JOURNAL OFVIROLOGY, Oct. 2009, p. 10322–10335 Vol. 83, No. 20 0022-538X/09/$08.00⫹0 doi:10.1128/JVI.01186-09
Copyright © 2009, American Society for Microbiology. All Rights Reserved.
IRF3 Inhibition by Rotavirus NSP1 Is Host Cell and Virus Strain
Dependent but Independent of NSP1 Proteasomal Degradation
䌤
Adrish Sen,
1,2Ningguo Feng,
1,2Khalil Ettayebi,
3Michele E. Hardy,
3and Harry B. Greenberg
1,2*
Departments of Microbiology and Immunology1and Medicine,2Stanford University, Stanford, California 94305, and
Veterinary Molecular Biology, Montana State University, Bozeman, Montana 597173
Received 9 June 2009/Accepted 27 July 2009
Rotavirus host range restriction forms a basis for strain attenuation although the underlying mechanisms are unclear. In mouse fibroblasts, the inability of rotavirus NSP1 to mediate interferon (IFN) regulatory factor 3 (IRF3) degradation correlates with IFN-dependent restricted replication of the bovine UK strain but not the mouse EW and simian RRV strains. We found that UK NSP1 is unable to degrade IRF3 when expressed in murine NIH 3T3 cells in contrast to the EW and RRV NSP1 proteins. Surprisingly, UK NSP1 expression led to IRF3 degradation in simian COS7 cells, indicating that IRF3 degradation by NSP1 is host cell dependent, a finding further supported using adenovirus-expressed NSP1 from NCDV bovine rotavirus. By expressing heterologous IRF3 proteins in complementary host cells, we found that IRF3 is the minimal host factor constraining NSP1 IRF3-degradative ability. NSP1-mediated IRF3 degradation was enhanced by transfection of double-stranded RNA (dsRNA) in a host cell-specific manner, and in IRF3-dependent positive regulatory domain III reporter assays, NSP1 inhibited IRF3 function in response to pathway activation by dsRNA, TBK-1, IRF3, or constitutively activated IRF3-5D. An interesting observation arising from these experiments is the ability of transiently expressed UK NSP1 to inhibit poly(I:C)-directed IRF3 activity in NIH 3T3 cells in the absence of detectable IRF3 degradation, an unexpected finding since UK virus infection was unable to block IFN secretion, and UK NSP1 expression did not result in suppression of IRF3-directed activation of the pathway. RRV and EW but not UK NSP1 was proteasomally degraded, requiring E1 ligase activity, although NSP1 degradation was not required for IRF3 degradation. Using a chimeric RRV NSP1 protein containing the carboxyl 100 residues derived from UK NSP1, we found that the RRV NSP1 carboxyl 100 residues are critical for its IRF3 inhibition in murine cells but are not essential for NSP1 degradation. Thus, NSP1’s ability to degrade IRF3 is host cell dependent and is independent of NSP1 proteasomal degradation.
Rotavirus (RV) causes severe dehydrating diarrhea in the young of many mammalian species, and in humans the virus is responsible for approximately 600,000 deaths annually. RV is a member of theReoviridaefamily, and virus particles are non-enveloped and icosahedral and contain an 11-segmented dou-ble-stranded RNA (dsRNA) genome (13). As a consequence of high error rates of the viral RNA-dependent RNA polymer-ase and frequent gene reassortment, a large diversity of RV strains constantly circulates in nature. However, despite this diversity RVs are naturally replication restricted in heterolo-gous host species, generally resulting in poor replication (com-pared to homologous species RV replication) and the inability to cause diarrhea at low dosage in heterologous hosts (8, 14, 15, 23). This natural phenomenon of host range restriction formed the basis for development of several attenuated RV vaccine strains (1, 3, 18). Notably, the mechanistic basis for host range restriction is not understood, which impedes the rational design of additional RV vaccine strains.
We have recently demonstrated that primary mouse em-bryonic fibroblasts (MEFs) restrict the replication of differ-ent heterologous RV strains (such as the bovine UK strain),
and this restriction can be alleviated by the absence of an intact type I interferon (IFN) response (16). In these studies the simian RRV strain was unique among heterologous RVs and displayed an IFN-independent replication phenotype similar to homologous murine RVs in MEFs. This IFN-resistant replication phenotype could be conferred to UK RV by reassortment of the RRV NSP1-encoding gene seg-ment 5. The unusual nature of RRV compared to other heterologous RV strains is also underscored by its ability to spread systemically in vivo and cause a lethal phenotype in type I IFN signaling-deficient mice (14, 15).
RV NSP1 is a nonstructural protein that is poorly conserved across virus isolates and is of variable length (approximately 486 to 494 amino acids [aa]). Evolutionarily, NSP1 sequences tend to phylogenetically cluster with their cognate host species, suggesting host-specific selective constraints on NSP1 function (12). Notably, NSP1 can interact with and direct the degrada-tion of IFN regulatory factor 3 (IRF3), a host transcripdegrada-tion factor that is essential for early IFN secretion in response to virus infection although the precise mechanisms involved are still unknown (4, 22). In MEFs, restricted replication of het-erologous UK bovine RV correlates with a lack of IRF3 deg-radation, and this property segregates with the NSP1-encoding gene segment by reassortment analysis (16). Roles for NSP1 in cell-cell spread (4, 29), virus replication (5, 15, 16), and effi-cient spread from pup to pup within a mouse litter (8) have been reported. Thus, it seems likely that following infection of the host, heterologous RV restriction is linked, at least in part,
* Corresponding author. Mailing address: Departments of Medicine and Microbiology and Immunology, Stanford University School of Medicine, Always Building, Room M-121, 300 Pasteur Drive, Stanford CA 94305-5119. Phone: (650) 725-9722. Fax: (650) 852-3259. E-mail: hbgreen@stanford.edu.
䌤Published ahead of print on 5 August 2009.
10322
on November 8, 2019 by guest
http://jvi.asm.org/
to early IFN responses in the host and ineffective viral NSP1-mediated IRF3 degradation. Promiscuous (e.g., RRV) and restricted (e.g., UK) types of heterologous RV have been rec-ognized in the murine fibroblast model based on viral replica-tion that correlates with IFN sensitivity and IRF3 degradareplica-tion (16). The promiscuous RRV replicates efficiently in murine fibroblasts and degrades murine IRF3 (Mu-IRF3) while the restricted bovine UK RV does neither. These phenotypes seg-regate with RV gene 5, which encodes NSP1. While certain RVs might be inherently defective in their ability to degrade IRF3, thus representing candidate attenuated RV strains, it is likely that NSP1-directed IRF3 degradation and related repli-cation potential are also dependent on the host cell studied and not solely on intrinsic traits of a particular RV strain.
In this paper, we demonstrate that IRF3 degradation during RV infection and following transient expression of NSP1 oc-curs in a host cell-specific manner. The cell-dependent com-ponent of IRF3 degradation can be reconstituted with only the addition of ectopically expressed heterologous IRF3 proteins in reciprocal host cell environments, indicating that IRF3 is the minimal host factor involved in cell specificity. We also exam-ined specific stages in the IRF3 signaling pathway targeted by NSP1 in a host cell-dependent manner, observing that NSP1 degradation of IRF3 is enhanced by intracellular dsRNA-me-diated activation and that the IRF3 transcription function can be effectively blocked by NSP1 following pathway activation by either poly(I:C), TBK-1, IRF3 overexpression, or constitutively active IRF3. Analysis of full-length UK and RRV NSP1 ex-pression in cell culture indicates that, in contrast to the stable nature of UK NSP1, the RRV NSP1 protein is highly unstable and degraded by a polyubiquitin-dependent proteasomal path-way. Interestingly, NSP1 proteasomal degradation itself was not required for the IRF3 degradation function. Using do-main-swapping mutagenesis, we confirmed that the NSP1 car-boxyl-terminal 100 residues encode determinants of IRF3 in-hibition but not of NSP1 stability.
MATERIALS AND METHODS
Cells and viruses.Human embryonic kidney HEK 293 and 293T cells, mouse NIH 3T3 fibroblast cells, simian kidney COS7 fibroblast-like cells, and African green monkey kidney MA104 cells were purchased from the American Type Culture Collection (Maryland). Cells were routinely maintained in Dulbecco’s modified Eagle medium (DMEM; Cellgro) or M199 medium for MA104 cells containing 10% fetal calf serum (Invitrogen) and supplemented with penicillin and streptomycin (complete DMEM). Primary MEFs were prepared as de-scribed previously (16). RV strains ETD (a tissue culture-adapted derivative of the wild-type EW strain which was used for cloning the murine NSP1 as de-scribed below) (23), RRV, and UK were propagated, and titers were determined in MA104 cells by plaque assay as described previously (27). Recombinant adenovirus stocks (see below) were prepared by infection of 293T cells in T-175 flasks following a single round of virus amplification in T-25 flasks. Completely confluent monolayers of 293T cells were washed extensively with DMEM, and virus was adsorbed for 2 h before three washes and the addition of fresh com-plete DMEM. Cells were monitored daily for infection by expression of yellow fluorescent protein (YFP) by fluorescence microscopy and harvested following complete cytopathic effect. Infected cells were collected by centrifugation at
2,500⫻gfor 15 min and resuspended in fresh complete DMEM. The
resus-pended cell pellets were subjected to four freeze-thaw cycles to release virus and
clarified by centrifugation at 10,000⫻gfor 30 min at 4°C. Titers of the clarified
supernatants were determined in 293T cells grown in 24-well plates by end-point dilution and enumeration of YFP-expressing cells. Virus stocks were stored in
single-use aliquots at⫺80°C until used.
