0022-538X/84/100129-07$02.00/0
Copyright
© 1984, AmericanSociety
forMicrobiologyMapping and Expression of
a
Human
Cytomegalovirus Major Viral
Protein
MICHELLE GALL
DAVIS,'*
ENG-CHUNGMAR,1 YUAN-MINGWU,1t AND ENG-SHANGHUANG1'2'3
DepartmentofMedicine,2Departmentof Microbiology andImmunology,3 and Cancer Research Center,1 University of North Carolina, Chapel Hill, North Carolina27514
Received 12 March 1984/Accepted 23 June 1984
Weconstructed a DNAfragmentmap of low-passageTownestraincytomegalovirus by analyzingcross-blot hybridization and hybridizationsofisolated recombinant clones. Theabundant latetranscriptswere located on this map by hybridization oflabeled total RNA ofvirus-infected cells to blottedDNA fragments. The most abundantlatetranscript, carriedbythe 11.7-kilobase EcoRIfragment(EcoRI-G), wasprecisely mapped. The EcoRI fragmentwas fragmentedand subcloned in a plasmid carryingsimian virus 40sequences (pSV-OH, constructedby Chi-Bom Chae, Department of
Biochemistry,
Universityof NorthCarolina, Chapel Hill).One resultingrecombinantplasmid,pHD713SV2,
wastransferredtosimian virus 40-transformedmonkey kidney cells(COS-1) byDNAtransfection. Synthesisof acytomegalovirus-specific 67-kilodaltonproteinwasdetected in thesecells by reaction of blottedproteins withvirus-specificmonoclonalantibody.The 67-kilodaltonprotein isa major phosphorylated proteinfoundinvirions; it is not glycosylated. The location ofthegene for this 67-kilodalton protein is thereforeassigned to the center of the L-unique region of human cytomegalovirus, at 0.37 to 0.39map units.Humancytomegalovirus (HCMV) isaherpesvirus associ-ated withclinical manifestations ranging from asymptomatic infection to congenital abnormality, mental retardation, in-trauterine death, or, inorgantransplant patients, interstitial pneumonia. This virus can infect at multiple sites, and its antigenic diversity allowssuperinfection of seropositive indi-viduals (9). Recently, it has been suggested that this virus has oncogenic potential comparable with those of other herpesviruses. Cytomegalovirus(CMV)wascapableof stim-ulating cellular macromolecular synthesis (19) and trans-forming mammalian cells in vitro (14).
Physical maps of the double-stranded DNA genome of some
laboratory
strains have been constructed(3, 18; R. L. LaFemina and G. S. Hayward, personal communication). Inaddition,
recombinant DNA clones of these strains are available; this development will allow workers to define functional genes and compare specific portions of the genome from'different
strains to study gene function andexpression.
We have isolated recombinant DNAclones of CMV restriction fragments derived from Towne strain, passage 36, andconstructed the DNAfragmentmap shown in Fig. 1. The restriction enzyme sitesare not identical to those mapped by LaFemina and Hayward (Lafemina and Hayward, personal communication) for a high-passage Townestrain.In thiscommunication,wedescribeourstudy of abundant late transcripts. One of the most abundant late transcripts has been mapped to a small restriction
fragment,
and we have determined that this gene codes for one of the most abundant proteins in infected cells at the late stage of infection.MATERIALS ANDMETHODS
Virus and viral DNA. HCMV Towne
strain,
passage 36,was used for all
experiments.
Virus was grown in WI-38 * Correspondingauthor.tPermanent address: Wuhan Institute of
Virology, Academy
Sinica, Wuhan,China.
humanfibroblasts, and virusDNA wasisolatedas
previous-ly described (8).
Enzymatic fragmentation of virus DNA andgel electropho-resis. Restriction enzymes were purchased from Bethesda Research Laboratories (BRL), and digestions were per-formedasrecommendedby the
supplier.
