Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Induction and Bypass of p53 during Productive Infection
by Polyomavirus
Dilip Dey,† Jean Dahl, Sayeon Cho,‡ and Thomas L. Benjamin*
Department of Pathology, Harvard Medical School, Boston, Massachusetts 02115
Received 1 March 2002/Accepted 19 June 2002
Lytic infection by polyomavirus leads to elevated levels of p53 and induction of p53 target genes p21Cip1/ WAF1 (p21) and BAX. This is seen both in polyomavirus-infected primary mouse cell cultures and in kidney tissue of infected mice. Stabilization of p53 and induction of a p53 response are accompanied by phosphory-lation of p53 on serine 18, mimicking a DNA damage response. Stabilization of p53 does not depend on p19Arf
interaction with mdm2. Cells infected by a mutant virus defective in binding pRb and in inducing G1-to-S
progression show a greatly diminished p53 response. However, cells infected by wild-type virus and blocked from entering S phase by addition of mimosine still show a p53 response. These results suggest a role of E2F target genes in inducing a p53 response. Polyomavirus large T antigen coprecipitates with p53 phosphorylated on serine 18 and also with p21Cip1/WAF1. Implications of these and other findings on possible mechanisms of induction and override of p53 functions during productive infection by polyomavirus are discussed.
The tumor suppressor p53 plays a pivotal role in carcino-genesis and is the most frequently mutated gene in human cancers (31, 34). Most DNA tumor viruses have mechanisms to inactivate p53. The large T antigen (large T) of simian virus 40 (SV40) binds p53 and inactivates at least some of its functions (44). The E6 proteins of the highly oncogenic human papillo-mavirus type 16 (HPV-16) and HPV-18 promote the rapid degradation of p53 through a ubiquitin-dependent proteolytic pathway (47). The oncogenic human adenoviruses act to block p53 through dual functions of the E1B proteins acting directly on p53 and on its downstream targets (16).
Thus far, none of these mechanisms have been ascribed to the mouse polyomavirus. This is surprising in view of the effi-ciency and rapidity with which this virus induces tumors. p53 is not stably upregulated in polyomavirus tumors, and most tu-mor cell lines examined show a normal p53 response to DNA damage (18). Regulation of p53 occurs through a variety of mechanisms that may operate differently in different cell types. The possibility that polyomavirus may have some way of coun-teracting p53 in various target cells was tested by determining the effect of the absence of p53 on tumor induction by the virus. Tumors arose significantly more rapidly in p53⫺/⫺than
in p53⫹/⫺or p53⫹/⫹mice, supporting the generally held view
that this virus has no effective way of blocking p53 functions during the course of tumor development (18). This result con-trasts with those in the SV40 system, where the large T antigen clearly binds p53 and where the absence of p53 in the host can retard rather than accelerate tumor development (28).
A single cycle of polyomavirus growth in mouse cells re-quires roughly 48 h and is dependent on cell cycle progression
from G0/G1into S. In the absence of a counteracting
mecha-nism(s) by the virus, induction of a p53 response leading either to cell cycle arrest or apoptosis would be expected to block virus growth. Expression of polyomavirus large T in NIH 3T3 cells can overcome p53-dependent arrest by a mechanism de-pendent on large T interaction with pRb (20, 25). In REF52 established rat fibroblasts, which express normal p53, polyoma-virus large T and/or small T can block signaling between p19ARF and p53 (39, 43). Middle T by itself fails to override p53 (19) and also fails to transform REF52 cells unless accom-panied by large and/or small T (43). These studies were carried out with subviral constructs or in nonpermissive cells, i.e., under conditions where normal virus replication does not oc-cur. The present study was undertaken to gain a better under-standing of the effects of polyomavirus on p53 during lytic infection of mouse cells and of how the virus might override effects of p53.
MATERIALS AND METHODS
Cells and viruses.Primary baby mouse kidney (BMK) cells were prepared from 15-day-old baby mouse kidneys (63). INK-4a⫺/⫺mouse embryo fibroblasts
(MEF) (third passage) were a generous gift of Ron DePinho and Norman Sharpless of the Dana-Farber Cancer Institute, Boston, Mass. p53⫺/⫺MEF were
derived from p53-deficient mice from our colony (18) or from James DeCaprio of the Dana-Farber Cancer Institute. Polyomaviruses were the wild-type labo-ratory strain RA (24), the highly virulent strain LID (4), and the RB1 mutant encoding large T defective for binding of pRb (26). Monolayers were maintained in Dulbecco’s minimal essential medium supplemented with 1 and 10% fetal bovine serum for BMK and MEF, respectively. Cells were infected at a multi-plicity of infection (MOI) of 1 to 30 PFU. The percentage of cells infected was determined by nuclear staining for large T by indirect immunofluorescence, using rat polyclonal anti-T antigen (T Ag) (55) and fluorescein isothiocyanate-conjugated anti-rat immunoglobulin G (IgG) (Jackson ImmunoResearch).