Reagents, plasmids, and antibodies. MG132 and PYR-41 were purchased from Calbiochem, and poly(I:C)-LyoVec (transfection reagent) was from
InvivoGen. Plasmids encoding full-length Mu-IRF3 and human IRF3 (Hu-IRF3) were purchased from InvivoGen. Plasmids encoding the human TBK-1 and a green fluorescent protein-tagged IRF3-5D (GFP-IRF3-5D) were kind gifts from David Baltimore (MIT, Boston, MA) and Adolfo Garcia-Sastre (Mount Sinai Hospital, NY), respectively. The positive regulatory domain III-I [PRD(III)] luciferase reporter (17) was a gift of Tom Maniatis (Harvard University, Boston, MA). The following commercial antibodies were used: anti-IRF3 (FL-325 rabbit antibody; Santa Cruz Biotechnology), anti-phospho-S396 IRF3 (4D4G rabbit antibody; Cell Signaling Technologies), anti-actin (AC-15 mouse monoclonal; Sigma), anti-Myc (rabbit polyclonal; Invitrogen), and secondary horseradish per-oxidase (HRP)-conjugated anti-mouse and anti-rabbit polyclonal antibodies (Amersham).
Virus infections.NIH 3T3 or COS7 cells were seeded in six-well cluster plates 2 to 3 days prior to infection to allow cells to reach complete confluence. Cells were washed three times with DMEM without additives, and virus was added at the multiplicity of infection (MOI) specified in the figure legends and adsorbed at 37°C for 1 h. Cells were washed three times, and infection was allowed to proceed in DMEM lacking serum for the times indicated in the figure legends prior to further analysis. For quantifying virus replication in NIH 3T3 cells, infected cells were collected 24 h following adsorption, and virus was released by three freeze-thaw cycles. Clarified supernatants were used for infection following
trypsin activation (5g/ml; Sigma-Aldrich) for 1 h at 37°C. Virus titration was
performed using a fluorescent focus-forming assay in MA104 cells as described previously (27). For NSP1 expression experiments using adenovirus vectors, HEK293 cells or primary MEFs were grown in six-well plates and infected at an MOI of 5.0 with each recombinant adenovirus as described above. Cells were collected at 24 h postinfection (hpi) after visualization of infection by
fluores-cence microscopy and lysed in Laemmli sample buffer containing
-mercapto-ethanol prior to immunoblot analysis, as detailed below.
Cloning.MA104 cells were infected as described above with RRV and UK viruses at an MOI of 1.0, and total cellular RNA was isolated at 12 hpi with Trizol LS (Invitrogen) per the manufacturer’s protocol. RNA was used as a template for reverse transcription-PCR with NSP1-specific oligonucleotides containing restriction enzyme adaptor sequences using the SuperScript One-Step RT-PCR
with PlatinumTaq(Invitrogen) to generate full-length PCR products containing
the entire reading frames of the NSP1 genes. A full-length cDNA clone of EW NSP1 was used as a template for PCR of the EW NSP1 gene. PCR products containing RRV NSP1, UK NSP1, and EW NSP1 were cloned directionally into the XhoI-BamHI (RRV and EW NSP1s) and XhoI-EcoRI (UK NSP1) sites of the pEGFP-C1 vector (Clonetech). A domain-swap mutant of RRV GFP-NSP1 (dsC1 NSP1) was constructed by modified site-directed mutagenesis as described previously (30). Briefly, oligonucleotides flanking the C-terminal 100 aa of UK NSP1 and containing 21-base RRV GFP-NSP1 homologous adaptors were used to generate PCR products that were then used as primers for site-directed mutagenesis of the RRV GFP-NSP1 template using a commercial kit (QuikChange; Stratagene). Positive clones were sequence verified prior to further use. Plasmid DNA was prepared using an endotoxin-free plasmid preparation kit (Qiagen).
Construction of recombinant adenoviruses expressing NCDV or OSU NSP1.
Recombinant adenoviruses were constructed using the AdEasy system according to procedures described by the manufacturer (Stratagene) (6). Sequences en-coding enhanced YFP (EYFP) followed by a poliovirus internal ribosome entry site (IRES) and then either NCDV NSP1 or OSU NSP1 (Myc-tagged at the N terminus) were amplified by PCR from plasmids described previously (21). EYFP-IRES-NSP1 fragments were cloned into pShuttle-CMV (where CMV is cytomegalovirus). Constructs were cotransformed with supercoiled adenoviral DNA plasmid pAdEasy-1 into BJ5183 bacterial cells to generate recombinant adenoviral DNA containing EYFP-IRES-Myc-NSP1 inserts. Viral DNA was then purified and transfected into Ad-293 cells. Virus replication was monitored by fluorescence microscopy and by cytopathic effect. Titers of recombinant vi-ruses were determined in Ad-293 cells after three passages. NSP1 expression was confirmed by immunoblotting with a rabbit polyclonal antiserum to OSU NSP1 or anti-myc antibody to detect NCDV NSP1 (20).
Transfection, cell lysis, and immunoblotting.COS7 or NIH 3T3 cells were seeded in six-well plates 1 day prior to transfection and transfected with Fugene
6 (Roche) per the manufacturer’s protocol. For immunoblotting, 2g of each
relevant GFP plasmid was transfected per well, and medium was changed 14 to 16 h posttransfection (p.t.). Where indicated in the figure legends, cells were
transfected with 1g/ml of poly(I:C)-LyoVec complexes at 36 h following the
initial transfection. Transfected cells were observed for GFP-NSP1 expression 48 h p.t. by fluorescence microscopy and lysed in Laemmli buffer containing 2%
sodium dodecyl sulfate and 5%-mercaptoethanol after two washes in
phos-phate-buffered saline (pH 7.0). Cell lysis was performed at room temperature for
on November 8, 2019 by guest
http://jvi.asm.org/
20 min, and lysates were passed through a 25-gauge needle to reduce sample viscosity. Cell lysates were boiled for 5 min, and tubes were briefly spun at
10,000⫻gand separated on 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis gels. Following electrophoresis, proteins were transferred onto nitrocellulose membranes (Amersham Biosciences) and probed using the indi-cated antibodies. Blots were exposed to autoradiography film (Amersham) and developed using an enhanced chemiluminescence kit (GE Healthcare). Blots were subsequently stripped and reprobed using a commercially available kit, following the manufacturer’s instructions (Re-blot; Millipore). Protein band relative intensities (RIs) from scanned blots were determined using the public domain NIH Image program (http://rsb.info.nih.gov/nih-image/). For compari-son of expression levels of GFP plasmids in COS7 cells, fluorescent foci were counted in three to five fields containing equivalent cell densities at a
magnifi-cation of⫻10 under an inverted fluorescence microscope (Nikon).
Luciferase assay.COS7 or NIH 3T3 cells in 24-well plates were transfected in
duplicate with 4 ng ofRenillaluciferase reporter (pRL-TK; Promega), 200 ng of
the firefly PRD(III) reporter, and indicated amounts of activator and GFP alone or GFP-NSP1 plasmids. The total amount of DNA was normalized using empty vector. Where indicated in the figure legends, cells were transfected 36 h later with
1g/ml of poly(I:C)-LyoVec as described above. At 48 h p.t., cells were washed
in phosphate-buffered saline and lysed using Stop and Glo Dual Luciferase Assay
kit reagents as directed by the manufacturer (Promega). Renillaand firefly
luciferase activities were measured using a luminometer (Turner Designs), and
PRD(III) activation was normalized toRenillaluciferase levels and is reported as
normalized PRD(III) activity.
IFN measurement.NIH 3T3 cells were seeded in 24-well plates and infected in duplicate as described above at an MOI of 1.0 with the viruses indicated in the figure legends. Twenty-four hours later, the cell culture supernatants were
col-lected, and secreted mouse IFN-was measured in duplicate using a Verikine
mouse IFN-enzyme-linked immunosorbent assay kit (PBL Interferon Source).
Amounts of secreted IFN-were calculated from a standard curve created using
a mouse IFN-standard as per the manufacturer’s instructions.