Agarose gelswere run aspreviously described (17).Transferof DNA restrictionfragmentsingelsto nitrocellu-lose. DNA was transferred to nitrocellulose paper
by
the procedure by Southern (17). Gelsweresoakedon0.5NHCI solution for 15min and thenwere transferredto a solution containing0.5 MNaOH and1MNaClfor 15min todenature the DNA. Finally, gelswereneutralizedby soaking fortwo 15-min periods in a buffer solution containing 1 M Trishydrochloride
(pH 7.4) and 1.5 M NaCl. The DNA was transferred by blotting a transfer solution of6x SSC (lx SSC is 0.15MNaCl plus 0.015Msodiumcitrate) throughthe gels, nitrocellulose paper, and absorbentpaper.Nick-translation of DNA. DNA waslabeledfor hybridiza-tionprobe by incorporation of
[a-32P]deoxyadenosine
mono-phosphate. Each nick-translation mixture contained 0.5 ,ugofDNA,25
,uCi
of[a32P]-dATP
(ICNBiomedicals, Inc.),0.1mM dCTP, 0.1 mM dGTP, 0.1 mM TTP, 100 mM Tris hydrochloride (pH 7.5),20 mMNaCl,5 mM
P-mercaptoeth-anol,
5mMMgCl2,
2U ofPoll(BRL)and 0.01 ,ugofDNase (Worthington Biochemicals Corp.)per ml.After1h at12°C, reaction mixtures wereappliedtoSephadex G-100 columns eluted with10 mMTrishydrochloride buffer(pH8) contain-ing1 mM EDTA, and the excluded peakwaspooled.Nucleic acid hybridizations. Hybridizations were done either in 6x SSC-0.1% sodium dodecyl sulfate
(SDS-1Ix
Denhardt solution at65°C for20 h or in the same solution containing 10%dextran sulfateand 35%formamide at
45°C
for 20 h. Probe DNA wasdenatured
by
boiling along
with competing nucleotides, including (permilliliter)
10 ,ug ofpolyadenylic
acid, 50 ,ug of calf thymus DNA, 50 ,ug of salmon testis DNA, and 50p.g
ofyeastRNA; inhybridiza-tionsinwhich bacterialDNAwasadheredto
filters,
10p.g
of E. coliDNAper mlwasaddedas acompetitor.
Concentra-129on November 10, 2019 by guest
http://jvi.asm.org/
130 DAVIS ET AL.
tions of competing nucleotides are expressed as final
amounts in the hybridization solutions. All filters were
washed afterhybridization, first at roomtemperature for5 min in 2x SSC, and then three timesat 55°C for 20min in O.lx SSC with 0.1% SDS. X-ray exposures ofhybridized
filtersweresometimesenhancedbyaCronex Lightning-Plus
intensifying screenand exposureat -70°C.
Cloning of DNA fragments of HCMV. A collection of recombinantplasmids carrying HCMVDNAwasgenerated
by enzymatic recombinations of EcoRI-digested DNA of HCMV Towne strain, passage 36, and EcoRI-digested
pBR322 plasmid DNA. Ligation mixtures (20 jil)contained
A
B
0 b
C ...
0.5 jigof pBR322 plasmid DNA, 1 jigoffragmentedHCMV
DNA, 50 mMTris hydrochloride(pH 7.5), 10 mMNaCl, 10 mMdithiothreitol,50 jigofbovineserumalbuminperml,0.1
mM ATP, 25 mM MgCl2,and 1 U of T4 ligase (BRL). After incubation at 110C overnight, the mixture was used to transform E. coli LE392 to ampicillin resistance. Bacterial colonies were tested for hybridization to HCMV DNA by the procedure described by Grunstein and Hogness (6). Some fragments eluted from gels bya procedure described
by Maxam and Gilbert (12)were recombined similarly.
Subclonesof EcoRIrecombinantplasmidsweregenerated
by redigestion of the pBR322 recombinant plasmids and
0,0 0.7 02^ 0.3 0.4 0.5 0.6 0.7 0.8 0.9 .0
A 1.lA
A~~~p
" {VCK ...1ltJb'}UP'PLZXifflAlr:|LJ5*C\ (MO1. CIULAR
cc.sE I- RAV,MY. N I L.L'NG;'Il;
IIt 4
D A
C 18.4
1) 1.2
E 15.9
14.7
11 .5
1 .9
8.7
K; 6.4
1. 5.8
1: 5.8
5.6
O 5.1
P34.7
4.2
Kt 4.1
1[ 3 .5
2.9
W- 2.0
X 1.7
1' 1.6
7 1 .5 Si 7 .5
L.z ~~4.2
1?DNA EcoRX
Fragmsent
A B
C
D
E
F
pill1) PLASM1I)
3
4 5 6 7
1(1 9
10
11
1(0i
13
1 4
15
16
17
18
1 9
2 0 21
22
2 3
1(1
1 0 1
34
31
22 I 7 6
s . 9
.6 4.9
3e6
FIG. 1. Mapof therecognition sites forEcoRIendonucleaseonthe DNAofCMVTownestrain, passage36.(A)Fractionallengthofthe DNAmolecule. (B) Recognitionsites for EcoRIendonuclease.A,Sites which differ between thismapand thesitesdetermined by LaFemina
andHayward (Lafemina andHayward,personalcommunication),andA,DNAfragments which encode abundant latetranscripts. (C)X-ray
exposureofend-labeled fragments of ADNA (a)andHCMV DNA (b)digested with EcoRI endonuclease. (D) Molecular length in kilobases of eachEcoRI fragment and the designation ofthe recombinant plasmid carrying the fragment. Plasmid pHD101 is a pBR322 recombinant
carrying HindIII-A of HCMV.Fragmentswhichdonotappear onthismaparederived from the L-S junction (fragmentsPand Q)orappearto
hybridize with fragmentsXand Zoncross-blot hybridization(fragmentYandfragments smallerthan 1.5 kb)but donotoccurin plasmid
pHD101.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.167.458.219.663.2]EXPRESSION OF CYTOMEGALOVIRUS PROTEIN 131
(A.)
pBR322 Hind m + SV40 Hind m
ligation deletion Avol-,Pvu
1.0kb R
)1D 1.7 kb
pSV-OH (4.84kb)
EcoRI
1.8 kb HqRI
mS
~~~~~~~~(C
H RNA
H
1.45 kb
(B.)
