences associated with purified
polyribo-somes
behave like mRNA.
The
experiment also
shows that the
sedimentation
profile of
virus-specific
polyribosomes
is
very
similar
to
that
of
the
bulk
of
cellular
polyribosomes.
We then
examined
the
size of viral mRNA's.
For
this
purpose,polyribosomes
werepurified
from cells infected for 48 h and RNAs wereextracted from the
polysome pellet. The RNAs
were
sedimented
through
a 5 to 20% sucrosegradient
containing
SD&,and
the
hybridization
profile
with
[H]cDNA
wasdetermined
asde-scribed above.-Figure
6shows
atypical profile.
In all
polyribosomal RNA
preparationsexam-ined,
asharp
peaksedimenting
at35S
waspresent.
This discrete
RNA
species
wasalways
accompanied by
analmost
equal
amount ofon November 10, 2019 by guest
http://jvi.asm.org/
[image:6.504.285.427.60.198.2] [image:6.504.77.221.314.505.2]80
BRAHIC ET AL.smaller
RNAs
that
constantly
presented peaks
at
208
and
148.
The
exactprofile
inthis region of the gradient,
however, variedfrom
oneexperiment
toanother.DISCUSSION
Visna virus replication is restricted in the tissues
of
infected animals
(in vivo) (10), andthis
might explain the
persistenceof
theinfec-tion
and its slowness. The level
atwhich this
restriction is
exerted
in vivohas
not yetbeen
determined but could be that of
transcription,since
proviral
DNA is
readily detectable
inbrain cells
of
infected animals (10). We,
there-fore,
began investigation of the modalities of
proviral DNA expression
invivo
and
invitro.
The
main purposeof the
presentwork
was toobtain
base line information
onproviral DNA
transcription
incells infected
invitro.
We
have
shown
that, in
this
system,proviral
DNA is
extensively
trahribd and that
viralRSA
dimentsin
twodistinct
classes:
35S
RNA and
10-20S
RNA.
Figure 1
demonstrates that uninfected
sheepcells
do
notcontain RNA
sequencesrelated
tovisna virus genome.
This
result
extends
pre-viousobservations
(7)showing that visna
60-70S RNA
did
nothybridize
to avast excessof
DNA
extracted
from
uninfected sheep cells.
Both experiments
strongly suggest that visna
virus
is
apurely
exogenousvirus
of sheep.
Since there
was notranscription from
endoge-noussequences,
backgrounds
werelow
enough
toallow
us tofollow the fate of the few
copies of
RNA introduced in the cell.
Our
study
of the
time courseof
viral RNA
synthesis
revealed several
interesting
phenom-ena.
In
the
first
part of
the lytic cycle (from
0.5
h
to 10h),
wewere
folowig the
fate
of
paren-tal
RNA.
The
moststriking
observation
wasthat
pnal
RNA is
rapidly transported
tothe
nucleus,
aphenomenon that takes
place during
the
first 30
minof the infection. For
reasonsalready discussed
inMaterials and
Methods,
webelieve
that this
was notjust the
result of
contamination of nuclear RNA
by
cytoplasmic
RNA. A
rapid
transportof
parental
aviansar-coma
and leukosis RNA
to thenucleus of
chicken
embryo fibroblasts has also
beende-scribed (3).
Interestingly,
studies
fromthis
lab-oratory (B.Traynor et
al.,
inpreparation)
havedemonstrated
that visnaproviral
DNA isfirstdetected
exclusively
in thenucleus
approxi-mately
3 hafter
infectionand that
noviral
DNA is
found
inthe
cytoplasm
atany timeof
the
lytic
cycle.
Itis,therefore,
likely that
uponinfection of
sheep
cells,
visna virus RNAand
some
viral
structural
proteins,
including
theRNA-dependent DNA polymerase,
are rapidlytransported
to the nucleus, where proviralDNA
synthesis takes place. This is in contrastto
the
replication of
avian sarcoma viruses. Inthis
case,proviral DNA
isapparently
synthe-sized
inthe cytoplasm and transported
to thenucleus
(20).This
difference
in thesites ofpro-viral
DNA synthesis of visna and aviansar-coma viruses
could be related
tothe
typeof
tissue inwhich these
virusesnormally
repli-cate. Visna virus multipliesessentially innon-dividing cells
(for example,
brain
cells),
whereas avian
sarcoma orleukemia
virusesmultiply
inactively dividing cells. One
canspeculate
that
cellular proteins involved
inthe
replication of cellular DNA might be
required
for the
synthesis of
typeC
proviral DNA. These
proteins
would be
actively
synthesized
inthe
cytoplasm of
rapidly
growing
cells and would
therefore be available for the cytoplasmic
syn-thesis of
aviansarcoma-leukemia
proviral
DNA. These
proteins might be present only inthe nucleus
of
nondividing cells, which would
explain why
visnaproviral DNA synthesis
isexclusively nuclear.