RESULTS
Degradation of IRF3 during RV infection is host cell depen-dent. Recently, we found that infection with RRV (simian, heterologous virus) and EW (murine, homologous virus) RVs led to IRF3 degradation in murine (primary MEFs and NIH 3T3) fibroblast cells (16). In contrast, UK—a bovine RV—was unable to direct IRF3 degradation in these cells. Notably, in contrast to RRV, UK virus replicated poorly in wild-type MEFs, and its replication was greatly enhanced (⬎100-fold) in the absence of an intact type I IFN response. UK virus infec-tion was associated with IRF3 activainfec-tion, nuclear translocainfec-tion, and IFN-secretion. These contrasting RRV and UK pheno-types segregated with the parental origin of the NSP1-encoding gene 5 (16). These findings thus directly link RV strain-specific growth restriction in murine fibroblasts with NSP1-mediated inhibition of the type I IFN response in mouse fibroblasts.
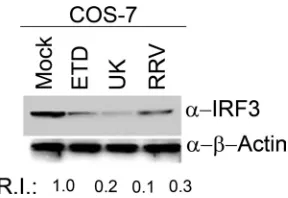
Several attenuated or nonpathogenic virus strains have been described that inherently lack the ability to counteract host innate immune responses (53). The inability of UK RV to degrade IRF3 in murine fibroblast cells may reflect an intrinsic property of the UK RV strain or may also depend on extrinsic factors such as the nature of the infected host cell. In order to examine this latter possibility, we next examined the effect of UK infection on levels of endogenous IRF3 in COS7 cells, a simian fibroblast-like line (Fig. 1). COS7 cells were infected with RRV, ETD, and UK viruses at an MOI of 1.0, and levels of endogenous IRF3 were examined at 24 hpi by immunoblot analysis of whole-cell lysates. We observed that similar to RRV and ETD, UK virus infection in COS7 cells resulted in signif-icant IRF3 degradation. Thus, in COS7 cells, UK virus de-grades IRF3 (Fig. 1) in contrast to observations in MEFs or NIH 3T3 cells, in which UK infection was not associated with
IRF3 degradation (16). Taken together, the results demon-strate that the ability to degrade host IRF3 by UK RV is host cell dependent.
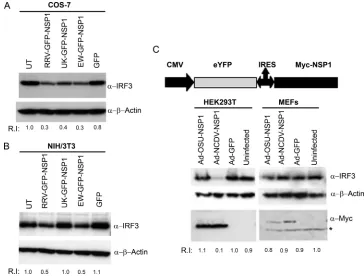
[image:3.585.350.494.67.166.2]RV NSP1 expression leads to host cell-specific IRF3 degra-dation.We demonstrated previously that both IFN sensitivity and growth restriction of UK virus in murine fibroblasts map to the NSP1-encoding gene (16), and studies from other labora-tories clearly established the ability of NSP1 to direct IRF3 proteasomal degradation (4, 5, 11, 21). Presently, it is unclear whether the inability of certain NSP1 proteins to degrade IRF3 proteasomally is dependent on host cell origin. In order to examine this question directly, we cloned the full-length NSP1 genes from RRV, EW, and UK RV into the mammalian expression vector pEGFP-C1 as carboxyl-terminal fusions to EGFP. COS7 or NIH 3T3 cells grown in six-well plates were transfected with the plasmids encoding GFP-NSP1 proteins or GFP alone, and total cellular proteins were examined for levels of endogenous IRF3 48 h later by immunoblot analysis. As shown in Fig. 2A, expression of RRV, EW, or UK NSP1 in COS7 cells led to IRF3 degradation, with comparable efficien-cies at 48 h p.t., just as observed during viral infection (Fig. 1). In contrast to the results in COS7 cells, we found that in NIH 3T3 cells only RRV and EW GFP-NSP1 proteins could direct IRF3 degradation while UK NSP1 expression was unable to mediate any detectable IRF3 degradation (Fig. 2B), again, consistent with findings made during viral replication (16). As described fully later, we verified NSP1 expression levels in these and subsequent experiments by fluorescence microscopy of transfected cells prior to cell lysis (see Fig. 8). The efficiency of IRF3 degradation by transiently expressed NSP1 was slightly lower than that obtained during virus infection (compare rel-ative IRF3 intensity differences in Fig. 1 and 2), and this is likely due to differences in efficiency of NSP1 expression fol-lowing transfection versus virus infection. In addition, the ef-ficiency by which expressed NSP1 blocks IRF3 functional ac-tivity as measured by PRD(III) acac-tivity (see Fig. 5 and 6 below) is substantially greater than the degree of degradation ob-served on Western blots. This may indicate that NSP1 encodes mechanisms capable of blocking IRF3 signaling that are not directly tied to its degradation. These results, in agreement with our observations following actual virus infection, demon-strate the functional integrity of the GFP-NSP1 fusion proteins as well as the fact that host cell-specific degradation of IRF3
FIG. 1. Degradation of IRF3 is host cell-dependent. COS7 cells were infected at an MOI of 1.0 with the indicated RV strains, and cell lysates were examined at 24 hpi for levels of endogenous IRF3. Blots were reprobed for actin, and the RIs of IRF3 were quantified after normalization to actin levels. Blots shown are representative of three independent experiments.␣, anti.
10324 SEN ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
during infection can be reproduced by the expression of NSP1 alone.
In order to confirm and extend the observation that RV NSP1-directed IRF3 degradation was mediated in a virus- and host cell-dependent manner, we used recombinant adenovi-ruses expressing two additional RV NSP1s, porcine OSU NSP1 and bovine NCDV NSP1, or an adenovirus GFP control. In these experiments, NSP1 was expressed by IRES-dependent translation. Human kidney 293T cells and primary MEFs were infected with recombinant adenovirus at an MOI of 5.0, and at 24 hpi cells were lysed and examined by immunoblot analysis (Fig. 2C). Adenovirus-mediated expression of Myc-tagged NCDV NSP1 resulted in efficient IRF3 degradation in 293T cells but not in primary MEFs. In contrast, OSU NSP1 was unable to degrade IRF3 in either 293T cells or MEFs. Degra-dation of IRF3 by NCDV but not OSU NSP1 was also ob-served in COS7 cells infected with recombinant adenoviruses (data not shown). Thus, the host cell-dependent degradation of IRF3 by UK GFP-NSP1 (simian COS7 versus murine NIH 3T3) represents a phenotype that is shared (in human 293T versus murine NIH 3T3 cells) with NCDV Myc-NSP1. Com-parison of NSP1 expression levels indicated that despite dif-ferences in their ability to cause IRF3 degradation, both OSU and NCDV Myc-NSP1 proteins were expressed to similar lev-els in 293T cells (Fig. 2C, anti-myc panel). Further, the use of an IRES-dependent expression system and myc-tagged NSP1
demonstrate that differences in NSP1 translation efficiency or the presence of the GFP tag are unlikely to account for the phenotypes observed. Taken together, our results demonstrate that the ability of NSP1 to direct IRF3 degradation is not invariant but, rather, can be both virus strain and host cell specific and independent of NSP1 expression levels.
Heterologous IRF3 is sufficient for NSP1-dependent IRF3 degradation in a restrictive host cell.Our results demonstrate that NSP1-mediated IRF3 degradation is dependent on host cell origin although which host-specific cellular factors deter-mine NSP1’s specific ability to degrade IRF3 in one host cell versus another is unknown. In order to investigate the basis for host cell-specific IRF3 degradation by NSP1, we expressed RRV and UK GFP-NSP1 proteins in NIH 3T3 cells together with a plasmid encoding the full-length Hu-IRF3 protein (96% iden-tical to the simian IRF3 [Si-IRF3]protein at the amino acid level and used as a surrogate for Si-IRF3 in these experiments) (Fig. 3). Cells were treated with 10M MG132 or dimethyl sulfoxide as shown in the figure, and IRF3 levels were deter-mined by immunoblotting. As shown in Fig. 3A, in NIH 3T3 cells expression of the both RRV and UK NSP1s resulted in degradation of ectopically expressed Hu-IRF3 (Fig. 3A, lanes 1 and 3) and, as expected, levels of Hu-IRF3 increased in the presence of the proteasomal inhibitor MG132 (lanes 2 and 4). However, the endogenous Mu-IRF3 was degraded only in NIH 3T3 cells transfected with RRV NSP1. These results
demon-FIG. 2. NSP1 expression results in degradation of endogenous IRF3 in a host cell-dependent manner. (A) COS7 cells were transfected with indicated GFP plasmids, and at 48 h p.t. whole-cell lysates were analyzed by immunoblotting for endogenous IRF3. Blots were reprobed for actin and RIs of IRF3 estimated after actin normalization. (B) NIH 3T3 cells were transfected with the indicated plasmids and analyzed as above. (C) Recombinant adenoviruses (Ads) expressing OSU or NCDV NSP1 or a GFP control were used to infect HEK293 and MEFs at an MOI of 5.0. The schematic shows the organization of the adenovirus constructs used. At 24 hpi, whole-cell lysates were prepared and examined for IRF3, actin, and Myc-tagged NSP1 by immunoblotting. The asterisk indicates a nonspecific band. The results shown are representative of at least three
independent experiments. UT, untransfected;␣, anti.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.585.108.472.68.344.2]strate that UK NSP1 is capable of efficiently degrading Hu-IRF3 in murine fibroblasts in the absence of any other heter-ologous human cellular factors. The presence of Hu-IRF3 in the murine cells did not, however, alter UK NSP1’s inability to degrade Mu-IRF3 in the NIH 3T3 cells. Conversely, COS7 cells express all the required factors for UK NSP1-mediated Si-IRF3 degradation. We therefore tested whether in such an environment UK NSP1 could degrade ectopically expressed Mu-IRF3. As shown in Fig. 3B, UK NSP1 was able to degrade endogenous Si-IRF3 but not the overexpressed Mu-IRF3 in COS7 cells. In contrast, RRV NSP1 expression resulted in the degradation of both endogenous Si-IRF3 and the overex-pressed Mu-IRF3 in COS7 cells. Taken together, these experi-ments indicate that IRF3 is the minimal cellular factor responsi-ble for the host cell-specific component of IRF3 degradation by NSP1.