III
pHD7(pBR322-EcoRI-G)
j Bgl 11
11.3 kb 0.7 kb 2.9 kb +M13mp7
(7.2kb) ligation *BomHl
R 2.9 kb R
ATCCGGAATTC GAATTCCCCGATCT
M 13-713 (10.1kb)
I*
Eco RI (M13mp7)7.2 kbR H + HR RNA
ltion 2.9 kb
2.15 kb
H.
ikb
XR /
l.1kb\ H
RNA isolation. Cells were lysed with 7 M guanidine hydrochloride and Sarkosyl and then layeredover 6 M CsCl solution and centrifuged to separate RNA and DNA, as described by Seeburg et al. (16). RNA was extracted from the pellets.
RNA gel electrophoresis. RNA was separated on 0.8% agarose gels containing 6% formaldehyde and 20 mM mor-pholinepropanesulfonic acid-acetic acid bufferat pH 7and 1 mM EDTA. After electrophoresis, RNA was blotted onto
a
b
i10..:
:*.
...
_-A S'
do.
Om
c
d
e
f
_i.
6-
-
4-
0
go~~
-_pHD713 SVI
[image:3.612.68.277.75.389.2](8.7kb) pHD713 SV2(8.7 kb)
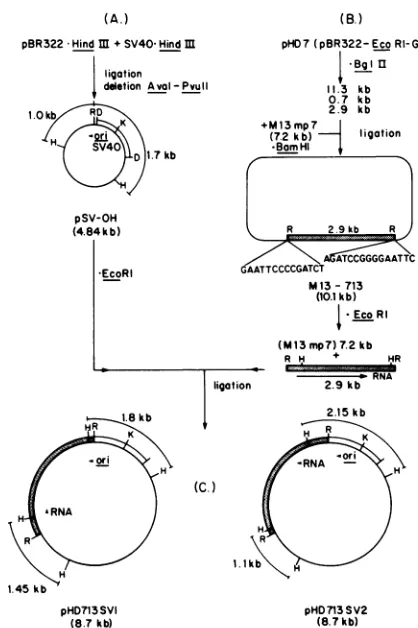
FIG. 2. Derivation of plasmids pHD713SV1 and pHD713SV2. (A) Plasmid pSV-OH was generated byrecombination of pBR322
( ~)andSV40(IZZ)DNAs.Thedirection of ori is thesame as
the early promoterof SV40. (B) Fragments resulting from BgIII
digestion ofplasmid pHD7wererecombined with M13mp7 replica-tiveform DNAdigested with BamHI. The EcoRI sites are 10bp
from the BamHI sites of the bacteriophage DNA.= l, HCMV DNA. (C) The 2.9-kb fragment resulting from EcoRI digestion of M13-713 DNA was recombined with EcoRI-digested pSV-OH
DNA.Recombinantplasmidsweredigested with Hinfltodetermine theorientation of theinserted CMV DNA (datanotshown). Enzyme sites indicatedare: R, EcoRI; D,HindIlI; K, KpnI;and H,Hinfl.
enzymatic recombination with other vectorDNAs (see the
legend toFig. 2for detaileddescription ofsubclones). The plasmid pSV-OH was isolated by C.-B. Chae of the Department of Biochemistry at the University of North CarolinaatChapelHill(unpublished data).Theplasmidwas
generated by enzymatic recombination ofHindlIl-digested simian virus 40(SV40) DNAand HindIll-digested pBR322 DNA. A deletion between the AvaI siteat 1,424 base pairs (bp) and the Pvull site at 2,065 bp on pBR322 was also introducedto generate a4.8-kilobase (kb) vector, including
SV40 sequences between the HindlIl site at 5,107 bp of SV40 and the origin ofSV40DNAreplication and between theKpnI site at230bpand theHindIII siteat 982bp(C.-B. Chae,personal communication). Adiagramof theplasmidis included in Fig. 2, indicating the direction of the SV40 sequencesandtheHinfl sitesofpBR322 whichwereusedto orient the CMVsequencesin therecombinants pHD713SV1 andpHD713SV2.
Otherrecombinantsweregenerated by enzymatic
recom-bination ofHindIII,BamHI,orBglII-digested HCMV DNA and plasmid pBR322 DNA or replicative form DNA of
[image:3.612.316.554.201.618.2]bacteriophage M13mp7.
