0022-538X/08/$08.00⫹0 doi:10.1128/JVI.00409-08
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Henipavirus
V Protein Association with Polo-Like Kinase Reveals
Functional Overlap with STAT1 Binding and Interferon Evasion
䌤
Louise E. Ludlow,
1,2Michael K. Lo,
3,4Jason J. Rodriguez,
1,2Paul A. Rota,
3and Curt M. Horvath
1,2*
Department of Medicine, and Department of Biochemistry, Molecular Biology, and Cell Biology, Northwestern University, Evanston, Illinois 60208-35001; Department of Medicine, Evanston Northwestern Healthcare, Evanston, Illinois 602082; Measles, Mumps,
Rubella, and Herpes Laboratory Branch, Centers for Disease Control and Prevention, 1600 Clifton Road, MS C-22, Atlanta, Georgia 303333; and Department of Microbiology and Immunology, Emory University School of Medicine,
1510 Clifton Road, Atlanta, Georgia 303224
Received 25 February 2008/Accepted 7 April 2008
Emerging viruses in the paramyxovirus genusHenipavirusevade host antiviral responses via protein inter-actions between the viral V and W proteins and cellular STAT1 and STAT2 and the cytosolic RNA sensor MDA5. Polo-like kinase (PLK1) is identified as being an additional cellular partner that can bind to Nipah virus P, V, and W proteins. For both Nipah virus and Hendra virus, contact between the V protein and the PLK1 polo box domain is required for V protein phosphorylation. Results indicate that PLK1 is engaged by Nipah virus V protein amino acids 100 to 160, previously identified as being the STAT1 binding domain responsible for host interferon (IFN) signaling evasion, via a Thr-Ser-Ser-Pro motif surrounding residue 130. A distinct Ser-Thr-Pro motif surrounding residue 199 mediates the PLK1 interaction with Hendra virus V protein. Select mutations in the motif surrounding residue 130 also influenced STAT1 binding and innate immune interference, and data indicate that the V:PLK1 and V:STAT complexes are V mediated yet indepen-dent of one another. The effects of STAT1/PLK1 binding motif mutations on the function the Nipah virus P protein in directing RNA synthesis were tested. Remarkably, mutations that selectively disrupt the STAT or PLK1 interaction site have no effects on Nipah virus P protein-mediated viral RNA synthesis. Therefore, mutations targeting V protein-mediated IFN evasion will not alter the RNA synthetic capacity of the virus, supporting an attenuation strategy based on disrupting host protein interactions.
The genus Henipavirus within the family Paramyxoviridae
was created in 2002 to accommodate the recently emerging and closely related Nipah and Hendra viruses (10, 28, 46, 47). Nipah virus emerged in Peninsular Malaysia in 1998 as a result of human contact with infected swine, and recent outbreaks have been reported in Bangladesh and India between 2001 and 2005 (7, 17, 20, 27). Hendra virus emerged in Brisbane, Aus-tralia, in 1994 as a result of human contact with infected horses and last appeared in Queensland, Australia, in 2004 (3, 32). The viruses were responsible for zoonotic respiratory disease and severe acute encephalitis in humans and livestock and exhibit respiratory and neurological tropism (24). These hu-man pathogens are set apart by their low homology to other paramyxoviruses, wide host range, and high level of virulence, which has limited investigations into the interaction of virally encodedHenipavirusproteins with their natural host and sus-ceptible livestock. Furthermore, these viruses caused signifi-cant economic losses for the Malaysian swine industry (30), and their potential use in bioterrorist scenarios (23), due to a lack of therapeutic intervention, warrants further investigation. The cellular response to type I interferons (IFNs) (IFN-␣ and IFN-, collectively referred to as IFN) results in the es-tablishment of a potent antiviral state in addition to regulating components of the adaptive immune response (21, 22). This
process is mediated by the signal transducer and activator of transcription (STAT) protein family. Following IFN exposure, STAT proteins translocate to the nucleus to directly regulate cellular gene transcription, creating a cellular antiviral state (1). Many viruses have evolved well-characterized adaptations to evade the IFN-induced antiviral responses of their hosts (15). The familyParamyxoviridae, comprised of enveloped neg-ative-strand RNA viruses in several distinct genera, has devel-oped unique molecular mechanisms of IFN signaling inhibi-tion, several of which have been attributed to a virus-encoded protein derived from a polycistronic gene (10, 18, 19).
As with other paramyxoviruses, the cotranscriptional insertion of nontemplated nucleotides generates alternative mRNAs from the HenipavirusP locus, encoding the P, V, and W proteins (45). A fourth protein, C, is generated by alternate translation initiation site selection from all these mRNAs and is unrelated to the other products. The P, V, and W proteins share 407 amino acids in their N termini but differ in their C termini as a result of alternate reading frames produced by the addition of nontemplated nucleotides at the “editing site” (Fig. 1A). The P protein contains an additional 301 amino acids to pro-duce a 78-kDa polypeptide. The P protein is highly phosphory-lated, albeit by unknown kinase(s) (43), and is an essential component of the RNA transcription and replication machin-ery (8). The V protein C-terminal domain (CTD) is 50 amino acids in length and encodes a zinc finger domain that is a highly conserved hallmark domain of paramyxovirus V proteins (26, 36). The Nipah virus and Hendra virus V proteins are well known to antagonize antiviral signaling by interaction and in-terference with STAT1 and STAT2 transcription factors (38,
* Corresponding author. Mailing address: Pancoe-ENH Research Pavilion, Northwestern University, 2200 Campus Drive, Evanston, IL 60208. Phone: (847) 491-5530. Fax: (847) 491-4400. E-mail: horvath @northwestern.edu.
䌤Published ahead of print on 16 April 2008.
6259
on November 8, 2019 by guest
http://jvi.asm.org/
39, 41, 42). The Nipah virus V protein shuttles between the nucleus and cytoplasm, which is mediated by a chromosomal region maintenance 1-dependent nuclear export signal at amino acids 174 to 192 (37), and this behavior and sequence are conserved in the Hendra virus V protein.HenipavirusV proteins inhibit IFN responses by sequestering STAT1 and STAT2 in high-molecular-weight cytoplasmic complexes (38, 39). As a result, IFN-induced STAT tyrosine phosphorylation is prevented, resulting in the inhibition of an antiviral state. The primary IFN evasion target of the Nipah virus V protein is STAT1, which is absolutely required for the STAT2
[image:2.585.66.513.63.413.2]associa-tion (37). Both IFN evasion and STAT1 binding were mapped to amino acids 100 to 160 in the N terminus of the Nipah virus V protein. In addition, the Henipavirus V protein CTD was found to interact with the RNA sensor MDA5, preventing double-stranded RNA signaling (2, 6). The W protein CTD is 44 amino acids long and contains a functional nuclear import signal (42). The Nipah virus W protein has been reported to modulate both STAT1 and Toll-like receptor 3-dependent IFN regulatory factor 3 signaling (41). All four products of the Nipah virus P locus have been demonstrated to antagonize cellular antiviral responses (34), and due to their identical
FIG. 1.HenipavirusP, V, and W proteins associate with PLK1. (A) Schematic representation of the Nipah virus P, V, and W proteins (NiP, NiV, and NiW, respectively) illustrating the STAT1 binding domain, the cysteine-rich CTD, and the nuclear localization signal (NLS). (B) Nipah virus V and Hendra virus V (HeV) but not measles virus V (MeV) proteins copurify with endogenous PLK1. 293T cells were transfected to express FLAG-tagged measles virus V, Nipah virus V, Hendra virus V, or GFP, followed by immunoprecipitation with FLAG affinity gel and elution with FLAG peptide. The FLAG eluate and lysate samples were subjected to immunoblot analysis to detect endogenous PLK1 or FLAG-V protein. The asterisk indicates the immunoglobulin G (IgG) heavy chain, which migrates near PLK1. (C) Expressed HA-tagged PLK1 copurifies with Nipah virus V and Hendra virus V but not measles virus V. 293T cells were transfected to express HA-PLK1 together with either FLAG-tagged measles virus V, Nipah virus V, Hendra virus V, or GFP, followed by HA immunoprecipitation and elution with HA peptide. The HA immune complexes and lysate samples were subjected to immunoblotting with the FLAG antibody to detect V or the anti-HA antibody to detect HA-PLK1. (D) Nipah virus P and W proteins copurify with endogenous PLK1. 293T cells were transfected to express FLAG-tagged Nipah virus P, Nipah virus V, Nipah virus W, or GFP, followed by immunoprecipitation with FLAG affinity gel and elution with FLAG peptide. The FLAG eluate and lysate samples were subjected to immunoblot analysis to detect endogenous PLK1 or FLAG-V protein. (E) Purified HA-PLK1 phosphorylates recombinant
HenipavirusV proteins in vitro. (i) Purification of PLK1. 293T cells were transfected to express HA-PLK1, followed by HA immunoprecipitation (I.P.) and elution with HA peptide. Purified PLK1 and the input samples were subjected to SDS-PAGE and silver staining. (ii) Bacterial expression of recombinantHenipavirusGST-V proteins. Recombinant GST-FLAG-Nipah virus V, GST-FLAG-Hendra virus V, and control GST-FLAG were purified from bacteria and analyzed by SDS-PAGE and Coomassie blue staining. (iii)HenipavirusV proteins are phosphorylated by purified HA-PLK1. Approximately 1, 5, or 10 ng of purified PLK1 was incubated with 5g of bacterially expressedHenipavirusV protein or dephosphor-ylated bovine casein in the presence of [␥32-P]ATP. Reaction mixtures were resolved by SDS-PAGE and visualized by autoradiography.