A
second observation
concerningthe
fate of
parental RNA
isthat
someviral RNA remains
in
the
cytoplasm and
isdegraded
after
6
h
(Fig.3).
It
ispossible that
these RNAmolecules
cor-respond
toviral
particles that failed
toreach
the nucleus
and whose infectious
cycle
had
been
aborted.
Alternatively, they
mayfunction
as
mRNA
for
the
synthesis
of
early essential
viral
protein(s). Such
aphenomenon
has been
recently
described
inthe
caseof
murinesar-coma
leukemia
viruses (17).In
agreementwith
this, preliminary
experiments
from
this
labora-tory
(J.
c,personal
comm
ucation)
have
shown
aburst of
synthesis
of the
major
visnavirus
structural protein
during the first hours
of
the
lytic cycle.
The
second
partof the
lytic cycle (between
10and 72 h
postinfection) corresponds
tothe
phase
of active RNA
synthesis.
The kinetics of
ap-pearance of viral RNA sequencesinthe nucleus
and the
cytoplasm
and
the kineticsof
release of
infectious
virions (Fig. 3) suggestthat
viral
RNA
istranscribed
inthe
nucleus,
exported
tothe
cytoplasm,
and
eventually
encapsidated
into
viral
particles. This general scheme
hasalso been
proposed
inthe
caseof the avianRNA
tumor viruses (14). From
several experiments
analogous
tothat
described
inTable 1,
weesti-mate
that cells which have
beeninfected
by
8to10
viral
genomes contain4,000
to5,000copies of
viral
genome at theend of
thelytic
cycle.
The main size
class of
virusRNAmin
lyticlUy
infected cells
cosediments
wikhkiome
OS-units
and has the
samepolarity
asthe
viral
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
genome.
Figure
4 shows that 35S
genomic RNA
is
found in the
nucleus and is therefore
amajor
transcription
product of proviral
DNA. Since
we
only
studied
steady-state
nuclear
RNA,
ourexperiments
do
notaddress the issue of
whether
this RNA arises from the
processing
of
a
large
precursormolecule.
We
could
notdetermine whether the
nonco-valent
linkage of 35S
subunits
intovirion
60-70S RNA
complexes
takes
place
in
infected
cells,
because viral RNA
aggregated during
ex-traction
and
had
tobe
denatured
before
sedi-mentation.
A
similar aggregation
phenomenon
has also
been
described
inthe
caseof
intracellu-lar
RNA of
murineRNA
tumorviruses
(see
Discussion
of
reference
11)and
avian
sarcoma viruses (4).Figure 5
demonstrates
that
ourprocedure for
purifying
polyribosomes
yielded almost
exclu-sively
ribonucleoprotein
which could be
disso-ciated with EDTA. Therefore the viral RNA
present inthis fraction has the characteristic
properties
of mRNA.
Figure 6
shows that
358
genomic
RNA
functions
asmRNA. Smaller
viral
RNA
molecules
arealsoI prsent
inpuri-fied
polyribosomes that
mayrepresentdiscrete
RNA species,
and
notjust
degradation products
of 35S
RNA, because: (i)
polyribosomes
wereprepared
inthe
presenceof
1mgof
heparin
perml,
agood RNase inhibitor; and (ii) all
prepara-tions
exhibited
peaks
orshoulders
at20S
and
14S.
Such small viral mRNA's
have also been
described
inthe
caseof murine
leukemia
vi-ruses(6, 19).
Their
possible
significance in the
regulation of the synthesis of the
variousstruc-tural viral polypeptides has been
discussed
re-cently (13).
ACKNOWLEDGMENTS
We gratefullyacknowledgeJ. Tamaletforhis support during the realization of thiswork,P.Ventura,N.Sauze, andG. Vestris for their excellenttechnicalassistance, and
H.Lukesforherhelpinthepreparationof themanuscript.
M.Brahic wassupported bytheCalifornia division ofthe AmericanCancerSocietyseniorfellowship D-281. We ac-knowledge grants A6512119 and995717 from Centre Na-tionaldela RechercheScientifique and Public Health Ser-vice grantNS11782from theNational Institute of Neuro-logical and Communicative Disorders and Stroke. This
project wasVAH MRIS 3367 from the Veterans
Administra-tion.