Immunoprecipitation and immunoblotting.Lysates of infected cells were pre-pared at the indicated times with NP-40 lysis buffer (20 mM Tris, pH 7.5, 135 mM NaCl, 1 mM MgCl2, 0.1 mM CaCl2, 10% glycerol, 1% NP-40, 0.1 mM Na3VO4, 50 mM-glycerophosphate, 10 mM NaF, and the protease inhibitor Complete Mini from Roche) and were used for immunoblotting. For immunoprecipitations cells were lysed in buffer containing 20 mM Tris, pH 7.5, 135 mM NaCl, 1% glycerol, 1% NP-40, 0.1 mM Na3VO4, 10 mM-glycerophosphate, 10 mM NaF, and the protease inhibitor Complete Mini from Roche. For immunoblotting, 70 g of protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel
elec-* Corresponding author. Mailing address: Department of Pathology, Harvard Medical School, 200 Longwood Ave. D2-230, Boston, MA 02115. Phone: (617) 432-1960. Fax: (617) 277-5291. E-mail: thomas_benjamin@hms.harvard.edu.
† Present address: Division of Biology, University of California at San Diego, La Jolla, CA 92093-0366.
‡ Present address: Proteome Research Laboratory, Korea Research Institute of Bioscience and Biotechnology, Taejon 305-33, Korea.
9526
on November 8, 2019 by guest
http://jvi.asm.org/
trophoresis. For immunoprecipitations, extracts (0.5 to 1 mg of protein) were incubated with antibody at 4°C overnight and the immune complexes were recovered with protein A-Sepharose CL-4B (Amersham Pharmacia). The pro-teins were resolved on 10 or 12.5% polyacrylamide gels and were transferred to nitrocellulose membrane. Antibodies for immunoblotting were rabbit polyclonal anti-p53 (CM5) from Novocastra; monoclonal anti-p53 (Ab-3, Pab 240) from Oncogene Research; rabbit polyclonal p21 (C-19), rabbit polyclonal anti-BAX (N-20), goat polyclonal anti-p19Arf (M-20), rabbit polyclonal anti-mdm2 (H-221), mouse monoclonal antiactin (C-2) (all from Santa Cruz), and rat poly-clonal anti-T Ag ascites (55). Antibodies for immunoprecipitation were rabbit polyclonal anti-p21 (C-19) from Santa Cruz, rabbit polyclonal anti-p53 or rabbit polyclonal phosphoserine-15 p53 from Cell Signaling, and monoclonal anti-p53 (Ab-1, Pab 421) from Oncogene Research.
To block immunoprecipitation by anti-phosphoserine-15 p53, 2.5l of anti-body and 2.5l of phosphoserine-15 blocking peptide (Cell Signaling) in 95l of lysing buffer were incubated on ice for 2 h before addition of cell extract.
Immunocytochemistry.Infected cells were fixed with 3.7% neutral buffered formalin at room temperature for 20 min. After permeabilization with 0.3% Triton X-100 in phosphate-buffered saline (PBS) for 20 min, the cells were blocked with 5% normal donkey serum (Jackson ImmunoResearch) in PBS. Cells were stained for T Ags with polyclonal anti-T ascites (55), followed by fluorescein isothiocyanate-conjugated anti-rat IgG (Jackson ImmunoResearch), and were stained for phosphoserine-15 p53 with a rabbit polyclonal antibody from Cell Signaling, followed by tetramethyl rhodamine isocyanate-conjugated anti-rabbit IgG (Jackson ImmunoResearch). Cells were counterstained with 4⬘,6⬘-diamidino-2-phenylindole (Sigma) and were analyzed with a Nikon TE300 fluorescence microscope. To block immunoreactivity, 2l of a 1:10 dilution of the phosphoserine-15 p53 antibody and 1l of blocking peptide (Cell Signaling) in 97l of Tris-buffered saline containing 0.1% Tween and 5% bovine serum albumin were incubated at room temperature for 1 h before being added to slides.
Mice and virus injections.Newborn C3H/BiDa mice less than 18 h of age were injected intraperitoneally with the LID strain of polyomavirus at approximately 106PFU/animal. After 8 days, mice were sacrificed, kidney homogenates were prepared in NP-40 lysis buffer, and 180 to 200g of protein was used for immunoblotting.