6260 LUDLOW ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
N-terminal domains, the P, V, and W proteins all share the 60-amino-acid STAT1 binding site between residues 100 and 160, which may account for much of their observed IFN sig-naling evasion capabilities. The formation of protein com-plexes is crucial for V-mediated IFN evasion, but more infor-mation regarding the interface with the host cell is essential for understandingHenipavirus pathogenesis and designing thera-peutic or antiviral reagents. Affinity purification was used to identify additional host factors that might contribute to Heni-pavirus biology. This approach identified polo-like kinase 1 (PLK1) as being aHenipavirusP, V, and W protein-interacting partner. Data indicate that PLK1 is recruited by the 60-amino-acid STAT1 binding domain and phosphorylates the Nipah virus V protein. Molecular analysis demonstrates the impor-tance of consensus phosphopeptide polo box domain (PBD) binding motifs with overlapping functions for both PLK1 and STAT1 interactions.
MATERIALS AND METHODS
Cell culture.Human embryonic kidney 293T, human fibrosarcoma 2fTGH, 2fTGH-derivative U3A:STAT1-deficient (29), and U6A:STAT2 deficient (25) cell lines were maintained in Dulbecco’s modified Eagle medium supplemented with cosmic calf serum (10%; HyClone), penicillin (100 U/ml), and streptomycin
(100g/ml; Gibco-BRL).
Plasmids, transfections, and luciferase assay. Expression plasmids pEF-FLAG, pEF-FLAG-measles V, pEF-FLAG-Nipah V, and pEF-FLAG-Hendra V were described previously (33, 38, 39). Nipah virus V truncation vectors and Rev1.4-green fluorescent protein (GFP) fusion vectors were previously described (37). FLAG-tagged Nipah virus and Hendra virus V cDNAs were amplified by PCR and subcloned in frame into vector pGEX-6P-1 (GE Healthcare) to allow
the production of bacterially expressed glutathioneS-transferase (GST) fusion
proteins. Nipah virus P and W cDNAs were subcloned in frame by PCR into vector pEF-FLAG using templates pTM1-NiP and pCAGGS-NiW (kindly pro-vided by Michael K. Lo and Paul A. Rota, CDC, Atlanta, GA). Human PLK1 cDNA (GenBank accession number NM_005030.3; OriGene) was amplified by PCR and cloned into pcDNA3 (Invitrogen) with an N-terminal hemagglutinin (HA) epitope sequence. PLK1 fragments encoding the N terminus (bp 1 to 1034) and C terminus (bp 1035 to 1809) were generated by PCR and subcloned in frame into plasmid pcDNA3-HA. Single point mutations in Nipah virus V, Nipah virus P, and PLK1 were introduced using the QuikChange II site-directed mu-tagenesis kit (Stratagene) according to the manufacturer’s recommendations. All cDNA constructs and point mutations were verified by DNA sequencing.
High-efficiency transient transfection of 293T cells for affinity purification was
carried out by use of a standard calcium phosphate method where 3g of
FLAG-tagged V protein was transfected per 100-mm plate. For cotransfection experiments, FLAG-V and HA-PLK1 were expressed at a 1:1 ratio. U3A and U6A cells were transfected using SuperFect (Qiagen). For luciferase assays,
2fTGH or 293T cells in six-well plates were transiently transfected with 2g of
reporter gene (5⫻ IFN-stimulated response element (ISRE)-luciferase for
IFN-␣and 4⫻M67 IFN-␥-activated site (GAS)-luciferase for IFN-␥) and 6g
of FLAG-tagged V or plasmid pEF-FLAG alone using SuperFect (Qiagen).
IFN-␣(1,000 U/ml) or IFN-␥(5 ng/ml) was added for 10 h, and analysis was
performed using the dual-luciferase assay system (Promega); as an internal
control, theRenillaluciferase construct was used to normalize activity.
Cell extracts, immunoprecipitation, and immunoblotting. Cells transfected with expression plasmids were lysed in whole-cell extract buffer (50 mM Tris-HCl [pH 8.0], 280 mM NaCl, 0.5% NP-40, 0.2 mM EDTA, 0.2 mM EGTA, 10% glycerol, 1 mM dithiothreitol supplemented with protease inhibitor cocktail
[Complete; Boehringer Mannheim], and 1 mM Na3VO4). The clarified cell lysate
was precleared with Sepharose 6B (Sigma-Aldrich) and then incubated with anti-FLAG M2 affinity agarose (Sigma-Aldrich) or EZview Red anti-HA affinity gel (Sigma-Aldrich) to purify FLAG- or HA-tagged protein by immunoaffinity. Immunoprecipitation of the GFP fusion protein was previously described (37). Agarose beads with immune complexes were washed five times with whole-cell extract buffer and once with PEB:300 buffer (20 mM Tris-HCl [pH 7.5], 300 mM NaCl, 0.2 mM EDTA, 0.1% NP-40, 15% [vol/vol] glycerol), and bound proteins
were competitively eluted in PEB:300 buffer using 150g of 3⫻FLAG peptide
(Sigma-Aldrich) or influenza virus HA peptide (Sigma-Aldrich), boiled in
pro-tein loading buffer, and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for immunoblot analysis. Protein was transferred
onto nitrocellulose, and antibodies were as follows: rabbit anti-STAT1␣(1:4,000)
(sc-345; Santa Cruz), rabbit anti-STAT2 (1:4,000) (sc-476; Santa Cruz), mouse anti-PLK1 (1:2,000) (ab17056; AbCam), rabbit anti-FLAG tag (1:4,000) (F7425; Sigma-Aldrich), and rabbit anti-HA tag (1:4,000) (H6908; Sigma-Aldrich). Pri-mary antibody incubations were done overnight at 4°C. Secondary antibodies used were goat anti-mouse and goat anti-rabbit conjugated to horseradish per-oxide (VWR). Detection was performed with enhanced chemiluminescence re-agent (Perkin-Elmer).
Proteomics analysis.Following large-scale immunoprecipitation of FLAG-Nipah virus V and FLAG-Hendra virus V, copurified proteins were separated by SDS-PAGE, and large gels were stained using Coomassie brilliant blue R-250 (0.1% [wt/vol] R-250 in 40% [vol/vol] methanol–10% [vol/vol] acetic acid; Bio-Rad) and destained in 40% (vol/vol) and 10%(vol/vol) acetic acid. Selected protein bands were excised for identification using the thermo-LTQ-FT mass spectrometer at the CBC-RRC Proteomics Core Laboratory (University of Illi-nois at Chicago). Peptides with multiple hits were assessed using the Mascot 2.1 software platform and Bioworks 3.2 Turbosequest.