NSP1 degradation of IRF3 is enhanced by dsRNA activation in a host cell-specific manner.Phosphorylation-dependent IRF3 degradation occurs as a consequence of infection with several viruses (32), and IRF3 proteasomal degradation requires polyubiquitination and occurs via multiple regulatory events involving distinct cellular kinases, adaptors, and ubiquitin li-gases within cytoplasmic or nuclear compartments (7, 9, 10, 17, 26, 31, 32, 35, 36, 40, 45, 46, 48). However, both the mechanism and specific stage of the IRF3 signaling pathway involved in NSP1-mediated IRF3 degradation are presently unknown (e.g., basal versus activated IRF3 or IRF3 present in specific
signaling complexes). Here, we examined if stimulation with intracellular poly(I:C), a synthetic dsRNA ligand for TLR3/ MDA5 in cultured cells (28, 34), resulted in IRF3 degradation by NSP1 proteins of RRV, EW, and UK viruses. COS7 and NIH 3T3 cells were transfected with plasmids expressing RRV, EW, and UK GFP-NSP1 proteins, and at 36 h p.t., cells were transfected with 1g/ml of a poly(I:C)-cationic liposome com-plex. The ability of NSP1 protein to degrade endogenous IRF3 was determined 12 h later by immunoblot analysis of whole-cell protein lysates. Under these conditions, we observed ro-bust IRF3 activation in both types of host cells by functional reporter assays (see Fig. 7 and 8; also data not shown), indi-cating that poly(I:C) triggered IRF3 activation. As shown in Fig. 4A and confirming findings shown in Fig. 2A, expression of RRV, EW, and UK NSP1 proteins in unstimulated COS7 cells resulted in IRF3 degradation (RI of 0.5 to 0.6) compared to either untransfected or GFP controls (Fig. 4A, lanes 1 and 9 versus lanes 3, 5, and 7). Of note, in cells transfected with poly(I:C), an approximately twofold increase in IRF3 degra-dation was observed in the presence of RRV, EW, or UK NSP1 proteins [Fig. 4A, for lanes 2 and 10 versus 4, 6, and 8, compare normalized RI levels with and without poly(I:C)]. Thus, poly(I:C) activation appears to enhance NSP1-medi-ated IRF3 degradation in COS7 cells.
In NIH 3T3 cells expressing GFP-NSP1 and treated with poly(I:C), we similarly observed that expression of both RRV and EW NSP1 proteins resulted in approximately twofold
in-FIG. 3. Effect of NSP1 on ectopically expressed heterologous IRF3 in reciprocal types of host cells. (A) Murine NIH 3T3 cells were transfected with indicated NSP1 and full-length Hu-IRF3 plasmids, and cells were treated with MG132 as indicated. Whole-cell lysates were examined for levels of endogenous murine and overexpressed Hu-IRF3 at 48 h p.t. The two IRF3 types are indicated by arrows. Blots were reprobed for actin. Relative actin-normalized levels of Mu-IRF3 and Hu-IRF3 in the presence (⫹) and absence (⫺) of MG132, and the ratio of each IRF3 type (⫹MG132/⫺MG132), a measure of IRF3 degradation, is plotted in the lower graph. (B) Simian COS7 cells were transfected with NSP1 expression plasmids and a full-length Mu-IRF3 and analyzed as described in panel A for levels of endogenous Si-IRF3 and overexpressed Mu-IRF3 (arrows). Blots are representative of two independent experiments.␣, anti.
10326 SEN ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.100.484.67.333.2]creases in IRF3 degradation in the presence of poly(I:C) (Fig. 4B, compare lane 3 with 4 and 7 with 8). In contrast, in NIH 3T3 cells UK NSP1 was unable to degrade IRF3 either in the presence or absence of poly(I:C) activation (Fig. 4B, lanes 5 and 6). Since NSP1 degrades a portion of the endogenous IRF3 in the absence of any exogenous stimulation (Fig. 3), we surmise that residual IRF3 that is activated by poly(I:C) treat-ment at 36 h p.t. is efficiently targeted for degradation by NSP1 in these experiments. These results demonstrate that NSP1 degradation of IRF3 is enhanced by poly(I:C)-mediated acti-vation in a host cell-specific manner.
Pathway-specific analysis of IRF3 inhibition by NSP1.Both transcriptional activity and proteasomal degradation of IRF3 are dependent on several upstream events including formation of signaling complexes with cellular factors including TRIF, IPS-1, TRAF3, TBK-1/IKKε, stepwise IRF3 phosphorylation, dimerization, and nuclear translocation (26). Notably, the spe-cific stage in the IRF3 activation process targeted by NSP1 is unclear, and this information is likely to be useful to under-stand the basis for the host cell-specific NSP1 inhibition observed here. To determine the pathway-specific location of inhibition of IRF3 function by NSP1 in COS7 cells, we expressed UK NSP1 in COS7 cells since UK NSP1 could ef-fectively degrade IRF3 in this host cell. In order to directly examine inhibition of IRF3 function, we coexpressed an IFN- PRD(III)-firefly luciferase [PRD(III)] functional reporter whose expression is dependent on an IRF3-sensitive promoter element (17) and aRenillaluciferase internal control plasmid to normalize differences in transfection efficiencies. First, we determined the ability of NSP1 expression to block PRD(III) activation in response to intracellular poly(I:C). As shown in Fig. 5A, poly(I:C) transfection in cells expressing GFP alone resulted in robust activation of PRD(III) activity (⬃450-fold) 16 h after poly(I:C) exposure. Expression of RRV, EW, or UK NSP1 strongly inhibited PRD(III) activity to nearly basal lev-els, indicating that in COS7 cells, UK NSP1, similar to the RRV and EW NSP1 proteins, is highly effective at blocking IRF3 transcriptional activity in response to dsRNA. Of note, the inhibition of PRD(III) luciferase activity appears to be a
more sensitive indicator of NSP1 inhibitory function than the simple degradation of IRF3 as measured by Western blotting (compare Fig. 4 and 5). Further, immunoblot analysis of cells similarly transfected indicated that phosphorylation of IRF3 at S396, an event that is required for its transcriptional activation (45), was significantly reduced by NSP1 expression. This inhi-bition correlates well with the ability of UK NSP1 to degrade IRF3 in these cells.
The result obtained above demonstrates the ability of NSP1 to effectively inhibit IRF3-mediated function in response to a dsRNA trigger but do not reveal whether NSP1 is also able to inhibit IRF3 downstream of its activation by kinases such as TBK-1. In order to examine this, we performed PRD(III) activity assays as described above with the notable difference that IRF3 pathway activation was triggered at points progressively down-stream by overexpression of TBK-1, IRF3, or IRF3-5D (a phos-phomimetic form that is constitutively activated) (32) in each assay. As demonstrated in Fig. 5B, C, and D, expression of NSP1 from RRV, EW, or UK RV in COS7 cells resulted in highly efficient inhibition of PRD(III) activity in response to pathway stimulation at points progressively proximal to the PRD(III) re-porter, namely, at TBK-1, IRF3, and IRF3-5D, respectively. These findings demonstrate that in COS7 cells, the NSP1 protein of UK as well as RRV and EW RVs is able to inhibit IRF3 function following activation of the IRF3 C-terminal cluster of five phospho-acceptor sites. Thus, our results indicate that NSP1 inhibits IRF3 function, presumably through degradation by tar-geting the hyperphosphorylated/dimerized form of IRF3.