Cell cycle analysis.Cells were harvested by trypsinization and frozen at⫺80°C in 250 mM sucrose–40 mM sodium citrate–5% dimethyl sulfoxide at 2⫻106 cells/ml (8). For analysis, thawed cells (200l) were washed in PBS containing 2% calf serum and incubated with 300l of PBS–0.5% Triton X-100–0.5 mM EDTA containing 100g of propidium iodide/ml for 20 min. Eighty microliters of 2 M HCl was added, incubation was continued for 30 min, and 300l of 1 M Tris base was added. Ten thousand cells per sample were analyzed for total DNA on a FACSCalibur flow cytometer (Becton Dickinson) using the programs Cell Quest and ModFit.
RESULTS
Stabilization of p53 and induction of p53 target genes
dur-ing productive infection by polyomavirus. Extracts of
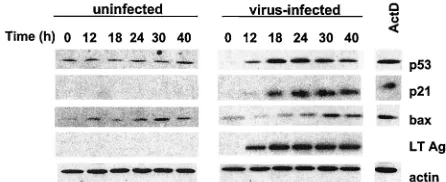
wild-type-polyomavirus-infected BMK cells were prepared at vari-ous times after infection, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and blotted with antibodies to p53 and p53 target genes p21Cip1/WAF1 (p21) and BAX (Fig. 1). Levels of p53 in uninfected cultures were low and remained constant over the 40-h time course examined; p21 was barely detectable. In infected cultures, levels of p53 and p21 began to rise around 12 h, coincident with the appearance of large T. By 24 h postinfection, the amounts of p53 and p21 rose 10- to 20-fold or more compared to those in uninfected cultures. The slight declines in infected cultures at the later time points coincide with the development of cytopathic effects and loss of cells from the monolayers. Levels of BAX were low and somewhat variable in uninfected cultures but were seen to rise upon infection in most experiments but to a lesser degree than those of p21. Induction of p53 and its targets following polyomavirus infection resembled that seen in uninfected cells treated with 5 nM actinomycin D (Act D) for 24 h. When a
rabbit polyclonal antibody to mdm2 capable of recognizing the 90-kDa form and the p60 cleavage product was used, little or no change in levels was seen in either infected cultures or in Act D-treated cells (not shown). The rise in levels of p53 in infected BMK cells is thus accompanied by induction of some but not all p53-responsive genes. Different patterns of p53 response may occur among the many different cell types that the virus is known to lytically infect in vivo (15).
p21 is known to be induced at the transcriptional level by p53 (23, 38) but may also be regulated through E2F-1 in a p53-independent manner (30). To determine if the increase in p21 in virus-infected cells is p53 dependent, experiments were carried out with p53⫹/⫹and p53⫺/⫺MEF. Results in Fig. 2
show that induction of p21 following polyomavirus infection is dependent on p53. The induction of p21 by Act D is also p53 dependent in these cells.
The p53 response in polyomavirus-infected cells is accom-panied by phosphorylation of p53 on serine 18 and does not
depend on the mdm2/p19Arf pathway.Regulation of p53 levels
occurs primarily through posttranslational mechanisms (1). One of these depends on mdm2 binding to p53, leading to p53 degradation via ubiquitin-dependent proteolysis (45, 52). This pathway is opposed by the p19Arf product of INK-4a, which binds and sequesters mdm2 in nucleoli leading to accumula-tion of p53 (62). To determine if p53 accumulaaccumula-tion following polyomavirus infection depends on p19Arf, normal and
[image:2.587.308.531.73.164.2]INK-FIG. 1. Induction of p53 and its target genes. Confluent BMK cells were either uninfected or infected with wild-type polyomavirus at an MOI of 20 to 30. Extracts were prepared at the times indicated and were analyzed by immunoblotting. p53 was detected with anti-p53 (CM5 from Novocastra). At 12 and 24 h postinfection, approximately 20 and 85% of the cells were T Ag positive. Uninfected cells treated with 5 nM Act D for 24 h served as a control. LT, large T.
FIG. 2. Induction of p21 is p53 dependent. Early-passage p53⫹/⫹
and p53⫺/⫺MEF were either uninfected or infected with wild-type
polyomavirus at an MOI of 5 to 10 and were incubated for 24 h. Approximately 75% of the cells were T Ag positive. The conditions for treatment with Act D are given in the legend for Fig. 1A. Extracts were prepared and analyzed by immunoblotting. p53 was detected with anti-p53 (Ab-3 from Oncogene Research).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.587.361.480.573.661.2]4a⫺/⫺MEF were infected and analyzed. Levels of p53 and p21
were seen to increase in the same manner in both sets of cultures (Fig. 3). These results, as well as the finding of little or no change in levels of mdm2 and p19Arf (not shown), indicate that p53 accumulation in polyomavirus-infected BMK cells is not primarily dependent on p19Arf.