In vitro kinase assay.To obtain purified PLK1, 293T cells were transfected using the calcium phosphate transfection method with pcDNA3.1-HA-PLK1. Cells were lysed in TBSN buffer [20 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.5%
NP-40, 5 mM EGTA, 1.5 mM EDTA supplemented with 0.5 mM Na3VO4, 20
mMp-nitrophenyl phosphate (Sigma-Aldrich), 1 mM
4-(2-aminoethyl)-benzene-sulfonyl fluoride (AEBSF) (Pefabloc; Roche) supplemented with protease in-hibitor cocktail (Complete; Boehringer Mannheim)]. Immunoprecipitation was carried out using HA affinity gel, and bound complexes were eluted using HA
peptide in TBMD buffer (50 mM Tris-Cl [pH 8.0], 10 mM MgCl2, 5 mM
dithiothreitol, 2 mM EGTA, 0.5 mM Na3VO4, and 20 nMp-nitrophenyl
phos-phate [Sigma-Aldrich]), and purified PLK1 was visualized using silver stain and quantified using Bradford estimation (4a). Kinase activity assays were carried out
in TBMD buffer; approximately 5g of bacterially produced V protein or 3g
of dephosphorylated bovine casein (Sigma-Aldrich) substrate was combined with
either 1, 5, or 10 ng of purified HA-PLK1 in the presence of 50M ATP and 5
Ci of [␥-32P]ATP. Following incubation at 30°C for 30 min, the reaction was
terminated by the addition of protein gel loading buffer to the reaction mixture, which was separated by SDS-PAGE and visualized by autoradiography.
Immune complex kinase assay.Immunoprecipitation of FLAG-tagged V pro-teins was carried out as described above, and bound propro-teins were eluted in TBMD buffer supplemented as described above. Eluates were incubated in the
presence of 50M ATP and 5Ci of [␥-32
P]ATP at 30°C for 30 min; the reaction was terminated by the addition of protein gel loading buffer to the reaction mixture, which was separated by SDS-PAGE and visualized by autoradiography.
Indirect immunofluorescence.2fTGH cells were grown to 60 to 80% conflu-ence in Permanox chamber slides (Nalgene) and transfected with FLAG-tagged V protein using SuperFect (Qiagen). Indirect immunofluorescence was per-formed as described previously by Rodriguez et al. (38) using mouse anti-FLAG M2 monoclonal antibody (Sigma), followed by Alexa 546-conjugated anti-mouse secondary antibody (Invitrogen). The second stain for STAT1 and STAT2 was detected with anti-rabbit fluorescein isothiocyanate (Invitrogen). Images were obtained using a Leica confocal microscope at the Biological Imaging Facility, Northwestern University, Evanston, IL.
Antiviral assay.Pools of 2fTGH cells stably expressing Nipah virus V protein, Hendra virus V protein, phosphopeptide binding motif mutations, and the
pEF-FLAG control were generated by the cotransfection of 10g of V DNA and 1
g pBABE-puro (31) using SuperFect (Qiagen). Resistant pools were selected
for 2 weeks using 1g/ml puromycin (Sigma), and expression was verified using
immunoblotting. Following an 8-h treatment with IFN-␣or medium alone, cells
were infected with recombinant vesicular stomatitis virus (VSV) harboring a GFP transgene (VSV-GFP) (a gift of John Hiscott, Montreal, Canada). Infec-tions were performed at a multiplicity of infection of 0.6 PFU/ml, and virus was diluted in serum-free medium and added to the cells for 1 h to allow adsorption. Virus was then removed, and Dulbecco’s modified Eagle medium with 2% cosmic calf serum was added for the remaining time. After 22 h, GFP levels were analyzed by flow cytometry (BD FACSCalibur; BD Biosciences), and data were assessed using CELLquest (BD Biosciences). Percent GFP was calculated in comparison to uninfected 2fTGH cells. Cells were photographed using a Zeiss inverted fluorescence microscope.
Minigenome assay and CAT ELISA.BHK/sr/T7 cells were transfected with
1.75g of Nipah virus N, 0.8g of Nipah virus P (or Nipah virus P point
mutations), 1.2g of Nipah virus L, and 3.5g of Nipah virus chloramphenicol
acetyltransferase (CAT) minigenome plasmid constructs in Opti-MEM medium (Life Technologies, Invitrogen) using LT-1 transfection reagent (Mirus)
on November 8, 2019 by guest
http://jvi.asm.org/
ing to the manufacturer’s instructions. Total amounts of transfected DNA were kept constant by the addition of pTM1 empty vector DNA where applicable. For the negative control, the Nipah virus L plasmid was replaced with an equivalent amount of pTM1 empty vector. Details of the Nipah virus minigenome replica-tion assay were described elsewhere previously (16). Cytoplasmic extracts were
prepared⬃42 h posttransfection in 500l of lysis buffer from the CAT
enzyme-linked immunosorbent assay (ELISA) kit (Roche). To ensure comparable re-sults, the amounts of harvested cytoplasmic extracts used per sample were ad-justed for protein concentration using the BCA protein assay reagent (Pierce). The total protein concentration of the lysate supernatants and the concentration of CAT reporter protein in the samples were determined according to protein/ enzyme standards used for each respective assay. Twenty-five micrograms of total protein per sample lysate was used for the CAT ELISA (Roche), which was performed according to the manufacturer’s protocols. The assay was performed three separate times, in which each sample was assayed in duplicate.
RESULTS
Henipavirus P, V, and W proteins copurify with PLK1.
Large-scale immunoprecipitation and mass spectrometry
[image:4.585.111.465.65.446.2]iden-tified PLK1 as being aHenipavirusV protein-interacting part-ner (Fig. 1). To confirm this association, FLAG-tagged V pro-teins expressed in human embryonic kidney 293T cells were immunoprecipitated using FLAG affinity agarose and assessed for endogenous PLK1 binding. Trace amounts of PLK1 were found to coprecipitate with control GFP or FLAG-tagged measles virus V protein, but the Nipah virus and Hen-dra virus V proteins strongly copurified endogenous PLK1 (Fig. 1B). The protein interaction was independently con-firmed in human fibrosarcoma 2fTGH cells and 2fTGH cells stably expressing the FLAG-Nipah virus V protein (data not shown). In a complementary experiment, 293T cells expressing both HA-tagged PLK1 and FLAG-tagged V protein were an-alyzed by HA affinity purification (Fig. 1C). The expressed HA-PLK1 copurified with the Nipah virus and Hendra virus V proteins but not the measles virus V protein or control FLAG-GFP. To determine if the Nipah virus P and W proteins (Fig.
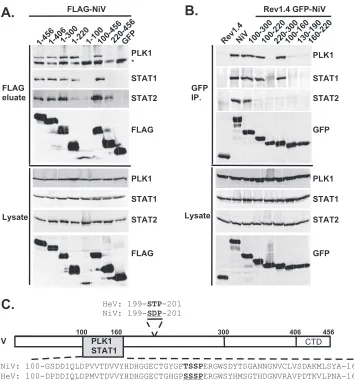
FIG. 2. PLK1 binds Nipah virus V protein via the STAT1 binding domain. (A) Selective PLK1, STAT1, and STAT2 binding to Nipah virus V (NiV) fragments. 293T cells were transfected to express FLAG-tagged Nipah virus V, GFP control, and Nipah virus V truncation constructs (37), followed by FLAG immunoprecipitation and elution with FLAG peptide. The FLAG eluate and lysate samples were analyzed by immunoblotting using antibodies to detect endogenous PLK1, STAT1, STAT2, and FLAG-V proteins. (B) Interaction of Nipah virus V protein with PLK1, STAT1, and STAT2. 293T cells were transfected to express the Rev1.4-GFP fusion vectors as indicated (37), and lysates were immunoprecipitated (IP) with antisera to GFP. Immune complexes were probed for PLK1, STAT1, STAT2, and GFP. (C) Schematic representation of Nipah virus V protein illustrating the PLK1 and STAT1 interaction domains. The consensus S-[pS/pT]-P motifs for Nipah virus and Hendra virus V (HeV) proteins are indicated by boldface type and double underlining.
6262 LUDLOW ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
1A) also copurified with endogenous PLK1, FLAG-tagged P and W proteins expressed in 293T cells were immunoprecipi-tated and assessed for endogenous PLK1 binding (Fig. 1D). In addition to the V protein, both the P and W proteins copre-cipitated with PLK1. These results confirm PLK1 to be a He-nipavirusprotein partner.