FIG. 3. Hybridization of 32P-labeled RNA isolated from cells infected with HCMV with nitrocellulose filters carrying HCMV
DNA. Cells were labeled with [32P]orthophosphate 60 h after infection, and RNAwasisolatedasdescribed in thetext. Nitrocellu-lose strips containing EcoRI fragments (lanes a and d), XbaI
fragments (lanes b and e), andHindIllfragments (lanescand f)were
hybridized with nick-translated HCMVDNA(lanesa toc)or32p labeledRNA(lanes dtof). Thefragments indicatedareEcoRI-G,
M, and LT (lanes a and d), Xba-B and LT (lanes b and e), and HindIII-Cand LT (lanescandf). The major fragments correspondto
themaplocation intheL-unique region indicated in Fig.1,and the LT fragments and EcoRI-Mcorrespondtotheterminal and junction regions ofcytomegalovirus DNA.
am
I us
-1
VOL. 52, 1984
on November 10, 2019 by guest
http://jvi.asm.org/
132 DAVIS ET AL.
TABLE 1. Hybridization ofRNA versusplasmidDNA immobilized onfilters'
Plasmid RIfragment cpm
pHD7 G 3,825
pHD9 I 504
pHD14 N 1,400
pHD15 0 302
pHD20 T 748
pHD22 V 242
pHD13 M 2,291
a 2 ,ug of plasmid DNA was immobilized onnitrocellulose filters, and these werehybridized with 3.4 x 107cpmof RNA isolated from WI-38 cells 60 h after infection with HCMV. Duplicate samples were averaged, and the background radioactivity from filters carrying pBR322 DNA (168 cpm) was subtractedfrom each sample.
nitrocellulose in the same manner as described for DNA, with lOx SSC as atransfer solution.
Transfer of DNA to cells in tissue culture. Cellsgrownin 5-cm plastic wells were treated with CMV DNA by the technique described by Graham and Van der Eb (5) as modified by Stowe and Wilkie (20). The DNA-calcium phosphateprecipitates containingS ,ugofplasmidDNAand 10 ,ugof calf thymus DNA were added to subconfluentcell monolayers,and the treatedcellswereincubatedat36°Cina CO2 incubator with occasional agitation for 40 min. Ten milliliters of minimal essential medium (MEM) containing 10% fetal calfserum was added, and after4 h the medium wasreplaced with 2mlof15%
dimethyl
sulfoxide in MEM. After 4 min at room temperature,dimethyl
sulfoxide was removed, andthecellswerewashedwith MEM.Finally,
10a b c d e f 9 h i k
*041(
,-WwIll,0w
probe: rDNA
406o
HCMV
1.9kb
pHD7
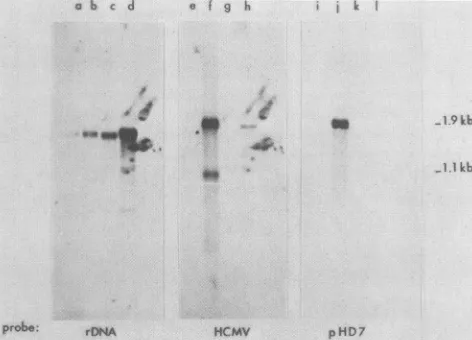
FIG. 4. Hybridization of 32P-labeled DNA with RNA
immobi-lized onfilters. RNAwas separated onagarose-formaldehyde gels, blottedontonitrocellulosefilters, and hybridizedwith
nick-translat-edDNA.RNAwasisolatedfrom uninfectedcells(lanesa, e,andi),
cells infected with HCMV 60h previously(lanes b, f, and j), cells infected with HCMV and treated with phosphonoacetic acid (100 p.g/ml)for10h(lanesc, g,andk), and cellsinfectedwith HCMVand
treated with cycloheximide (50 p.g/ml) for 4 h (lanes d, h, and 1). Nick-translated DNAprobesare:rDNA, plasmidpSF2124carrying
DNAfrom theseaurchin Arbaciapunctulata specificfor 18SRNA (M. Davis, unpublished results); HCMV, total virus DNA;pHD7, plasmid pBR322 carrying EcoRI-G of HCMV. The molecular weights indicatedwere estimated from the size of18S RNA(1.869 kb)and other gels in which pBR322 DNAfragmentswereappliedto
the gels.
ml ofMEM
containing
10%fetalcalf serum wasadded, and the cells wereincubatedfor 24 h and then harvested.Detection of HCMV-specific antigens. Proteins from the treated cells were
analyzed by
SDS-polyacrylamidegel
electrophoresis by
the method ofLaemmli(10),asprevious-ly
described(11).