Phosphorylation of p53 occurs at multiple sites in response to various stimuli, and these modifications are important in mediating p53’s stability and functions (1, 5, 21, 33, 37, 40, 52–54). DNA damage from UV or ionizing radiation leads to phosphorylation on serines 18 and 23 of murine p53 involving ATM, ATR, CHK1/2, or other kinases (9, 29, 50, 51, 59). Phosphorylation of p53 at serines 15 and 20 (serines 18 and 23 in mouse p53) is known to prevent the binding of mdm2 to p53 and the ability of mdm2 to inhibit p53 transactivation (12, 52). Mutation of serines 15 and 20 to alanine prevents full stabili-zation of p53 in vivo (10, 60), and mutation of mouse serine 18 to alanine in murine embryonic stem cells reduces p53 accu-mulation in response to DNA damage (9). A phosphopeptide antibody specific for p53 phosphorylated on serine 18 was used in an immunoblot of extracts from uninfected, infected, and Act D-treated BMK cells. Polyomavirus infection clearly leads to phosphorylation of p53 on serine 18, as does the DNA damage induced by Act D (Fig. 4).
Induction of a p53 response in vivo.To determine whether
lytic infection in the intact host is also accompanied by a p53 response, extracts of kidneys of 8-day-old neonatally infected mice were prepared and analyzed. The virulent LID strain of polyomavirus was used because of its ability to cause rapid and extensive lytic damage in the kidney (4, 6). Extracts of kidneys from LID-infected mice showed significant elevations of p53 compared to extracts from kidneys of uninfected mice (Fig. 5). Stabilization of p53 in the virus-infected kidney is accompanied by induction of p21 and by phosphorylation of p53 on serine 18. The p53 response induced by polyomavirus lytic infection in vitro and in vivo resembles that induced by DNA damage in all respects examined thus far.
Induction of a p53 response does not require entry of cells
into S phase and synthesis of viral DNA.In spite of induction
of p53 and p21, polyomavirus drives cells into S phase during lytic infection in a manner dependent on large T binding to pRb (20, 25). To determine whether p53 induction by the virus requires override of the G1block and synthesis of viral as well
as cellular DNA, levels of p53 and p21 were compared in cells infected by wild-type virus and the RB1 virus mutant. RB1 encodes a large T defective in binding to pRb, is inefficient in inducing S-phase entry, and shows delayed and reduced levels of viral DNA synthesis (25). BMK cultures were infected by wild-type or mutant virus at MOIs of around 1, leading to roughly two-thirds of the cells becoming T Ag positive in each set of cultures. Cells infected by the wild-type virus progressed into S phase, accompanied by a roughly 18-fold increase in the level of phosphorylated p53 and a 10-fold increase in p21. In contrast, cells infected by RB1 at a matched MOI showed increases of less than twofold in the same parameters (Fig. 6). At considerably higher MOIs, the RB1 mutant was able to induce a larger p53 response. Binding of pRb by large T is therefore essential for efficient induction of a p53 response. These results are consistent with those reported earlier, indi-cating p21 induction by wild-type large T but not by a pRb binding mutant (49).
[image:3.587.360.486.72.141.2]To determine the efficiency of induction of a p53 response by wild-type virus at the cellular level and to confirm that the RB1 mutant is impaired in inducing this response, infected BMK cells were examined by double indirect immunofluorescence
FIG. 3. Accumulation of p53 in polyomavirus-infected cells does not depend on the p19Arf/mdm2 pathway. Early-passage wild-type MEF and INK-4a⫺/⫺ MEF were infected as described for Fig. 1A.
Extracts were prepared at the times indicated and were analyzed by immunoblotting. p53 was detected with anti-p53 (CM5 from Novocas-tra).
[image:3.587.44.284.75.126.2]FIG. 4. Phosphorylation of p53 on serine 18 (Pser18) is induced by infection of BMK cells with polyomavirus. BMK cells were infected or treated with Act D as described for Fig. 1A, and lysates were prepared at the times indicated. Immunoblot analysis was performed with a polyclonal antibody specific for p53 phosphorylated on serine 15 (serine 18 in mouse).