[image:6.585.126.454.66.221.2]In order to further substantiate the results obtained, COS7 cells were transfected with a constant amount (200 ng) of UK NSP1-encoding plasmid in the presence of increasing amounts of a plasmid encoding the full-length Hu-IRF3 protein (50, 100, or 150 ng) (Fig. 5E). Cells were subsequently transfected with poly(I:C), and the ability of increasing amounts of ectopic Hu-IRF3 to abrogate UK NSP1-mediated PRD(III) inhibition in response to poly(I:C) was examined 6 h following poly(I:C) treatment. As shown, in the absence of any ectopic IRF3, UK NSP1 expression resulted in a significant inhibition of poly(I:C)-directed PRD(III) activity compared to GFP alone
FIG. 4. Effect of intracellular poly(I:C) activation on NSP1-mediated IRF3 degradation in COS7 and NIH 3T3 cells. (A) COS7 cells were transfected with the indicated NSP1 expression plasmids and with poly(I:C) as described in Materials and Methods. Levels of endogenous IRF3 in the presence (⫹) and absence (⫺) of poly(I:C) were examined in whole-cell lysates 48 h p.t. IRF3 RIs normalized to internal actin loading controls are indicated. (B) NIH 3T3 cells were transfected and analyzed exactly as described for panel A; a nonspecific cellular band (ns) is indicated by the asterisk. Blots shown are representative of at least two independent experiments. UT, untransfected.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 5. Pathway-specific dissection of NSP1 mediated inhibition of IRF3 function in COS7 cells. (A to D) COS7 cells transfected with NSP1 or GFP expression plasmids, PRD(III) luciferase reporter [PRD(III)], and 1g of the following activators: human TBK-1 (B), h-IRF3 (C), or IRF3-5D (D). In panel A, cells were transfected with 1g/ml of poly(I:C) at 36 h following transfection, and one set of cells was used for immunoblotting. PRD(III) activation is shown after normalization to internalRenillaluciferase. (E) Cells were transfected as indicated, poly(I:C) was added as described for panel A, and 6 h later, the ability of increasing doses of Hu-IRF3 to alleviate NSP1 inhibition of the poly(I:C)-directed PRD(III) activity was examined. Bars indicate the standard error of duplicate wells. Experiments were repeated at least two times with similar results.␣, anti.
10328
on November 8, 2019 by guest
http://jvi.asm.org/
(Fig. 5E, lane 2 versus lane 3). This inhibition could be allevi-ated in a dose-dependent manner by ectopic expression of Hu-IRF3 (lanes 4 to 6). Similar results were obtained using RRV GFP-NSP1 or EW GFP-NSP1 (data not shown). We interpret these results to indicate that NSP1 inhibition of the poly(I:C) response occurs primarily by degradation of IRF3 since overexpressing IRF3 partially restored PRD(III) activity in a dose-dependent manner (Fig. 5E, lanes 3 to 6). Despite this relative dose-dependent abrogation of NSP1 inhibition, compared to GFP alone, UK NSP1 still strongly inhibited PRD(III) activity (⬃20-fold activity with UK NSP1 compared to⬃250-fold activity with GFP alone) (Fig. 5E, lanes 6 versus lane 7), supporting the enhanced ability of UK NSP1 in these cells to inhibit activated IRF3 (Fig. 4). Nevertheless, increasing IRF3 expression was sufficient to partially overcome UK NSP1 inhibition of IRF3 function in response to poly(I:C) stimula-tion in COS7 cells in a dose-dependent manner, supporting the hypothesis that IRF3 depletion by UK NSP1 in these cells is an important mechanism for subverting dsRNA-directed IRF3 responses.
Effect of UK NSP1 expression on IRF3 functions in murine NIH 3T3 cells.The results obtained thus far demonstrate that UK NSP1 is highly efficient at inhibiting IRF3 functions in COS7 cells and that this inhibition is associated with degrada-tion. However, UK virus infection as well as UK NSP1 ex-pressed in NIH 3T3 cells did not result in IRF3 degradation and did induce robust IFN- secretion (16). We therefore examined the ability of UK NSP1 to antagonize IRF3 in the restrictive NIH 3T3 cell line using an IRF3 functional assay. We found that overexpression of Mu-IRF3 in NIH 3T3 cells strongly induced PRD(III) activation, and expression of RRV and EW NSP1 proteins concomitantly resulted in a significant inhibition of this IRF3-mediated PRD(III) activation (Fig. 6A). In contrast, UK NSP1 expression was unable to signifi-cantly inhibit PRD(III) activity. Thus, unlike in COS7 cells, UK NSP1 is unable to inhibit IRF3 function in NIH 3T3 cells. In order to validate these results in the context of virus infec-tion, IFN-secretion in response to poly(I:C) was measured in NIH 3T3 cells infected with UK, RRV, or ETD RV at 16 hpi. As shown in Fig. 6B, infection with UK virus resulted in a strong activation of the IFN response in the absence of any exogenous pathway stimulation. We have shown earlier that this response occurs in a replication-dependent manner during viral infection of primary murine fibroblasts (16). Notably, in response to intracellular poly(I:C), RRV and ETD continued to actively suppress the secretion of IFN-. In contrast, UK virus infection could not block poly(I:C)-directed IFN- induc-tion, and the amount of secreted IFN was additive to the amounts measured in response to UK virus or poly(I:C) ligand alone. Unexpectedly, and in contrast to these results, in NIH 3T3 cells stimulated with poly(I:C) the expression of UK NSP1 alone resulted in inhibition of the PRD(III) luciferase reporter with an efficiency (⬃66% inhibition) comparable to the inhi-bition obtained by expression of EW NSP1 (⬃58% inhibition) or RRV NSP1 (⬃79% inhibition) (Fig. 6C). Similar inhibition of IRF3 function was also observed in cells transfected with increasing amounts of UK, RRV, and EW NSP1 expression plasmids (Fig. 6D). Thus, although UK NSP1 failed to inhibit IRF3-dependent responses in the context of virus infection (Fig. 6B) as well as in response to IRF3 overexpression (Fig.
6A), UK NSP1 expression could inhibit IRF3 activation in response to poly(I:C) in NIH 3T3 cells. These results raise the interesting possibility that NSP1 encodes additional mecha-nisms to inhibit IRF3 function that are independent of IRF3 degradation but instead depend on the stimulus-specific man-ner in which IRF3 is activated.
Host cell-specific IRF3 inhibition is independent of the polyubiquitinylation-mediated proteasomal degradation of NSP1.Interestingly, NSP1 expression itself is reportedly reg-ulated by proteasomal degradation (21, 39). Low levels of NSP1 expression are a result of both degradation and poor translation from viral transcripts during infection (29, 38), and it is possible that low NSP1 levels may be predictive of the cumulative levels of their IRF3 substrates (21). Our results indicated that in COS7 cells, RRV, UK, and EW NSP1 pro-teins efficiently direct IRF3 degradation. In order to examine NSP1 stability, COS7 cells grown in six-well plates were trans-fected with plasmids encoding GFP-NSP1 proteins or GFP alone, and 36 h later cells were treated with the proteasomal inhibitor MG132 before analysis by fluorescence microscopy (Fig. 7A). Transient expression of RRV and EW GFP-NSP1 proteins in COS7 cells resulted in poor expression levels in contrast to relatively higher expression of the UK GFP-NSP1 protein (Fig. 7A). Inhibition of the proteasome using MG132 resulted in significantly higher expression levels of both RRV and EW GFP-NSP1s (⬃4.2- and 3.3-fold, respectively), indi-cating that both of these NSP1 proteins are highly unstable and barely detectable in untreated COS7 cells due to proteasomal degradation. In contrast to RRV and EW NSP1s, UK GFP-NSP1 was stable and expressed to similar levels in the absence or presence of MG132 (⬃1.1-fold difference). Thus, in contrast to the unstable nature of RRV and EW NSP1s, UK NSP1 does not appear to be susceptible to proteasomal degradation in COS7 cells although it can efficiently degrade IRF3 in these cells (Fig. 1 and 2A).
Proteasomal degradation of several proteins has been re-ported to occur in the absence of polyubiquitinylation (37). In order to determine whether NSP1 proteasome-dependent deg-radation requires polyubiquitin, we used PYR-41, an E1 ligase inhibitor that blocks the activation and subsequent transfer of polyubiquitin to substrates (52). COS7 cells were transfected with RRV or UK GFP-NSP1 plasmids as described above and treated with 10M PYR-41 for 8 h. PYR-41 treatment re-sulted in a significant enhancement of RRV NSP1 expression levels (approximately fourfold), comparable to the increase observed in the presence of MG132 (Fig. 7B). As with the lack of increased expression of UK NSP1 with MG132, PYR-41 treatment did not augment UK NSP1 expression (⬃1.2-fold difference). These results substantiate the role of proteasomal degradation in NSP1 expression and indicate that RRV-NSP1 degradation by the proteasome requires modification by E1 ligase-dependent polyubiquitinylation. In all experiments with GFP-NSP1 constructs in this study, we verified the expression of GFP-NSP1 constructs by fluorescence microscopy of live cells using controls treated with MG132 prior to cell lysis for immunoblotting or luciferase activity estimation (data not shown). Notably, since UK NSP1 expression in COS7 cells is not proteasomally regulated (Fig. 7) despite efficient IRF3 degradation (Fig. 2, 3, and 4), we conclude that NSP1 (poly-ubiquitin dependent) proteasomal degradation is not essential
on November 8, 2019 by guest
http://jvi.asm.org/
for IRF3 degradation, at least in the case of UK NSP1 in COS7 cells.