Proteinswereelectroblottedonto nitrocel-lulosesheets,
as describedby
Towbin et al. (21). The identification ofHCMV-specific antigens
wasaccomplished
by
exposing the nitrocellulose sheets to monoclonalanti-body
(the detailsof theproductionofmonoclonalantibodies to HCMV Towne virionproteins
will be published else-where)and then stainingthemwithhorseradish peroxidase-conjugated immunoglobulin G (IgG), 4-chloronapthol, andH202,
as described by Hawkes et al. (7). A blue color reactionindicated the presence ofantigen.Immunoprecipitationof CMVproteins. Mock-or HCMV-infectedcellswerelabeledfor 20 hat72 hpostinfection with [35S]methionine, [3H]glucosamine, or [32P]orthophosphate as described by Mar et al. (11). Cells were harvested
by
scraping, washed three times withTris-buffered saline, and solubilized with RIPA buffer (0.05 M Tris
hydrochloride,
0.15 M NaCl, 0.1% SDS, 1% sodium deoxycholate, 1% Triton X-100,0.1 mMphenylmethyl sulfonyl fluoride) for30 min on ice. The lysate was sonified and centrifuged in a microcentrifugefor15 min. One-half milliliter of supernatant wasmixed with 10 pu1 of ascites fluid from hybridoma clone 185E, 132-5, H-644, orH176 and with 100 p.l ofanti-mouse IgG (whole molecule)-agarose (Sigma Chemical Co.) and gently agitated at 4°C overnight. The agarose beads were collected bycentrifugation, washed five times with 1 ml of RIPAbuffer, and suspended in 50 p.1 ofsamplebuffer(0.37 M Tris hydrochloride [pH 6.8], 15% glycerol, 5% ,3-mercap-toethanol, 1.5% SDS) for polyacrylamide gel electrophore-sis. Afterheating at90°C for5 min, the sample wascooled, thebeads werepelleted, and the supernatant proteins were loaded onto a9% polyacrylamide gel. Detection of [3H]glu-cosamineand35S-labeled proteinswasdoneby
fluorography
(1),anddetection of32P-labeledproteins wasdonebydirect exposure of gels to X-ray film, enhanced by a Cronex Lightning-Plus intensifying screen.
RESULTS
Physical map of HCMV Towne strain. Each recombinant plasmid was nick translated and hybridized to blotted re-striction
fragments
of total viral DNA. Theresults ofthese experiments were compared with data obtained by cross-blothybridization. Inaddition, restrictionenzyme sites were mapped on the recombinant plasmids. Physical maps of strains Davis (3), AD169 (18), and Towne (LaFemina and Hayward, personal communication) are similar to the ones showninFig. 1, but the regions at eachendand near the L-S junction vary. Thesepointsofvariability betweenthe maps of high- and low-passage Towne strain are indicated by asterisks.Mapping regionsoftranscriptiononthegenomeofHCMV. Hybridizationof32P-labeled RNA isolated from cellsatany time between 14 and 96 h after infection with filters contain-ing restriction enzyme fragments ofHCMV DNA yields a complex pattern (Fig. 3), indicating that many regions are beingtranscribed (22). Wefocused onthe sites ofabundant transcription at the late stage of infection as shown by the intensity of the hybridization of labeled RNA withblotted DNA fragments (Fig. 3 and Table 1) and by the reverse method, hybridization of nick-translated DNA fragments with RNA immobilized on nitrocellulose filters
(Fig.
4). In Fig. 4, the hybridized filters weredeveloped
after shortJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.66.301.99.182.2] [image:4.612.65.301.428.598.2]A a b c d e a b c d e
_
9
/
:FIG. 5. Physical map of plasmid pHD7. Anagarosegel(left) carrying restriction enzymefragmentsofplasmid pHD7 was blotted onto nitrocellulose paper whichwashybridized with 32P-labeledRNAisolated from cells60 hafter infection with HCMV(right). Lanes: a,XDNA digested withHindlll;b,undigestedplasmidpHD7; c, pHD7digestedwithEcoRI andBamHI; d, pHD7digestedwithEcoRIandBglII;ande, pHD7digestedwithEcoRl,BamHI,andBgIll. (B)AmapofplasmidpHD7indicatingtheorientation ofpBR322 (4.4kb) and sites for EcoRi (R),BamHI(B),BglII (G), and Hinfl (H). The orientation shown is the orientation offragmentEcoRI-Gonthemap shown inFig. 1, and the transcription of1.9kb RNA is from lefttoright.TheHinflsites have beenmappedby DNA sequencing experiments (data not shown). Only the terminalHinflsitesareindicatedonthe 2.9-kbBglII fragment.
exposure, to show only the most intense
labeling.
Two distinct regions oftranscription
locatedby
these experi-ments are indicated below theEcoRI restriction map (Fig. 1), one of these near the L-Sjunction and the otherat the middleof theL-unique region. Much of the complexpattern ofhybridization ofRNAwithEcoRIdigestion fragments
can be attributed to themultiple
L-Sjunction
andL-repeat
fragments
which are distributedthroughout
the size range (LaFemina andHayward, personal
communication)
and which hybridize to an abundant 1.1-kb RNA(Fig.