Purified PLK1 phosphorylates recombinant HenipavirusV proteins in vitro.To determine if PLK1 is able to phosphory-late theHenipavirus V proteins, an in vitro kinase assay was performed. PLK1 was purified by immunoprecipitation of HA-tagged PLK1 expressed in human 293T cells, and silver stain-ing revealed a highly enriched PLK1 preparation (Fig.1Ei). Recombinant GST-FLAG-Nipah V, GST-FLAG-Hendra V, and control GST-FLAG were expressed and purified from
Escherichia colicells (Fig. 1Eii). Approximately 1, 5, or 10 ng of purified PLK1 was incubated with 5 g of bacterially ex-pressedHenipavirusV protein or a control, dephosphorylated bovine casein in the presence of [␥32-P]ATP. Reaction
mix-tures were resolved by SDS-PAGE and visualized by autora-diography (Fig.1Eiii). Like the positive casein control, Henipa-virus V proteins were phosphorylated by PLK1 in a dose-dependent manner, but GST alone was not. This result
suggests that the PLK1-V protein interaction can lead to V protein phosphorylation.
PLK1 binds Nipah virus V protein via the STAT1 binding domain.To determine the V protein region required for the PLK1 association, two sets of previously described tagged Nipah virus V protein fragments were used for immunopre-cipitation experiments (37). The immune complexes were tested for the presence of PLK1, STAT1, STAT2, and FLAG-V protein (Fig. 2A and B). In agreement with previ-ously published results, STAT1 was found to interact with Nipah virus V amino acids 100 to 160, and STAT2 binding was confirmed for residues 100 to 300, with the fragment at resi-dues 220 to 456 retaining a partial STAT2 association (37). Endogenous PLK1 was detected in the exact same pattern as STAT1, mapping its interaction to Nipah virus V protein res-idues 100 to 160 (Fig. 2A and B). This finding revealed a shared 60-amino-acid region at residues 100 to 160 that is necessary and sufficient for both PLK1 and STAT1 interactions with the Nipah virus V protein (Fig. 1A).
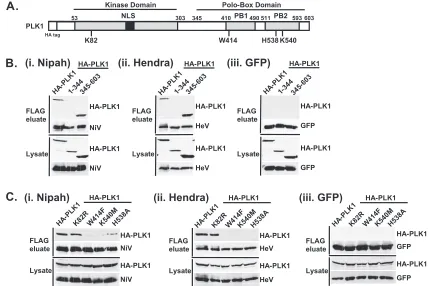
The PLK1 PBD mediates interactions with bothHenipavirus
[image:5.585.76.509.69.355.2]V proteins.PLK1 is a member of the polo kinase family char-acterized by a bipartite domain structure. The N-terminal 344
FIG. 3. The PLK1 PBD is necessary and sufficient to mediateHenipavirusV interactions. (A) Illustration of PLK1 domain structure. The PLK1 N-terminal kinase domain containing residue K82, which is required for ATP binding, and the C-terminal PBD containing residues W414, H538, and K540, which are required for contact with phosphopeptide substrates, are overlined. (B) The PBD mediatesHenipavirusV protein interactions. 293T cells were cotransfected to express HA-PLK1, the HA-PLK1 N-terminal kinase domain (residues 1 to 344), or the C-terminal PBD (residues 345 to 603) together with either FLAG-tagged Nipah virus V (NiV) (i), Hendra virus V (HeV) (ii), or GFP (iii), followed by FLAG immuno-precipitation and elution. The FLAG immune complexes and lysate samples were subjected to immunoblot analysis to detect FLAG-V, HA-PLK1, or HA-PLK1 fragments. (C) PBD contact residues mediate V protein interactions. 293T cells were cotransfected to express HA-PLK1 or HA-PLK1 point mutations together with either FLAG-tagged Nipah virus V (i), Hendra virus V (ii), or GFP (iii), followed by FLAG immuno-precipitation and elution. The FLAG immune complexes and lysate samples were subjected to immunoblotting to detect FLAG-V, HA-PLK1, or HA-PLK1 point mutations.
on November 8, 2019 by guest
http://jvi.asm.org/
amino acids form the catalytic kinase domain, while the C-terminal 259 amino acids form the PBD (Fig. 3A). This bipar-tite structure is involved in substrate selection and kinase ac-tivation. Current evidence supports a model that PLK1 is inactive in the steady state, but substrate proteins containing a phosphopeptide PBD binding motif trigger a kinase associa-tion. Proteins containing the consensus sequence Ser-[Ser/ Thr]-Pro, or SSP, are phosphorylated by priming kinase activity, converting the motif to Ser-[pSer/pThr]-Pro, which represents a high-affinity PBD recognition sequence. When the PBD binds the substrate, the kinase domain is activated to phosphorylate
cisortranstargets (44).
To test if the V protein association is mediated by the PBD, two PLK1 fragments representing the kinase domain (amino acids 1 to 344) or the PBD (amino acids 345 to 603) were expressed along with theHenipavirusV proteins (Fig. 3B). The V proteins effectively precipitated both full-length and PBD fragments but not the kinase domain fragment (Fig. 3B). These results confirm the importance of the PBD in directing the PLK1 association with the V proteins.
Structural studies indicated that the specific coordination of the phosphopeptide motif relies on PBD residues W414, K540, and H538 (5, 12, 14). The W414F, K540M, and H538A muta-tions are known to abolish PLK1 binding to Cdc25C (12, 14).
Immunoprecipitation was carried out as described above using the PBD W414F, K540M, and H538A mutations and the ki-nase-inactive N terminus K82R mutant as a control (40) (Fig. 3A). As expected,HenipavirusV proteins bound well to PLK1 and the K82R N-terminal kinase domain mutation. In contrast, all the PBD mutations resulted in defective PLK1 recognition (Fig. 3C). Together, these findings implicate the PBD as being the V protein interaction module.
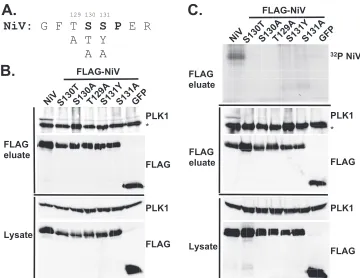
Distinct PLK1 binding motifs mediate Nipah virus and Hendra virus V protein association and phosphorylation.The PBD specifically binds to phosphopeptide motifs, and inspec-tion of the 60-amino-acid STAT1 binding domain identified an SSP motif at residues 130 to 132 of bothHenipavirusV proteins (Fig. 2C). To test the importance of this motif in PLK1 re-cruitment, selected amino acids were substituted with putative inactivating residues (Fig. 4Aand 5A) based on mutations de-scribed previously (11). The conserved serine at position 130 was mutated to both threonine, to preserve character, and alanine, and serine 131 was converted to tyrosine and to ala-nine. In addition, residue 129, which lies outside the SSP motif, was converted from threonine (in Nipah virus) or serine (in Hendra virus) to alanine.
[image:6.585.111.471.66.344.2]FLAG-tagged wild-type andHenipavirusV proteins contain-ing mutations or control FLAG-GFP were expressed in 293T
FIG. 4. Nipah virus V protein PLK1 interaction and phosphorylation require a consensus motif surrounding serine 130. (A) Selected residues of the consensus TSSP motif were mutated as indicated. (B) The TSSP motif is necessary for PLK1 binding to the Nipah virus V (NiV) protein. 293T cells were transfected to express FLAG-tagged Nipah virus V, the GFP control, or Nipah virus V point mutations, followed by FLAG immunoprecipitation and elution with FLAG peptide. The FLAG eluate and lysate samples were analyzed by immunoblotting using antibodies to detect associated PLK1 or FLAG-V protein. (C) Contact-dependent phosphorylation of Nipah virus V. 293T cells were transfected to express FLAG-tagged Nipah virus V, the GFP control, or Nipah virus V mutations, followed by FLAG immunoprecipitation and elution. Immune complexes were incubated in the presence of [␥32-P]ATP, and reaction mixtures were resolved by SDS-PAGE and visualized by autoradiography.
The FLAG eluate was further assessed by immunoblotting using antibodies detecting endogenous PLK1 and FLAG-V protein.*, immunoglobulin G heavy chain.