The carboxyl-terminal 100 residues of NSP1 encode IRF3 inhibitory determinants but do not influence NSP1 stability.
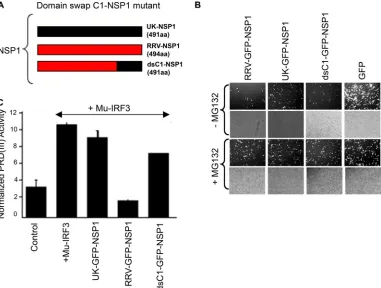
The carboxyl-terminal 164 aa of NSP1 are reported to be essential for IRF3 interaction based on analysis of truncation mutants (22), and the NSP1 C terminus is involved in IRF3 degradation (5). In our studies, we found that NIH 3T3 cells present a host cell with a marked phenotypic difference between UK and RRV NSP1 proteins. Here, we examined whether swap-ping the carboxyl-terminal 100-aa region of UK NSP1 with the homologous RRV NSP1 domain (mutant dsC1 NSP1) would destroy the ability of RRV NSP1 to inhibit Mu-IRF3 function (Fig. 8A). NIH 3T3 cells were transfected with RRV, UK, or
dsC1 GFP-NSP1 plasmids, and stability of the dsC1 mutant protein was examined in the presence of MG132 as described above. Fluorescence microscopy examination of transfected cells revealed that dsC1 NSP1 was a highly unstable protein similar to the full-length RRV NSP1 protein (Fig. 8B). Thus, the presence of the UK NSP1 C-terminal 100 residues does not alter the stability of RRV NSP1. Next, we examined the ability of dsC1 NSP1 to inhibit Mu-IRF3-directed PRD(III) lucifer-ase activity. NIH 3T3 cells were transfected as indicated in Fig. 8B, and activation of the PRD(III) reporter in response to overexpressed Mu-IRF3 was examined in the presence of UK, RRV, or dsC1 NSP1 proteins. We found that in contrast to the parental RRV NSP1 protein, dsC1 NSP1 displayed a signifi-cant loss of IRF3 inhibition. Thus, transposing the
carboxyl-FIG. 6. Effect of NSP1 expression and RV infection on IRF3 function in NIH 3T3 cells. (A) NIH 3T3 cells were transfected as described in the legend of Fig. 5, and the ability of NSP1 to inhibit Mu-IRF3-triggered PRD(III) activity was examined. (B) Cells were infected with the RV strains indicated at an MOI of 2.0 and transfected with poly(I:C) as described in the text. Culture supernatants were examined for secreted murine IFN-at 16 hpi by enzyme-linked immunosorbent assay. (C) NIH 3T3 cells were transfected as described in the legend of Fig. 5 with the plasmids indicated and subsequently transfected with poly(I:C) for 16 h before measurement of luciferase activity. (D) Cells were transfected with increasing amounts of the NSP1 expression plasmids indicated as follows: 1⫻, 100 ng; 2⫻, 200 ng; and 4⫻, 400 ng. Subsequently, cells were transfected with poly(I:C), and luciferase activity was measured as described above. Bars indicate standard errors of duplicate wells. Experiments were repeated at least twice with similar results.
10330 SEN ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:9.585.115.471.66.470.2]terminal 100 residues of UK NSP1 onto the RRV NSP1 pro-tein was sufficient to significantly abolish the IRF3 inhibitory phenotypic difference between the two NSP1 proteins in NIH 3T3 cells. Taken together, these results demonstrate that the carboxyl-terminal 100 residues are important in determining the ability of RRV NSP1 to block IRF3 function in NIH 3T3 cells and that NSP1 stability and its IRF3 inhibitory ability are functionally independent.
DISCUSSION
In this report we demonstrate that RV NSP1-mediated deg-radation of IRF3 occurs in a host cell-dependent manner. Expression of NSP1 proteins from heterologous UK and NCDV strains resulted in a lack of IRF3 degradation in NIH 3T3 cells although both UK and NCDV NSP1 proteins could effectively direct IRF3 degradation in COS7 and 293T cells, respectively. In contrast, expression of the NSP1 proteins en-coded by RRV and EW resulted in IRF3 degradation in both COS7 and NIH 3T3 cells. Recently, we established that the NSP1 inability to degrade IRF3 correlated with elevated levels of IFN- secretion and type I IFN-restricted replication of heterologous RVs in murine cells (16). In these studies, unlike other heterologous RVs studied (UK, NCDV, and OSU), the
RRV strain was unique in its ability to degrade Mu-IRF3, suppress IFN secretion, and replicate efficiently in wild-type MEFs with an intact IFN signaling response. Thus, based on the evidence obtained so far, heterologous (nonmurine) RVs can be classified on the basis of their IFN-sensitive replication and IRF3-degradative ability in murine cells as restricted (UK, NCDV, and OSU) and promiscuous (RRV), which is defined functionally as possessing the ability to degrade IRF3, suppress IFN-secretion, and replicate in the presence of an intact type I IFN signaling system in the heterologous mouse host. Al-though restricted heterologous RV strains (e.g., UK) were unable to direct IRF3 degradation in mouse cells (16), it was not clear whether this was due to an inherent defect in the UK NSP1 protein or whether IRF3 degradation depends on exter-nal host-encoded factors in a cell-specific manner. Other stud-ies have documented the inability of RV strains to degrade IRF3, but with the exception of the porcine OSU RV (21), such strains have encoded truncated NSP1 proteins (4, 5). Notably, it has not been examined whether the inability of certain NSP1 proteins to degrade IRF3 may be a host cell-specific phenomenon. Our findings clearly demonstrate that the IRF3-degradative phenotype of several full-length NSP1s is host cell dependent and emphasize the need to carefully evaluate RV IFN-inhibitory properties in cells of different host
FIG. 7. NSP1 expression is regulated by polyubiquitin-dependent proteasomal degradation. (A) COS7 cells were transfected with the GFP-NSP1 constructs indicated and treated with MG132 (⫹) or the solvent dimethyl sulfoxide (⫺) as described in Materials and Methods. At 48 h p.t., cells were examined by fluorescence microscopy, and the increase in expression on MG132 treatment compared to untreated cells was used to obtain stable and unstable phenotypes. (B) COS7 cells were transfected as above and treated with PYR-41, an E1 ligase inhibitor, as described in Materials and Methods. Micrographs are low-magnification (⫻10) images of fluor- and phase-filtered fields. Bar diagrams indicate the number of fluorescent cells along theyaxis, obtained by counting fluorescent cells from photomicrographs of cell monolayers at equal magnification and cell density; inhibitor treatments (⫺, with;⫹, without) are indicated alongxaxis.
on November 8, 2019 by guest
http://jvi.asm.org/
origins. Currently, it is not clear as to whether the OSU NSP1 is also conditionally defective in its IRF3-degradative ability and an appropriately permissive host cell for degradation has not been identified or whether, for this strain, NSP1 simply lacks the capacity, under any circumstance, to degrade IRF3. However, preliminary results suggest that in porcine cells, OSU NSP1 is unable to degrade IRF3 (unpublished observa-tions), indicating the likely existence of other mechanisms to attenuate the host IFN response to OSU infection.
The host cell-specific IRF3-degradative function of NSP1 observed in this study suggests a role for host-specific factors that determine the IFN-inhibitory role of NSP1 within a par-ticular host cell. We found that expression of IRF3 alone from the appropriate host could result in its degradation by NSP1 in a restrictive cellular environment. As shown in Fig. 3A UK NSP1 degraded Hu-IRF3 in murine fibroblasts; conversely, UK NSP1 could not degrade ectopically expressed Mu-IRF3 within fully permissive COS7 cells (Fig. 3B). Taken together, these findings indicate that IRF3 is likely the minimal host factor involved in host cell-specific NSP1 function. Of course, it must be stressed here that other host cell components may well be involved in the destructive interaction between NSP1 and IRF3, but all of these other factors appear to be fully functionally redundant between simian and murine cell lines, at least for UK NSP1. Additional NSP1s will need to be studied to determine whether this finding is universal or whether, in other cases, additional host proteins other than IRF3 are
re-quired to specify degradation. Within MA104 cells, NSP1 pro-teins from B641 and OSU strains differ in their IRF3 degra-dation abilities, and this difference correlates with their relative IRF3 binding affinities (21). Based on these findings, the like-lihood exists that NSP1-IRF3 binding characteristics in a par-ticular cell primarily determine or play a key role in determin-ing IRF3 degradation, and the unique ability of certain NSP1 proteins to recognize several different host IRF3 proteins re-sults in a promiscuous phenotype. We are currently testing these possibilities experimentally using different NSP1-IRF3 combinations.