4 and Table 1). This transcription region is represented at least twicein every virusmolecule,attheL-Sjunctionandatthe L terminus. At longer exposure, probably all restriction fragments of HCMV hybridizeswith32P-labeledRNA isolat-ed fromcells late ininfection.A comparison of the abundance of these two major transcripts with others was performed by
hybridizing
32p_ labeledRNAinsolutionwith filterscontainingrecombinant plasmidDNA(Table 1).The results are comparable with those
previously
deter-minedfortotal viralDNAblots, although neither experiment is correctedtomolarity byestimating the sizes of theRNA. Fragment G carried by plasmid pHD7 was labeled 15-fold more intensely than fragments V or 0, and fragment M, which carries part ofthe L-repeat DNA, wasninefoldmore radioactive thanfragment V. Asdemonstrated in Fig. 4, the RNAhybridizing to fragment G is a latetranscript of 1.9 kb. ThisRNA was not present in cells treated with cyclohexi-mide orphosphonoacetic acid after virus infection.
Isolation of one region of transcription. The most intensely labeled fragment, EcoRI-G carried by plasmid pHD7, was fragmented by enzymeBglIIinto fourportions,oneof which hybridizedtoRNA isolated frominfected cells(Fig. 5). This 2.9-kbBglIIfragmentis theonlytranscribedregionwehave detected in theoriginal 11.7-kb EcoRIfragment.This obser-vationwasconfirmedbyelectronmicroscopeobservation of R-loops, which showasingleuninterruptedregion of hybrid-ization (L. Loh and E.-S. Huang, unpublished data). The 2.9-kbBglII fragmentwassubcloned into the BamHI site of bacteriophage M13mp7, and the fragment which was
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.148.462.78.442.2]134 DAVIS ET AL.
leasedafterEcoRI digestionof thereplicative-formDNA of the recombinant M13-713 was recombined into the EcoRI site of plasmid pSV-OH (Fig. 2).
Plasmidsresultingfrom the recombination between M13-713 and plasmid pSV-OH were divided into two groups, based on digestion patterns with enzyme Hinfl. Plasmid pHD713SV1 carries the inserted region in the orientation opposite to that inplasmid pHD713SV2 (Fig. 2).
Expression of the 67K protein in tissue culture cells. Cul-tures of HEL cells and COS-1 cells were treated with plasmidDNA asdescribed above,andproteinwasextracted and applied toacrylamide gels asdescribed. Electroblotted protein was reacted with monoclonal antibody prepared in thislaboratory specificfor a 67,000-molecular-weight (67K) protein of HCMV. A photograph of the stained nitrocellu-losepaper is shown inFig.6. The 67K protein wasproduced in COS-1 cells carrying pHD713SV2 DNA but not in cells carryingpHD713SV1 DNA. Theorientation ofthe transcrip-tion ofthe 67K proteinsequence in plasmid pHD713SV2is downstream from the major early promoter of SV40. In plasmid pHD713SV1, this orientation is reversed.
Characterization of the 67Kproteinininfected cells. WI-38 cells infected with HCMV were labeled with [3H]glucos-amine,
[35S]methionine,
or[32P]orthophosphate
72 h after infection. Afterextraction of totalprotein, the 67K protein was immunoprecipitated with monoclonalantibody, andthe protein was analyzed by gel electrophoresis. The gel was exposed to X-ray film(Fig.
7). The 67Kprotein
was not labeled by[3H]glucosamine,
but it was labeled by 32P and 35s.DISCUSSION
The 67Kproteinis astructural protein of HCMV,infact, oneofthemajor proteins foundinisolated virions
(Fig.
6 andcellsweretreated with theplasmid DNAindicatedorinfected with HCMV. Protein was extractedfrom the cultures and from isolated virions, isolatedas previouslydescribed (8), appliedtoacrylamide gels, and separated by electrophoresis. Electroblotted protein was reacted with monoclonal antibody and stained with horseradish
f5fel..,
1 2
'200K
15 0K " "
n.'. .'f
68K.
5x
3
32ID /;riSK?
4 5 6
67K |..
inls
FIG. 7. Immunoprecipitationofpolypeptides ofHCMV-infected cells labeled with [35S]methionine, [3H]glucosamine, or [32P]ortho-phosphate byamonoclonalantibody. Theimmunoprecipitateswere subjectedtoSDS-polyacrglamidegelelectrophoresisasdescribed in the text. Lanes 1, 6, and 9 contain host cell proteins labeled with
[35S]methionine, [32P]orthophosphate, and [3H]glucosamine, re-spectively. Lanes 2, 5, and 8 containinfectedcellproteins labeled with [35S]-methionine, [32P]orthophosphate, and [3H]glucosamine, respectively.Lanes 3, 4, and 7 containlabeledpolypeptides precip-itated from HCMV-infected cellextracts by monoclonalantibody. No detectable [3H]glucosamine-labeled protein was precipitated (lane 7), althougha 67Kprotein was precipitatedfrom 35S-and 32p-labeled cultures.