6264 LUDLOW ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
cells, followed by FLAG immunoprecipitation. As expected, wild-type Nipah virus V protein bound PLK1, but all mutations including threonine 129 strikingly abolished the ability of PLK1 to bind the Nipah virus V protein (Fig. 4B). Therefore, the peptide129TSSP132 is essential for PLK1 binding to the
Nipah virus V protein. The phosphorylation statuses of the substrate recognition motif mutations were tested by an im-mune complex kinase assay (Fig. 4C). Briefly, 293T cells were transfected with FLAG-tagged V, point mutations, and control GFP, followed by FLAG immunoprecipitation and elution with FLAG peptide. Immune complexes were incubated in the presence of [␥32-P]ATP, and reaction mixtures were resolved
by SDS-PAGE and visualized by autoradiography. The FLAG eluate was further assessed by immunoblotting using antibod-ies to detect endogenous PLK1 and FLAG-V protein. Wild-type Nipah virus V protein was phosphorylated, while all the TSSP motif mutations that failed to bind PLK1 were not phos-phorylated (Fig. 4C). Therefore, the phosphorylation of Nipah virus V by PLK1 is contact dependent.
The identical experiments carried out with the Hendra virus V protein mutations to the peptide129SSSP132(Fig. 5A)
re-sulted in a dramatically different phenotype (Fig. 5B). None of the mutations in the Hendra virus V motif were deleterious for
the PLK1 interaction (Fig. 5B). Therefore, the analogous SSP-containing peptide is not the PLK1 recruitment motif for the Hendra virus V protein. In agreement with this, wild-type Hendra virus V protein and all mutations retained phosphor-ylation in the immune complex kinase assay (Fig. 5C). In con-clusion, the twoHenipavirusV proteins differ in their modes of PLK1 recruitment.
Identification of the Hendra virus V protein PLK1 recruit-ment motif.Inspection of the Hendra virus V protein sequence identified a distinct consensus motif,199STP201, that was not present in the Nipah virus V protein (Fig. 2C and 5D). To test this motif as a potential PLK1 recruitment site, mutagenesis was performed. Threonine 200 was converted to alanine, and PLK1 binding and phosphorylation were tested (Fig. 5E). As expected, wild-type Hendra virus V protein bound PLK1, but the mutation abolished the interaction. Thus, threonine 200 is required for PLK1 binding to the Hendra virus V protein. Accordingly, in immune complex kinase assays, wild-type Hen-dra virus V was phosphorylated, but the HenHen-dra virus V T200A mutation was not (Fig. 5E).
[image:7.585.73.508.71.388.2]Separation of V-PLK1 and V-STAT complexes. The ability of the Nipah virus V protein to associate with both PLK1 and STAT1 via a single 60-amino-acid region invited inquiry into
FIG. 5. Hendra virus V protein PLK1 interaction and phosphorylation require an alternate consensus motif surrounding threonine 200. (A) Selected residues of the conserved SSSP motif were mutated as indicated. (B) Hendra virus V (HeV) protein mutations retain PLK1 binding. 293T cells were transfected to express FLAG-tagged Hendra virus V, the GFP control, or Hendra virus V point mutations and analyzed as described in the legend of Fig. 4. (C) Hendra virus V mutations retain phosphorylation. Immune complex kinase assays were carried out as described in the legend of Fig. 4. (D) An STP motif that is not conserved with Nipah virus surrounding T200 in the Hendra virus V protein mediates the PLK1 interaction. T200 was mutated to A as indicated. (E) T200 is required for PLK1 binding to the Hendra virus V protein. Proteins were expressed and analyzed for PLK1 associations and phosphorylation as described above.
on November 8, 2019 by guest
http://jvi.asm.org/
the relationship between the two cellular proteins and their interactions with the V protein. First, the V protein could be coprecipitating an extant PLK1-STAT complex from the cell. Second, V could nucleate the formation of a higher-order complex including both PLK1 and STAT1. Third, the V pro-tein could assemble with PLK1 and STAT1 independently.
To test the possibility that PLK1 might be able to associate with STAT1 in the absence of the V protein, 293T cells were transfected with HA-tagged PLK1 followed by HA immuno-precipitation and peptide elution (Fig. 6A). The lysate, sequen-tial HA eluates (E1 and E2), and the supernatant following immunoprecipitation were evaluated by immunoblotting using antibodies detecting HA-PLK1, STAT1, and STAT2. PLK1 did not copurify with STAT1 and STAT2 in the absence of V protein expression, suggesting that there is little or no pre-formed PLK1-STAT association.
To confirm that the V-PLK1 interaction occurred indepen-dently of STAT1 and STAT2, parental human fibrosarcoma 2fTGH and derivative cells lines with single gene defects in STAT1, U3A (29), or STAT2, U6A (25), were utilized (Fig. 6B). Cells were transfected with FLAG-taggedHenipavirusV protein vectors or GFP followed by FLAG immunoprecipita-tion. Immune complexes and lysates were analyzed to detect PLK1, STAT1, and STAT2. In parental 2fTGH cells, Henipa-virusV protein copurified with endogenous PLK1, and binding patterns for STAT1 and STAT2 matched previous findings; in particular, we confirmed thatHenipavirus V proteins require cellular STAT1 to interact with STAT2 (37). Notably, in the absence of STAT1 or STAT2, V proteins retained the ability to copurify endogenous PLK1. These results indicate that PLK1:V interactions do not require STAT proteins.
To establish if the V-induced association with STATs and PLK1 produces a single tripartite complex, a sequential immu-noprecipitation assay was used. HA-tagged PLK1 was ex-pressed together with FLAG-taggedHenipavirusV proteins or GFP followed by FLAG immunoprecipitation and elution with FLAG peptide. The FLAG eluates were then reimmunopre-cipitated with HA affinity gel and eluted using HA peptide. Lysate, FLAG eluate, and HA eluate fractions were assessed by immunoblot analysis to detect STAT1, STAT2, or PLK1 (Fig. 6C). No background signals were observed with control FLAG-GFP.HenipavirusV proteins copurified with endoge-nous STAT1, STAT2, and HA-tagged PLK1, but reimmuno-precipitation with HA affinity gel separated HA-PLK1 from STAT1 and STAT2. This result demonstrates the segregation of the V:STAT and V:PLK1 complexes.
HenipavirusSSP motifs are involved in STAT1 binding.The location of the PLK1 binding motif in the center of the STAT1 binding region of the Nipah virus V protein suggests a con-served function that might be related to IFN evasion. To eval-uate the effects of the Henipavirus SSP motif mutations on STAT1 and STAT2 interactions, eluates from FLAG-V immu-noprecipitations were analyzed by immunoblotting with anti-sera for STAT1 or STAT2 (Fig. 7A). As expected, wild-type
HenipavirusV proteins bound STAT1 and STAT2, but some of the proteins containing mutations were defective for STAT interactions. The conservative substitution of serine with threonine at position 130 did not affect STAT binding, but the substitution to alanine significantly reduced the interaction. Changing Nipah virus V protein T129 or its Hendra virus counterpart S129 to alanine did not affect STAT binding. In contrast, the substitution of S131 with tyrosine or alanine
dra-FIG. 6. Independence of STAT and PLK1 complexes. (A) PLK1 does not associate with STAT1 and STAT2 in the absence of V protein. 293T cells were transfected to express HA-tagged PLK1, followed by HA immunoprecipitation and elution. The lysate, sequential HA peptide eluates (E1 and E2), and the supernatant following immunoprecipitation (I.P) were assessed by immunoblotting to detect HA-PLK1, STAT1, or STAT2. (B) The PLK1 and V protein interactions are independent of STAT1 or STAT2. Parental 2fTGH cells, STAT1-deficient U3A, or STAT2-deficient U6A derivatives were transfected to express FLAG-tagged V proteins or GFP, followed by FLAG immunoprecipitation. Immune complexes and lysates were analyzed by immunoblotting to detect PLK1, STAT1, STAT2, and FLAG-V. (C) V:STAT and V:PLK1 exist as distinct complexes. 293T cells were transfected to express HA-PLK1 together with either FLAG-tagged Nipah virus V (NiV), Hendra virus V (HeV), or GFP, followed by FLAG immunoprecipitation and elution with FLAG peptide (FLAG eluate). The FLAG immune complexes were then reimmunoprecipitated with HA affinity gel and eluted using HA peptide (HA eluate). Lysate (input), FLAG, and HA eluates were assessed by immunoblot analysis to detect endogenous PLK1, STAT1, or STAT2 and then reprobed to detect HA-PLK1 or FLAG-V.