The type I IFN host response is the first line of defense against virus infections, and in epithelial and fibroblast cells, an antiviral state is mediated following secretion of IFN- from infected cells (41). Early IFN induction in infected cells depends on recognition of virus-encoded molecular patterns leading to the activation of IRFs, most notably IRF3, which is essential for the transcription and secretion of IFN- (41). IRF3 is constitutively expressed in the host cells studied here and resides primarily in the cytoplasm in a latent form (31). Virus-induced signaling events initiated from pathogen recognition receptors (PRRs) result in the recruitment of IRF3 into PRR-specific signaling complexes and its subsequent ac-tivation by TBK-1/IKKi kinase, and complete IRF3 acac-tivation likely requires phosphorylation by other kinases including phosphatidylinositol 3-kinase (43). These events are believed to trigger IRF3 conformational changes resulting in
dimeriza-FIG. 8. The carboxyl-terminal 100 residues of NSP1 encode IRF3-inhibitory determinants but do not influence NSP1 stability. (A) Schematic of the domain swap mutant (dsC1 GFP-NSP1) containing the C-terminal 100 amino acids of UK NSP1 on an RRV NSP1 backbone. (B) NIH 3T3 cells that support contrasting UK and RRV NSP1 phenotypes were transfected with the indicated plasmids, and protein expression levels were compared in the presence (⫹) and absence (⫺) of MG132, as described in the legend of Fig. 7. (C) NIH 3T3 cells were transfected as described in the legend of Fig. 6, and PRD(III) activity in response to overexpressed Mu-IRF3 was determined.
10332 SEN ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:11.585.102.483.69.357.2]tion and nuclear translocation (26, 40). Interestingly, IRF3 degradation also occurs following IRF3 phosphorylation in response to virus infection (32); it also occurs as part of a likely signal autoregulatory mechanism (7, 9, 19, 24, 32, 42, 51, 54). RV NSP1 has been demonstrated to interact with IRF3 (22) and mediate its degradation (4, 5), and the results presented here indicate that NSP1-mediated degradation of IRF3 is host cell specific. However, despite these developments, several as-pects of RV NSP1’s ability to inhibit IRF3-dependent IFN responses remain unclear including the mechanism of IRF3 degradation and the exact stage(s) in the IRF3 signal trans-duction pathway targeted by NSP1. Using IRF3 functional reporter assays, we examined the ability of NSP1 to block IRF3 activity following pathway activation by different known trig-gers. NSP1 expression results in robust inhibition of IRF3 function in COS7 cells following activation with poly(I:C), a widely used synthetic mimic of dsRNA, and functional inhibi-tion is reflected by significantly reduced phospho-S396-acti-vated IRF3 in the presence of RRV, UK, and EW NSP1 proteins (Fig. 5A). We also demonstrate that NSP1 protein expression results in strong inhibition of the constitutively ac-tivated IRF3-5D phosphomimetic mutant (Fig. 5D). Thus, from our data it is possible to pinpoint the NSP1-mediated inhibition of IRF3 function in permissive cells to a stage be-tween IRF3 activation and its subsequent transcriptional ac-tivity. Notably, recent studies indicate that proteasomal degra-dation of IRF3 occurs subsequent to phosphorylation events associated with transcriptional activity, and discrete IRF3 phospho-acceptor sites may be involved in a switch-like mech-anism that directs IRF3 degradation (9). In this respect, it is interesting that NSP1 degradation of IRF3 is enhanced follow-ing poly(I:C)-mediated activation since this suggests that acti-vated IRF3 might be a better substrate for NSP1 inhibition. Infection with UK (16) and OSU (21) RVs has been reported to result in IRF3 activation although the signaling pathways involved are unknown. It will be interesting to resolve the mechanism by which NSP1 targets activated IRF3 for degra-dation in the appropriate host cell.
In contrast to the efficient IRF3 inhibition mediated by UK NSP1 in COS7 cells, expression of UK NSP1 is unable to exert a similar inhibitory effect in murine NIH 3T3 cells in response to pathway activation by IRF3 overexpression. Thus, UK NSP1 is unable to suppress IRF3 function in murine cells, supporting our earlier observation that UK virus replication is IFN sensi-tive in murine cells (16). A recent report demonstrated that OSU NSP1 lacks IRF3 degradation ability in MA104 cells but can nevertheless suppress IFN secretion by inhibiting the ac-tivation of NF-kB (20). However, UK appears to be distinct in this regard, at least in mouse fibroblast cells, and induces a robust IFN-secretory response (Fig. 6B) that we have shown earlier to be replication dependent (16). As expected, UK virus infection also fails to actively block IFN induction in response to dsRNA (Fig. 6B), supporting the results observed with tran-sient NSP1 expression. It remains to be seen whether UK virus encodes mechanisms that negate the antiviral state induced by secreted IFN in murine cells, but it is clear that UK replication is enhanced approximately 100-fold in the absence of an intact IFN signaling system in murine cells (16). An interesting ob-servation arising from these experiments is the ability of tran-siently expressed UK NSP1 to inhibit poly(I:C)-directed IRF3
activity in NIH 3T3 cells (Fig. 6C and D), an unexpected finding since UK virus infection was unable to block IFN se-cretion (Fig. 6B) and since UK NSP1 expression did not result in suppression of IRF3-directed activation of the pathway (Fig. 6A). While these results clearly demonstrate the ability of UK NSP1 to inhibit IRF3 function in response to intracellular poly(I:C) at a point upstream of IRF3, the biological signifi-cance of this property is unclear. However, it should be noted that in the absence of an exogenous trigger, the replication of UK virus in NIH 3T3 cells is sufficient to trigger robust IFN induction (16) although the specific virus-associated molecular pattern and the host pathogen recognition receptor involved in this phenomenon are unknown. Recently, it was demonstrated that infection of intestinal epithelial cells by the mutant RV SA11-5S encoding a truncated NSP1 protein results in IRF3 activation through the RIG-I mediated pathway (25). Interest-ingly, activation of the IFN response by RV also occurs in other cell types such as in human plasmacytoid dendritic cells that lack RIG-I (unpublished observations), indicating a cell-type-specific role of discrete PRRs in RV recognition. Thus, the ability of transiently expressed UK NSP1 to specifically inhibit IRF3 functions in response to stimulation with poly (I:C), a ligand mainly for MDA5/TLR3 (28, 34), may be inef-fective in situations where virus replication stimulates early IFN responses via alternate PRRs such as RIG-I and may instead be critical for virus interaction with cell types lacking RIG-I. Although further experiments are clearly needed to understand the significance of this finding, the results obtained demonstrate that NSP1 employs additional mechanisms for IRF3 inhibition that are independent of IRF3 degradation.