7). The monoclonal antibody which reacts with the 67K protein does not neutralize virusinfectivity, indicating that the proteinmay be internal. Itprobably corresponds tothe 66K phosphorylated protein described by Gibson (4), a tegument protein which interfaces thecapsid and the mem-brane of the virus. A 64Kglycoprotein recently described by Clarket al. (2) is unrelatedto the 67Kphosphoprotein(Fig. 7). The monoclonal antibody preparation which precipitates the 67K protein fails to precipitate a labeled protein from infected cell proteins labeled with [3H]glucosamine. The gene for the 64K glycoprotein of Clark et al. is carried by fragment EcoRI-A (H. Pande, S. Baak, J. A. Zaia, B. R. Clark, J. E. Shively,and A. D. Riggs, Eighth International Herpesvirus Workshop, Oxford, abstr. no. 272, 1983).
The RNA transcripts that we describe have also been identifiedby Wathen and Stinski (22). We do not detect the larger RNAs that they describe, but the smaller species at 1.9 and 1.2 kb correspond to the map locations that we report for 1.9- and 1.1-kb RNA.
The intense transcription of L-repeat regions was also found in cellsinfected with strain Ad169 of CMV (13). The 1.9-kbtranscript we describe probably corresponds to RNA
peroxidase-conjugated IgG, 4-chloronaphthol, and H202. Only COS-1 cellscarrying DNAofplasmid pHD713SV2had detectable amountsofa67Kprotein.
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.65.302.444.667.2]hybridizingtoEcoRI-IofAd169, althoughit isnot character-ized asthe most abundant transcriptfrom analysis of cyto-plasmic, polyadenylated RNA. McDonough and Spector (13) describe straindifferences in the pattern oftranscription in CMV-infectedcells.Spectoretal.(18)also havemappeda 3-kbBglII fragment (BglII-X) inEcoRI-I, flankedbya 0.78-kbfragment
(BglII-f).
The orientation of thesefragmentshas been determined by DNA sequencingandcross-hybridiza-tion ofinternalfragments in this laboratory (Fig. 5). The L segment ofHCMV in maps of Ad169 is inverted relative to theorientationillustrated inFig. 1.Itappears that theregion near0.4map units is thesamebetween thesestrains; indeed, it is likely that most of the genomes of cytomegalovirus strainsare
genetically
colinear(23).
Variationsareclustered attherepeattermini, andthese areprobably responsible
for nonhomologous sequences betweenstrains (15).We have not determined whether the 2.9-kb
BglII
frag-mentcarriesafunctional promoter,sincecellsexpressedthe 67K
protein only
when transfected with recombinantplas-midDNAofpHD713SV2whichcarriesHCMV DNA down-stream from an
early
promoter ofSV40,
andonly
COS-1 cells expressed theprotein
at adetectable level.Expression
of the 67K
protein
was not detected in humanembryonic
lung cells transfected with either
pHD713SV1
orpHD713SV2 DNAs. If a viral promoter is located on the BglII fragment, it does not promote the
expression
of the 67Kprotein
when dissected from the total HCMV viral DNA.Evidently,
anearly
orimmediateearly
viral function is required forexpression
of the 67Kprotein.
We caninvestigate this
requirement by
measuring protein
expres-sion in cells after transfer ofpHD713SV1
DNA in combina-tion with other recombinantplasmids
oraftermicroinjection
of HCMV macromolecules.
We identified a
putative
promoter on theBglII
fragment
byDNAsequenceanalysis
ofthe DNAinsertion ofM13-713 bacteriophage. The 5'end ofthe RNA islocated300bp
from oneBglII site,
andnointervening
sequencesweredetected. The DNA sequence 5' to the site of RNA initiation is abruptly different from thesurrounding
sequence. Five hundredbase pairs
fromtheinitiationsite,
theG+Ccontentof the DNA is 66%. In contrast, the 230
bp
before the initiation site contain 29% G+C. The entirecoding region
including 700 bp downstream is 50%
G+C,
and the most distant sequence that has beendetermined,
700 to 900bp
after the
polyadenylation
signal,
is 66% G+C. A definite gene structureis evidentfrom thisanalysis
(M. G.Davisand E.-S. Huang,manuscript
inpreparation).
Theentiresequenceconsists of
surrounding
viralDNAofhigh
G+C content,abruptly
interrupted by
an extended A+T-richregion,
followedby an informationsequence that contains maximum nucleotidevariability.
Weplan
similar analyses ofothercoding regions
of humancytomegalovirus
todeterminethefunction oftheseregions,their
distribution,
and their
controlling
elements.Analysis
of similarcoding
regions of other
herpesviruses,
identifiedby
cross-hybridiza-tion oftheCMV
coding region
and restrictionfragments
of herpesvirus DNA, may indicate similar functions in other membersofthe virus group.LITERATURE CITED
1. Bonner, W. M., and R. A. Laskey. 1974. A film detection methodfor tritium-labelled proteins andnucleicacids in poly-acrylamide gels. Eur. J. Biochem. 46:83-88.
2. Clark,B.R., J. A. Zaia, L. Balce-Directo, and Y.-P. Ting. 1984.
Isolation and partial chemical characterization of a 64,000-dalton glycoprotein of human cytomegalovirus. J. Virol. 49:279-282.