6266 LUDLOW ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.65.528.69.258.2]matically reduced the coprecipitation of STATs. Clearly, the serine residues at positions 130 and 131 in bothHenipavirusV proteins contribute to the STAT association. In addition, the T200A substitution in Hendra virus V protein that eliminated the PLK1 association did not influence the STAT1 interaction (Fig. 7Aiii), a finding that is in agreement with the mapping of the core STAT binding region defined for the Nipah virus V protein (37) (Fig. 2C).
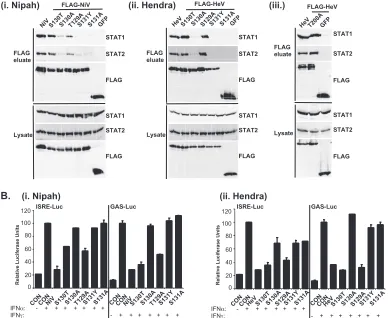
HenipavirusSSP motifs are required for STAT1 relocaliza-tion and IFN signaling evasion. To verify these results in a biological context, the abilities of the mutated proteins to pre-vent IFN signal transduction were tested in luciferase assays (Fig. 7B). Wild-typeHenipavirusV proteins prevented the in-duction of STAT-dependent IFN-␥-responsive and IFN-␣ -re-sponsive reporter genes. IFN signaling inhibition correlated exactly with STAT binding: only those V proteins that bound STATs were able to block IFN signaling. In addition, the Hendra virus V T200A substitution mutant that retained the STAT interaction also inhibited IFN signaling (not shown).
The mechanism of V protein inhibition involves the reten-tion of STATs in the cytoplasm. To confirm the consequences
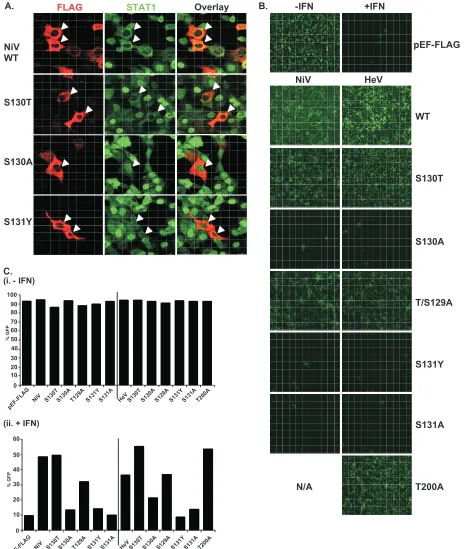
of the STAT interaction, indirect immunofluorescence was performed using 2fTGH cells transfected with FLAG-tagged Nipah virus V and the S130T, S130A, and S131Y point muta-tions followed by IFN-␥treatment to induce STAT1 translo-cation and accumulation in the nucleus (Fig. 8A). Both wild-type Nipah virus V and the S130T mutation retained STAT1 in the cytoplasm. In contrast, no STAT1 interference was de-tected in cells that expressed the S130A and S131Y mutations, as STAT1 still accumulated in the nucleus.
[image:9.585.99.487.79.397.2]To verify these results in the biologically meaningful context of virus infection, an antiviral assay was performed (Fig. 8B and C). Briefly, pools of 2fTGH cells expressing the control pEF-FLAG plasmid, wild-type V protein, or point mutations were treated with IFN-␣or medium alone and then infected with a reporter virus, VSV-GFP. Approximately 90% of un-treated cells became infected with VSV-GFP, but treatment with IFN-␣ resulted in cellular resistance to VSV infection (Fig. 8B and C). Wild-type V proteins and all variants capable of binding to STAT1 prevented the antiviral state, but the S130A, S131Y, and S131A mutations that failed to inactivate the STAT proteins did not (Fig. 8B and Cii).
FIG. 7. The conserved T/S SSP motif is necessary for interaction with STAT1 and STAT2. (A) T/S SSP motifs are required for STAT1 and STAT2 V protein interactions. 293T cells were transfected to express FLAG-tagged Nipah virus V (NiV) (i) or Hendra virus V (HeV) (ii), followed by FLAG immunoprecipitation and elution. The FLAG eluate and lysate samples were analyzed by immunoblotting using antibodies to detect endogenous STAT1, STAT2, or FLAG-V protein. (iii) Control analysis of the FLAG-Hendra virus V T200A point mutation. (B) IFN signaling inhibition correlates with STAT1 and STAT2 binding abilities. 2fTGH cells were transfected with the GAS- or ISRE-luciferase (Luc) reporter gene and Nipah virus V (NiV) (i), Hendra virus V (HeV) (ii), point mutations, or an empty vector (CON), as indicated. Cells were stimulated with IFN-␥ (5 ng/ml) or IFN-␣(1,000 U/ml) for 10 h or not stimulated (⫺) prior to lysis and luciferase assays. Data were normalized to cotransfectedRenilla
luciferase, and the bars indicate the averages (n⫽3)⫾the standard deviations.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 8. The conserved T/S SSP motif is essential for STAT1 relocalization and IFN signaling evasion. (A) A subset of mutated V proteins fail to retain STAT1 in the cytoplasm. 2fTGH cells were transfected with FLAG-tagged Nipah virus V (NiV) and point mutations, followed by indirect immunofluorescence to detect FLAG-V and STAT1. WT, wild type. (B) V proteins that fail to bind STAT1 do not disrupt the IFN antiviral state. Pools of 2fTGH cells stably expressing Nipah virus V protein, the Hendra virus V protein (HeV), phosphopeptide binding motif mutations, and the pEF-FLAG control were treated with medium alone or IFN-␣for 8 h, followed by VSV-GFP infection. (C) After 22 h, GFP levels were analyzed by flow cytometry, and the percentage of cells expressing GFP was calculated in comparison to uninfected 2fTGH cells. N/A, not applicable.
6268 LUDLOW ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
STAT1 and PLK1 interactions are dispensable for Nipah virus RNA transcription and replication. Our experimental procedures used Henipavirus V proteins to identify PLK1. However, the STAT1 and PLK1 binding region is shared among three proteins, P, V, and W. Both V and W have been referred to as accessory proteins required for host-suppressing actions and are not always required for virus replication in cell culture. In contrast, while expressed Nipah virus P protein can exhibit IFN signaling interference in vitro (42), it is better known to be an essential component of the viral RNA tran-scription and replication machinery (16). To study the effects of STAT1 or PLK1 binding deficiencies on the function of Nipah virus P protein, a Nipah virus minigenome replication assay that relies on the expression of the L, P, and NP proteins to replicate a CAT reporter gene was used (16). The assay was carried out with the wild type or TSSP mutations engineered in the context of the Nipah virus P protein. The substitution of mutated P proteins for the wild type did not alter replication activity, as all of the mutated proteins supported transcription and replication to the same extent as the wild-type P protein (Fig. 9).
DISCUSSION
PLK1 has essential roles in cell cycle regulation, controlling mitotic spindle formation and checkpoint responses to DNA damage. It has also been directly or indirectly linked to the replication of diverse viruses. For example, PLK1 interacts with and phosphorylates the human cytomegalovirus pp65 lower matrix protein and accompanies pp65 into budding par-ticles (13). In addition, deregulated levels of PLK1 have been associated with simian immunodeficiency virus (4), human T-lymphotrophic virus type I p30 protein (9), and human papil-lomavirus type 16 E6 and E7 proteins (35). The identification of PLK1 as a Nipah virus and Hendra virus P, V, and W protein binding partner and the demonstration of interaction-dependent V protein phosphorylation now connects this cel-lular Ser/Thr protein kinase to the emerging negative-strand RNA viruses in the paramyxovirus genusHenipavirus. As with
other PLK targets, the mode of interaction with both Nipah virus and Hendra virus V proteins was found to involve the PBD-dependent recognition of Ser-[pSer/pThr]-Pro peptides. The identification of this motif is significant because the He-nipavirusV proteins represent the first virally encoded proteins found to contain a functional PBD binding motif. However, several features that distinguish the two V proteins were iden-tified. For Nipah virus, it was found that PLK1 interacts with a Ser-Ser-Pro motif surrounding serine 130. Despite the absolute conservation of the core PLK1 triad of Ser-Ser-Pro between Nipah virus and Hendra virus V proteins, the ability of this peptide to mediate the interaction with PLK1 was not con-served. Instead, the Hendra virus V protein interaction with PLK1 was conferred by another consensus site, Ser-Thr-Pro, surrounding threonine 200 (Table 1). In both cases, the integ-rity of the sequence motif is essential for PLK1-dependent V protein phosphorylation. To account for the different binding sites, we speculate that differences in flanking sequences may determine the association with a V protein-specific priming kinase(s) used to create the essential pSer/pThr central to PBD recognition. One clue that may aid in the identification of the priming kinase is the observed critical importance, in the Nipah virus V protein, of Thr129 in the ⫺2 position (with respect to the pSer). When Thr129 is substituted, the mutated Nipah virus V protein no longer recruits or gets phosphory-lated by PLK1. The potential involvement of multiple or di-verse priming kinases may not be implausible, as the Henipa-virus P and V proteins are well known to be highly phosphorylated (43). In fact, the entire V protein N terminus, comprising over 75% of the sequence, including the PLK1 binding sites, is shared with the viral phosphoprotein, P. Our molecular studies demonstrate interaction-dependent phos-phorylation by PLK1, suggesting that the P, V, or W protein could represent a PLK1 substrate in vivo. However, the data indicate that the ability to associate with PLK1 does not alter the P protein’s function in RNA synthesis, as Nipah virus P proteins lacking the TSSP consensus were competent for mini-genome replication.