LITERATURE CITED
1. Brahic, M., and R. Vigne. 1975. Properties of visna virus particles harvested at short time intervals: RNA content, infectivity, and ultrastructure. J. Vi-rol. 15:1222-1230.
2. Britten, R. J., and J. Smith. 1970. A bovine genome. Carnegie Inst. Washington Yearb. 68:378-386. 3. Dales, S., and H. Hanafusa. 1972. Penetration and
intracellular release ofthe genome of avian RNA tumorviruses. Virology 50:440-458.
4. Deng, C. T., D. Stehelin, J. M. Bishop, and H. E. Varmus. 1977. Characteristicsof virus-specific RNA in avian sarcoma virus-transformed BHK-21 cells and revertants. Virology 76:313-330.
5. Fan, H., and D. Baltimore. 1973. RNA metabolism of murine leukemia virus: detection of virus-specific RNA sequences in infected and uninfected cells and identification of virus specific messenger RNA. J. Mol. Biol. 80:93-117.
6. Gielkens, A. L. J.,M. H. L. Salden, and H. Bloemen-dal. 1974. Virusspecific mRNA on free and mem-brane boundpolyribosomes fromcells infected with Rauscher leukemia virus. Proc. Natl. Acad. Sci. U.S.A. 71:1093-1097.
7. Haase,A.T.,andH. E.Varmus. 1973. Demonstration of a DNA provirus in thelyticgrowth of visna virus. Nature(London) New Biol. 245:237-239.
8. Haase, A. T., A.C.Garapin, A. J. Faras, H. E. Var-mus,and J. M. Bishop. 1974.Characterizationof the nucleicacid product of the visna virus RNA depend-entDNA polymerase. Virology 57:251-258. 9. Haase, A. T. 1975.The slow infectioncausedby visna
virus.Curr.Top. Microbiol. Immunol. 72:101-156. 10. Haase, A. T., L. Stowring,0. Narayan, D. Griffin, and
D. Price. 1977. Slow persistent infectioncaused by
visna virus:role of host restriction. Science 195:175-177.
11. Haseltine, W. A.,andD.Baltimore. 1976. Size of mu-rine RNA tumor virus-specific nuclear RNA mole-cules. J. Virol. 19:331-337.
12. Leong, J. A., A. C. Garapin, N. Jackson, L. Fanshier, W. Levinson, and J. M. Bishop.1972.Virus-specific ribonucleic acidincellsproducingRous sarcoma vi-rus:detection and characterization. J. Virol. 9:891-902.
13. Mueller-Lantzsch, N., and H. Fan. 1976. Monospecific immunoprecipitation of murineleukemia virus
poly-ribosomes: identificationof P30 protein-specific mes-sengerRNA. Cell 9:579-588.
14. Parsons, J. T.,J. M. Coffin, R. K. Haroz, P. A.
Brom-ley, and C.Weissmann. 1973. Quantitative determi-nationandlocation of newly synthesized virus-spe-cificRNA in chickencells infected with Rous sarcoma virus.J. Virol. 11:761-774.
15. Penman, S. 1969. Preparation of purified nuclei and nucleoli from mammalian cells, p. 35-48. In K. Habel and N. P.Salzman (ed.),Fundamental techniques in virology.Academic Press Inc., New York.
16. Perry, R. P., and D. E. Kelley. 1968. Messenger RNA-proteincomplexes and newly synthesized ribosomal subunits:analysis of free particles and components of
polyribosomes.J. Mol. Biol. 35:37-59.
17. Salzberg,S., M. S. Robin, and M. Green. 1977. A possi-ble requirement for protein synthesis early in the infectious cycle of the murine sarcoma-leukemia vi-rus.Virology 76:341-351.
18. Schincariol, A., and W. Joklik. 1973. Earlysynthesisof virus-specificRNA and DNA in cells rapidly trans-formed with Rous sarcoma virus. Virology 56:532-548.
19. Shanmugam, G., S. Bhaduri, and M. Green. 1974. The virus specific RNA species in free and membrane boundpolyribosomes of transformed cells replicating MSV-MLV. Biochem. Biophys. Res. Commun. 56:697-702.
20. Varmus, H. E., R. V. Guntaka, W. J. W. Fan, S.
Heasley, and J. M. Bishop. 1974.Synthesis of viral DNA inthecytoplasm of duck embryo fibroblasts and inenucleated cells after infection by avian sarcoma virus. Proc. Natl. Acad. Sci. U.S.A. 71:3874-3878. 21. Vigne, R., M. Brahic, P. Filippi, andJ. Tamalet. 1977.
Complexity and polyadenylic acid content of visna virus 60-70S RNA. J. Virol.21:386-395.