3. Demarchi, J. M. 1981. Human cytomegalovirus DNA: restric-tion enzyme cleavage maps and map locarestric-tions for immediate-early, immediate-early, and late RNAs. Virology 114:23-38.
4. Gibson,W.1981.Structural and nonstructural proteins of strain Colburncytomegalovirus. Virology 111:516-537.
5. Graham, F. L., and A. J. Van der Eb. 1973. A new technique for the assay of activity of human adenovirus 5 DNA. Virology 52:456-467.
6. Grunstein, M., and D. Hogness. 1975. Colony hybridization: a methodfor the isolation of cloned DNAs that contain a specific gene. Proc. Natl. Acad. Sci. U.S.A. 72:3%1-3965.
7. Hawkes, R., E. Niday, and A. Matus. 1982. Monoclonal antibod-ies identify novel neural antigens. Proc. Natl. Acad. Sci. U.S.A. 79:2410-2414.
8. Huang, E.-S., S.-T. Chen, and J. S. Pagano. 1973. Human cytomegalovirus. I. Purification and characterization of viral DNA. J.Virol. 12:1473-1481.
9. Huang, E.-S., S.-M. Huong, G. E. Tegtmeier, and C. Alford. 1980. Cytomegalovirus: genetic variation of viral genomes. Ann. N.Y. Acad. Sci.354:332-346.
10. Laeminli, U. K. 1970. Cleavage of structural proteinsduring the assembly of the head ofbacteriophage T4. Nature (London) 227:680-685.
11. Mar, E.-C., P. C. Patel, and E.-S. Huang. 1982. Effect of 9-(2-hydroxyethoxymethyl) guanine on viral-specific polypeptide synthesis in human cytomegalovirus-infected cells. Am. J. Med. 73:82-85.
12. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeled DNAwith base-specific chemical cleavages. Methods Enzymol. 65:49-560.
13. McDonough, S.H., and D. H.Spector. 1983. Transcription in human fibroblastspermissively infectedbyhuman cytomegalo-virus strain AD169. Virology 125:31-46.
14. Nelson, J. A., B. Fleckenstein, D. A. Galloway, and J. K. McDougall. 1982. Transformation of NIH 3T3 cells with cloned fragments ofhuman cytomegalovirus strain AD169. J. Virol. 43:83-91.
15. Pritchett,R. F. 1980. DNAnucleotide sequence heterogeneity
betweentheTowne andAD169 strains ofcytomegalovirus. J. Virol.36:152-161.
16. Seeburg,P.H.,J.Shine, J.Martial,A.Ullrich,J. D.Baxter,and H.M.Goodman.1977. Nucleotide sequence of part of the gene forhumanchorionicsomatomammotropin: purificationof DNA complementary to predominant mRNA species. Cell 12:157-165.
17. Southern, E. M. 1975. Detection ofspecificsequences among DNAfragments separatedby gelelectrophoresis. J.Mol. Biol. 98:503-517.
18. Spector,D.H., L. Hock,andJ. C. Tamashiro. 1982. Cleavage mapsfor humancytomegalovirus DNA strain AD169 for restric-tion endonucleasesEcoRI,
BgIII,
andHindlll. J. Virol. 42:558-582.19. St. Jeor,S. C., and R. Hutt. 1977. Cell DNAreplication asa
function in the synthesis ofhuman cytomegalovirus. J. Gen. Virol.37:65-73.
20. Stow,N.D.,and N. M. Wilkie.1976. Animproved technique for obtaining enhancedinfectivitywithherpessimplexvirus type1 DNA.J. Gen. Virol. 33:447-458.
21. Towbin, H.,T.Staehelin,andJ.Gordon. 1979. Electrophoretic transfer ofproteins from polyacrylamide gelsto nitrocellulose sheets:procedureandsomeapplications.Proc.Natl. Acad. Sci. U.S.A.76:4350-4354.
22. Wathen,M.W.,and M. F. Stinski. 1982.Temporalpatternsof humancytomegalovirustranscription: mappingtheviralRNAs synthesized at immediate early, early, and late times after infection.J. Virol. 41:462-477.
23. Westrate,M.W., J.L. M.C.Geelen,P. M.E.Wertheim,andJ. Van der Noordaa.1983.Comparisonofthephysicalmapsof the DNAsof twocytomegalovirusstrains. J. Gen. Virol. 64:47-55.


![FIG. 7.itated(lanewithrespectively.labeledthecellsNospectively.subjectedphosphate[35S]methionine, Immunoprecipitation of polypeptides of HCMV-infected labeled with [35S]methionine, [3H]glucosamine, or [32P]ortho- by a monoclonal antibody](https://thumb-us.123doks.com/thumbv2/123dok_us/1420594.94794/6.612.65.302.444.667/lanewithrespectively-labeledthecellsnospectively-subjectedphosphate-immunoprecipitation-polypeptides-methionine-glucosamine-monoclonal.webp)