[image:11.585.72.253.69.194.2]No alteration in cell cycle regulation was observed following the overexpression and stable expression of theHenipavirusP, V, or W protein and mutations defective for PLK1 binding (L. E. Ludlow and C. M. Horvath, unpublished observations). Analysis of these mutations in the context of virus infection is crucial to dissect the contribution of this interaction in the host
FIG. 9. Interaction of STAT1 and PLK1 is dispensable for P pro-tein-dependent Nipah virus RNA synthesis. BHK/sr/T7 cells were transfected to express Nipah virus N, Nipah virus P (or Nipah virus P point mutations), Nipah virus L, and the Nipah virus CAT minigenome plasmid. For the negative control (CON), the Nipah virus L plasmid was replaced with an equivalent amount of pTM1 empty vector. The assay was performed three separate times in which each sample was assayed in duplicate. Minigenome replication was assessed using a CAT ELISA system as described previously (16). WT, wild type.
TABLE 1. Properties of proteins containing T/S SSP mutations
Mutation
Consequence of mutationa
PLK1 STAT1 Replicon
Nipah virus V Hendra virus V Nipah virus V Hendra virus V Nipah virus P
Wild type ⫹ ⫹ ⫹ ⫹ ⫹
T/S129A ⫺ ⫹ ⫹ ⫹ ⫹
S130T ⫺ ⫹ ⫹ ⫹ ⫹
S130A ⫺ ⫹ ⫺ ⫺ ⫹
S131Y ⫺ ⫹ ⫺ ⫺ ⫹
S131A ⫺ ⫹ ⫺ ⫺ ⫹
T200A ND ⫺ ND ⫹ ND
a
ND, not determined.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:11.585.302.542.594.715.2]cells under natural conditions of infection. Determination of the precise role(s) for PLK1 duringHenipavirusinfections will require further studies using recombinant viruses grown under biosafety level 4 containment.
Equally interesting implications of this study relate to the observation that the Nipah virus V protein PLK1 interaction motif lies in the center of the previously characterized STAT1 interaction site. The interaction with STAT1 causes a cellular defect in IFN-mediated transcription and antiviral effects and is therefore fundamental to innate immune interference me-diated by both Nipah virus and Hendra virus. Evaluation of STAT binding to Hendra virus and Nipah virus V protein (T/S)SSP motif mutations revealed that Ser130 and Ser131 are both needed for binding to STAT1 and STAT2 and normal IFN signaling interference. Interestingly, the S130T mutant retained the ability to bind STATs, but the S130A mutant failed to bind STAT1 or block IFN signaling, revealing specific side-chain preferences for Henipavirus interference. Further insight was gained by examining PLK1 and STAT binding properties. PLK1 was not detected in association with STAT1 or STAT2, and the STATs were not required for V:PLK1 interactions. In agreement with this finding, the V:STAT and V:PLK1 subcomplexes exist independently within the cell.
The ability to interfere with host defense pathways is para-mount for virus pathogenesis, and we previously speculated that the ablation ofHenipavirusIFN signaling evasion would produce an attenuated virus and therefore a plausible vaccine candidate (38, 39). However, the V protein region mediating interactions with STAT1 is also present in the P protein, which is essential for viral RNA synthesis and must be functional to create a recombinant virus. S130 and S131 mutations represent attenuating point mutations, as these mutations are capable of dissociating V:STAT interactions. The analysis of P proteins containing these STAT1 and PLK1 binding mutations revealed no difference in their abilities to support minigenome tran-scription and replication. These findings reveal that it is indeed possible to dissociate the IFN signaling evasion functions of the V protein from the RNA synthesis functions of the P protein and specifically indicate that mutations of S130 or S131 are likely to generate viable viruses. We predict that these viruses would be attenuated due to the inability of the V or W protein to disrupt host cell STAT-dependent IFN antiviral immune responses and would represent vaccine candidates. This study has improved our understanding of the virus-host interface andHenipaviruspathogenesis, providing information for the design of therapeutic or antiviral reagents.
ACKNOWLEDGMENTS
We are grateful to members of the Horvath laboratory for helpful discussion and technical advice. Special acknowledgment goes to Lily Garza for expert technical support. We also thank Eric Weiss and Jennifer Brace for discussions and reading of the manuscript. Mass spectrometry-based protein identification was carried out by Alex-ander Schilling and Bao-Shang Lee at the CBC-RRC Proteomics Core Laboratory (University of Illinois at Chicago). We acknowledge Wil-liam Russin at the Biological Imaging Research Facility, Northwestern University.
This research was supported by NIH grant R01 AI055733 to C.M.H.
REFERENCES
1.Aaronson, D. S., and C. M. Horvath.2002. A road map for those who don’t
know JAK-STAT. Science296:1653–1655.
2.Andrejeva, J., K. S. Childs, D. F. Young, T. S. Carlos, N. Stock, S. Good-bourn, and R. E. Randall.2004. The V proteins of paramyxoviruses bind the inducible RNA helicase, mda-5, and inhibit its activation of the
IFN-beta promoter. Proc. Natl. Acad. Sci. USA101:17264–17269.
3.Anonymous.2004. Hendra virus—Australia (Queensland). ProMED archive number 20041214.3307. International Society for Infectious Diseases, Brookline, MA.
4.Bostik, P., G. L. Dodd, F. Villinger, A. E. Mayne, and A. A. Ansari.2004.
Dysregulation of the polo-like kinase pathway in CD4⫹T cells is
character-istic of pathogenic simian immunodeficiency virus infection. J. Virol.78:
1464–1472.
4a.Bradford, M. M.1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye
bind-ing. Anal. Biochem.72:248–254.
5.Cheng, K. Y., E. D. Lowe, J. Sinclair, E. A. Nigg, and L. N. Johnson.2003. The crystal structure of the human polo-like kinase-1 polo box domain and
its phospho-peptide complex. EMBO J.22:5757–5768.
6.Childs, K., N. Stock, C. Ross, J. Andrejeva, L. Hilton, M. Skinner, R. Randall, and S. Goodbourn.2007. mda-5, but not RIG-I, is a common target
for paramyxovirus V proteins. Virology359:190–200.
7.Chua, K. B., W. J. Bellini, P. A. Rota, B. H. Harcourt, A. Tamin, S. K. Lam, T. G. Ksiazek, P. E. Rollin, S. R. Zaki, W. Shieh, C. S. Goldsmith, D. J. Gubler, J. T. Roehrig, B. Eaton, A. R. Gould, J. Olson, H. Field, P. Daniels, A. E. Ling, C. J. Peters, L. J. Anderson, and B. W. Mahy.2000. Nipah virus:
a recently emergent deadly paramyxovirus. Science288:1432–1435.
8.Curran, J., R. Boeck, and D. Kolakofsky.1991. The Sendai virus P gene expresses both an essential protein and an inhibitor of RNA synthesis by
shuffling modules via mRNA editing. EMBO J.10:3079–3085.
9.Datta, A., L. Silverman, A. J. Phipps, H. Hiraragi, L. Ratner, and M. D. Lairmore.2007. Human T-lymphotropic virus type-1 p30 alters cell cycle G2
regulation of T lymphocytes to enhance cell survival. Retrovirology4:49.