NSP1 itself is subject to proteasomal degradation (21, 39), and NSP1 degradation may be linked to its functional ability to target IRF3 for degradation, with low levels of NSP1 correlat-ing with low IRF3 levels in cells (21). Earlier work has docu-mented differences in NSP1 expression levels depending on the RV strain (21, 38). Several proteins reportedly undergo pro-teasomal degradation without undergoing polyubiquitinylation (37). We found that RRV NSP1 proteasomal degradation is dependent on E1 ligase activity (Fig. 7B), indicating that NSP1 is likely subject to polyubiquitinylation leading to degradation. Interestingly, several cellular proteins involved in the IRF3 signal transduction pathway are also regulated by polyubiquitin including TRAFs, TBK-1, RIG-I, and E3 ligases (2, 7, 19, 24, 33, 42, 50, 51, 54, 55). Virally encoded IFN antagonists such as the hantavirus G1 glycoprotein (44) and the influenza virus NS1 protein (49), like NSP1, are also proteasomally degraded. In 293T cells infected with NSP1-encoding adenoviruses, NCDV Myc-NSP1 and OSU Myc-NSP1 expression levels were comparable despite their contrasting IRF3 degradation abili-ties. Stability of UK GFP-NSP1 was significantly greater than that of the NSP1 proteins of RRV or EW in COS7 cells (Fig. 7), yet UK NSP1 could degrade IRF3 efficiently in this host cell. Thus, while NSP1-mediated IRF3 degradation does not necessarily correlate with NSP1 degradation, the exact role of NSP1 degradation in IRF3 inhibition remains to be identified. Mutations in the N-terminal RING finger domain of NSP1 reportedly result in increased NSP1 expression and a reduced IRF3 degradation function (21). However, it is not clear whether these mutations of the RING finger, a structurally important domain, also caused disruption of NSP1
on November 8, 2019 by guest
http://jvi.asm.org/
tion and function at other distal domains. The chimeric NSP1 protein dsC1 NSP1 contains the entire RING finger domain from RRV and is unstable, like the RRV NSP1 protein, but displays a significant loss of its IRF3-inhibitory property, indi-cating that the carboxyl-terminal 100 residues of RRV NSP1 encode determinants of IRF3 degradation but not of stability. Interestingly, an earlier study showed that introduction of a single H136L mutation in the B641 NSP1 protein resulted in impaired NSP1 degradation but not the IRF3-degradative function (21), supporting our observations that NSP1 degra-dation may not necessarily be linked to its IRF3-inhibitory function. It is possible that the N terminus of NSP1 is critical for its stability and overall structural integrity, and the highly variable carboxyl terminus determines IRF3 interaction and inhibition. Due to the presence of zinc binding domains in E3 ligases, it is attractive to speculate that the NSP1 RING finger may encode a similar function and thus determines IRF3 deg-radation (39). However, this is not always the case when other examples of viral IFN antagonists are examined. For example, the hantavirus G1 glycoprotein contains an N-terminal atypical RING finger and, similar to NSP1, is proteasomally degraded while inhibiting host IFN responses in the absence of known E3 ligase function (44), whereas pestivirus Npro can mediate
IRF3 degradation in the absence of a RING finger (47). Ad-ditional studies will be required in order to examine whether RV NSP1 recruits the cellular E2-ubiquitin complex and en-codes E3 ligase enzymatic activity. Nevertheless, one plausible working model at this stage includes the following elements: (a) the highly conserved N-terminal RING finger domain of NSP1 functions as an E3 ubiquitin ligase following host cell-specific recruitment of IRF3 by the variable C-terminal binding domain; (b) IRF3 inhibition occurs despite activation of the IRF3 C-terminal domain cluster, and activated IRF3 may be a better substrate for NSP1-mediated inhibition; (c) NSP1 deg-radation itself is not a prerequisite for IRF3 inhibition to occur; (d) depending on the host cell and nature of pathway activation, NSP1 can also inhibit IRF3 function in the absence of detectable IRF3 degradation by an unknown mechanism; and (e) cellular factors other than IRF3 are apparently unnec-essary for mediating the host cell-specific component of IRF3 degradation. We are currently performing further mutagenesis and domain swapping of NSP1 in order to better resolve these questions and elucidate the relative roles of NSP1 N- and C-terminal domains in NSP1 stability and host cell-specific IRF3 inhibition.
ACKNOWLEDGMENTS
We thank Phuoc Vo for technical assistance, Joyce Troiano for administrative support, and Mario Barro and Nandini Sen for helpful discussions.
This study was supported in part by a VA Merit Award and NIH grants RO1 AI021362-26 and P30DK56339.
REFERENCES
1.Angel, J., M. A. Franco, and H. B. Greenberg.2007. Rotavirus vaccines:
recent developments and future considerations. Nat. Rev. Microbiol.5:529–
539.
2.Arimoto, K., H. Konishi, and K. Shimotohno.2008. UbcH8 regulates
ubiq-uitin and ISG15 conjugation to RIG-I. Mol. Immunol.45:1078–1084.
3.Arvin, A. M., and H. B. Greenberg. 2006. New viral vaccines. Virology
344:240–249.
4.Barro, M., and J. T. Patton.2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3.
Proc. Natl. Acad. Sci. USA102:4114–4119.
5.Barro, M., and J. T. Patton.2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors
IRF3, IRF5, and IRF7. J. Virol.81:4473–4481.
6.Berkner, K. L.1988. Development of adenovirus vectors for the expression
of heterologous genes. BioTechniques6:616–629.
7.Bibeau-Poirier, A., S. P. Gravel, J. F. Clement, S. Rolland, G. Rodier, P. Coulombe, J. Hiscott, N. Grandvaux, S. Meloche, and M. J. Servant.2006.
Involvement of the IB kinase (IKK)-related kinases tank-binding kinase
1/IKKi and cullin-based ubiquitin ligases in IFN regulatory factor-3
degra-dation. J. Immunol.177:5059–5067.
8.Broome, R. L., P. T. Vo, R. L. Ward, H. F. Clark, and H. B. Greenberg.1993. Murine rotavirus genes encoding outer capsid proteins VP4 and VP7 are not
major determinants of host range restriction and virulence. J. Virol.67:2448–
2455.
9.Clement, J. F., A. Bibeau-Poirier, S. P. Gravel, N. Grandvaux, E. Bonneil, P. Thibault, S. Meloche, and M. J. Servant.2008. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of
dimer-ization and CBP association. J. Virol.82:3984–3996.
10.Collins, S. E., R. S. Noyce, and K. L. Mossman.2004. Innate cellular re-sponse to virus particle entry requires IRF3 but not virus replication. J. Virol.
78:1706–1717.
11.Douagi, I., G. M. McInerney, A. S. Hidmark, V. Miriallis, K. Johansen, L. Svensson, and G. B. Karlsson Hedestam.2007. Role of interferon regulatory factor 3 in type I interferon responses in rotavirus-infected dendritic cells
and fibroblasts. J. Virol.81:2758–2768.
12.Dunn, S. J., T. L. Cross, and H. B. Greenberg.1994. Comparison of the rotavirus nonstructural protein NSP1 (NS53) from different species by
se-quence analysis and northern blot hybridization. Virology203:178–183.
13.Estes, M. K.2001. Rotaviruses and their replication, p. 1747–1785.InD. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Strauss (ed.), Fields Virology, 4th ed. Lippincott, Williams and Wilkins, Philadelphia, PA.
14.Fenaux, M., M. A. Cuadras, N. Feng, M. Jaimes, and H. B. Greenberg.2006. Extraintestinal spread and replication of a homologous EC rotavirus strain
and a heterologous rhesus rotavirus in BALB/c mice. J. Virol.80:5219–5232.
15.Feng, N., B. Kim, M. Fenaux, H. Nguyen, P. Vo, M. B. Omary, and H. B. Greenberg.2008. Role of interferon in homologous and heterologous rota-virus infection in the intestines and extraintestinal organs of suckling mice.
J. Virol.82:7578–7590.
16.Feng, N., A. Sen, H. Nguyen, P. Vo, Y. Hoshino, E. M. Deal, and H. B. Greenberg.2009. Variation in rotavirus NSP1 antagonism of the IFN re-sponse results in differential infectivity in mouse embryonic fibroblasts. J.
Vi-rol.83:6987–6994.
17.Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis.2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat.
Immu-nol.4:491–496.
18.Franco, M. A., and H. B. Greenberg.2001. Challenges for rotavirus vaccines.
Virology281:153–155.
19.Goutagny, N., M. Severa, and K. A. Fitzgerald.2006. Pin-ning down immune
responses to RNA viruses. Nat. Immunol.7:555–557.
20.Graff, J. W., K. Ettayebi, and M. E. Hardy.2009. Rotavirus NSP1 inhibits
NFB activation by inducing proteasome-dependent degradation of
beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog.5:e1000280.
21.Graff, J. W., J. Ewen, K. Ettayebi, and M. E. Hardy.2007. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent
degrada-tion of IRF3 and autoregulatory NSP1 stability. J. Gen. Virol.88:613–620.
22.Graff, J. W., D. N. Mitzel, C. M. Weisend, M. L. Flenniken, and M. E. Hardy.
2002. Interferon regulatory factor 3 is a cellular partner of rotavirus NSP1.
J. Virol.76:9545–9550.
23.Greenberg, H. B., P. T. Vo, and R. Jones.1986. Cultivation and
character-ization of three strains of murine rotavirus. J. Virol.57:585–590.
24.Higgs, R., J. Ni Gabhann, N. Ben Larbi, E. P. Breen, K. A. Fitzgerald, and C. A. Jefferies.2008. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated
degradation of IRF3. J. Immunol.181:1780–1786.
25.Hirata, Y., A. H. Broquet, L. Menchen, and M. F. Kagnoff.2007. Activation of innate immune defense mechanisms by signaling through RIG-I/IPS-1 in
intestinal epithelial cells. J. Immunol.179:5425–5432.
26.Hiscott, J. 2007. Triggering the innate antiviral response through IRF-3
activation. J. Biol. Chem.282:15325–15329.
27.Hoshino, Y., R. G. Wyatt, H. B. Greenberg, J. Flores, and A. Z. Kapikian.
1984. Serotypic similarity and diversity of rotaviruses of mammalian and avian origin as studied by plaque-reduction neutralization. J. Infect. Dis.
149:694–702.
28.Kato, H., O. Takeuchi, E. Mikamo-Satoh, R. Hirai, T. Kawai, K. Matsushita, A. Hiiragi, T. S. Dermody, T. Fujita, and S. Akira.2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible
gene-I and melanoma differentiation-associated gene 5. J. Exp. Med.205:
1601–1610.
29.Kearney, K., D. Chen, Z. F. Taraporewala, P. Vende, Y. Hoshino, M. A. Tortorici, M. Barro, and J. T. Patton.2004. Cell-line-induced mutation of
10334 SEN ET AL. J. VIROL.