10.Eaton, B. T., C. C. Broder, D. Middleton, and L. F. Wang.2006. Hendra and
Nipah viruses: different and dangerous. Nat. Rev. Microbiol.4:23–35.
11.Elia, A. E., L. C. Cantley, and M. B. Yaffe.2003. Proteomic screen finds
pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science299:
1228–1231.
12.Elia, A. E., P. Rellos, L. F. Haire, J. W. Chao, F. J. Ivins, K. Hoepker, D. Mohammad, L. C. Cantley, S. J. Smerdon, and M. B. Yaffe.2003. The molecular basis for phosphodependent substrate targeting and regulation of
Plks by the Polo-box domain. Cell115:83–95.
13.Gallina, A., L. Simoncini, S. Garbelli, E. Percivalle, G. Pedrali-Noy, K. S. Lee, R. L. Erikson, B. Plachter, G. Gerna, and G. Milanesi.1999. Polo-like kinase 1 as a target for human cytomegalovirus pp65 lower matrix protein.
J. Virol.73:1468–1478.
14.Garcia-Alvarez, B., G. de Carcer, S. Ibanez, E. Bragado-Nilsson, and G. Montoya.2007. Molecular and structural basis of polo-like kinase 1 substrate recognition: implications in centrosomal localization. Proc. Natl. Acad. Sci.
USA104:3107–3112.
15.Goodbourn, S., L. Didcock, and R. E. Randall.2000. Interferons: cell sig-nalling, immune modulation, antiviral response and virus countermeasures.
J. Gen. Virol.81:2341–2364.
16.Halpin, K., B. Bankamp, B. H. Harcourt, W. J. Bellini, and P. A. Rota.2004. Nipah virus conforms to the rule of six in a minigenome replication assay.
J. Gen. Virol.85:701–707.
17.Harcourt, B. H., L. Lowe, A. Tamin, X. Liu, B. Bankamp, N. Bowden, P. E. Rollin, J. A. Comer, T. G. Ksiazek, M. J. Hossain, E. S. Gurley, R. F. Breiman, W. J. Bellini, and P. A. Rota.2005. Genetic characterization of
Nipah virus, Bangladesh, 2004. Emerg. Infect. Dis.11:1594–1597.
18.Horvath, C. M.2004. Silencing STATs: lessons from paramyxovirus
inter-feron evasion. Cytok. Growth Factor Rev.15:117–127.
19.Horvath, C. M.2004. Weapons of STAT destruction. Interferon evasion by
paramyxovirus V protein. Eur. J. Biochem.271:4621–4628.
20.Hsu, V. P., M. J. Hossain, U. D. Parashar, M. M. Ali, T. G. Ksiazek, I. Kuzmin, M. Niezgoda, C. Rupprecht, J. Bresee, and R. F. Breiman.2004.
Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis.10:
2082–2087.
21.Isaacs, A., and J. Lindenmann.1957. Virus interference. I. The interferon.
Proc. R. Soc. Lond. B Biol. Sci.147:258–267.
22.Kawai, T., and S. Akira.2006. TLR signaling. Cell Death Differ.13:816–825. 23.Lam, S. K.2003. Nipah virus—a potential agent of bioterrorism? Antivir.
Res.57:113–119.
24.Lee, B.2007. Envelope-receptor interactions in Nipah virus pathobiology.
Ann. N. Y. Acad. Sci.1102:51–65.
25.Leung, S., S. A. Qureshi, I. M. Kerr, J. E. Darnell, Jr., and G. R. Stark.1995. Role of STAT2 in the alpha interferon signaling pathway. Mol. Cell. Biol.
15:1312–1317.
26.Liston, P., and D. J. Briedis. 1994. Measles virus V protein binds zinc.
Virology198:399–404.
27.Luby, S. P., M. Rahman, M. J. Hossain, L. S. Blum, M. M. Husain, E. Gurley, R. Khan, B. N. Ahmed, S. Rahman, N. Nahar, E. Kenah, J. A. Comer, and T. G. Ksiazek.2006. Foodborne transmission of Nipah virus,
Bangladesh. Emerg. Infect. Dis.12:1888–1894.
6270 LUDLOW ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
28.Mayo, M. A.2002. A summary of taxonomic changes recently approved by
ICTV. Arch. Virol.147:1655–1663.
29.McKendry, R., J. John, D. Flavell, M. Muller, I. M. Kerr, and G. R. Stark.
1991. High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc. Natl. Acad.
Sci. USA88:11455–11459.
30.Mohd Nor, M. N., C. H. Gan, and B. L. Ong.2000. Nipah virus infection of
pigs in peninsular Malaysia. Rev. Sci. Tech.19:160–165.
31.Morgenstern, J. P., and H. Land.1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a
complementary helper-free packaging cell line. Nucleic Acids Res.18:3587–
3596.
32.Murray, K., P. Selleck, P. Hooper, A. Hyatt, A. Gould, L. Gleeson, H. Westbury, L. Hiley, L. Selvey, B. Rodwell, et al.1995. A morbillivirus that
caused fatal disease in horses and humans. Science268:94–97.
33.Palosaari, H., J. P. Parisien, J. J. Rodriguez, C. M. Ulane, and C. M. Horvath.2003. STAT protein interference and suppression of cytokine
sig-nal transduction by measles virus V protein. J. Virol.77:7635–7644.
34.Park, M.-S., M. L. Shaw, J. Mun˜oz-Jordan, J. F. Cros, T. Nakaya, N. Bouvier, P. Palese, A. Garcı´a-Sastre, and C. F. Basler.2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol.
77:1501–1511.
35.Patel, D., A. Incassati, N. Wang, and D. J. McCance.2004. Human papillo-mavirus type 16 E6 and E7 cause polyploidy in human keratinocytes and
up-regulation of G2-M-phase proteins. Cancer Res.64:1299–1306.
36.Paterson, R. G., G. P. Leser, M. A. Shaughnessy, and R. A. Lamb.1995. The paramyxovirus SV5 V protein binds two atoms of zinc and is a structural
component of virions. Virology208:121–131.
37.Rodriguez, J. J., C. D. Cruz, and C. M. Horvath.2004. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V
protein reveals mechanisms underlying interferon evasion. J. Virol.78:5358–
5367.
38.Rodriguez, J. J., J. P. Parisien, and C. M. Horvath.2002. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and
STAT2 activation and nuclear accumulation. J. Virol.76:11476–11483.
39.Rodriguez, J. J., L. F. Wang, and C. M. Horvath.2003. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2
nu-clear accumulation. J. Virol.77:11842–11845.
40.Seong, Y. S., K. Kamijo, J. S. Lee, E. Fernandez, R. Kuriyama, T. Miki, and K. S. Lee.2002. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem.
277:32282–32293.
41.Shaw, M. L., W. B. Cardenas, D. Zamarin, P. Palese, and C. F. Basler.2005. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and Toll-like receptor 3-triggered signaling pathways. J. Virol.
79:6078–6088.
42.Shaw, M. L., A. Garcı´a-Sastre, P. Palese, and C. F. Basler.2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively.
J. Virol.78:5633–5641.
43.Shiell, B. J., D. R. Gardner, G. Crameri, B. T. Eaton, and W. P. Michalski.
2003. Sites of phosphorylation of P and V proteins from Hendra and Nipah
viruses: newly emerged members of Paramyxoviridae. Virus Res.92:55–65.
44.Sillje, H. H., and E. A. Nigg.2003. Signal transduction. Capturing polo
kinase. Science299:1190–1191.
45.Thomas, S. M., R. A. Lamb, and R. G. Paterson.1988. Two mRNAs that differ by two nontemplated nucleotides encode the amino coterminal
pro-teins P and V of the paramyxovirus SV5. Cell54:891–902.
46.Wang, L., and B. T. Eaton.2002. Henipavirus (Paramyxoviridae,
Paramyxo-virinae), p. 641–644.InC. A. Tidona and G. Darai (ed.), The Springer index
of viruses. Springer, Berlin, Germany.
47.Wang, L., B. H. Harcourt, M. Yu, A. Tamin, P. A. Rota, W. J. Bellini, and B. T. Eaton.2001. Molecular biology of Hendra and Nipah viruses. Microbes
Infect.3:279–287.