FIG. 5. Induction of a p53 response in vivo. Newborn C3H/BiDa pups were inoculated with a lethal dose of LID. After 8 days, pups were sacrificed and kidney homogenates were prepared for immuno-blot analysis. p53 was detected with anti-p53 (CM5 from Novocastra) and p21 with C-19 (Santa Cruz). Immunoblot analysis for phospho-serine-18 (Pser18) p53 was performed in a separate experiment using anti-phosphoserine-15 p53 from Cell Signaling.
FIG. 6. Induction of phosphoserine-18 (Pser18) p53 and p21 is dependent on binding of large T (LT) to pRb. Confluent BMK cells were either uninfected or infected with wild-type polyomavirus or mutant RB1 at an MOI of 1. Cells were harvested at the times indi-cated and were analyzed by immunoblotting, T Ag staining, and fluo-rescence-activated cell sorting.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.587.320.518.569.671.2] [image:3.587.50.275.637.671.2]staining using anti-T and anti-phosphoserine-18 p53 antibod-ies. Images are shown in Fig. 7, and quantitation is given in Table 1. It is clear that a majority of wild-type-virus-infected cells that become T Ag positive at 18 h postinfection also show induction of phosphoserine-18 p53. The percentage of T Ag-positive cells that are also phosphoserine-18 p53 Ag-positive rises with time after infection, from 62% at 18 h to 88% at 31 h. With the RB1 mutant at 18 h, only 3.4% of cells are doubly positive. The percentage rises with time but remains well below that achieved by the wild-type virus, consistent with the known defects and leakiness of this mutant (25).
Induction of p53 by the virus may depend on function(s) of one or more E2F family members that are activated following large T:pRb interaction but preceding actual S-phase entry. To test this possibility, BMK cells were infected by wild-type virus
allowing release and activation of E2F and were incubated with mimosine to impose a block at the G1/S boundary (35). Under
[image:4.587.53.539.67.377.2]these conditions, p53 and p21 were induced normally, along with phosphorylation of p53 on serine 18 (Fig. 8). Entry of infected cells into S phase and synthesis of viral DNA are therefore not required for induction of a p53 response. Rather, the response depends on large T interaction with pRb and
[image:4.587.323.516.543.663.2]FIG. 7. Immunofluorescence staining of BMK cells infected with wild-type or mutant RB1 virus for T Ag and phosphoserine-18 (Pser18) p53. Confluent BMK cells were infected with wild-type (WT) or RB1 virus at an MOI of 1 or 2 and incubated for 18 h. To block possible nonspecific staining, antibody to phosphoserine-18 p53 was incubated with the antigenic peptide for 1 h before being applied to cells on coverslips. DAPI, 4⬘,6⬘-diamidino-2-phenylindole.
[image:4.587.43.283.653.729.2]FIG. 8. Induction of phosphoserine-18 (Pser18) p53 and p21 occurs in cells blocked in S-phase entry by mimosine. Confluent BMK cells were either uninfected or infected with wild-type polyomavirus at an MOI of 1. At 4 h postinfection mimosine was added to a concentration of 0.2 or 0.5 mM. Cells were harvested at 24 h postinfection and were analyzed by immunoblotting, T Ag staining, and fluorescence-activated cell sorting.
TABLE 1. Incidence of T Ag and phosphoserine-18 p53 (Pp53) in wild-type-virus-infected and mutant-RB1-infected BMK cells
No. of h postinfection
Results for:
Wild-type Mutant RB1
% T Ag⫹ % T Ag/Pp53⫹ % T Ag⫹ % T Ag/Pp53⫹
18 70 62 57 3.4
24 88 74 63 12
31 91 88 70 18
on November 8, 2019 by guest
http://jvi.asm.org/
most likely activation of E2F function(s). Cellular genes re-quired for viral DNA synthesis are known to be induced by E2F-1. Beside G1cyclins, cyclin-dependent kinases, and
vari-ous enzymes needed for DNA synthesis, these include DNA polymerase␣(“primase”) and PCNA (17, 22). The latter as-semble along with RPA, topoisomerase I, and large T at the viral replication origin and are essential for the initiation re-action and replication (56).
Polyomavirus large T coprecipitates with phosphorylated
p53 and with p21. Earlier attempts at demonstrating direct
interaction between p53 and polyomavirus large T were nega-tive using a monoclonal antibody that effecnega-tively brings down SV40 large T:p53 complexes (61). To determine if polyomavi-rus might interact with phosphorylated p53, immunoprecipita-tion of an infected cell extract was carried out using serine 18 phosphopeptide antibody followed by blotting with anti-T. This antibody brought down significantly more large T than either control or other anti-p53 antibodies (Fig. 9A). The ability of the phosphopeptide antibody to coprecipitate large T was ef-fectively inhibited by preincubation with phosphopeptide (Fig. 9B). Polyclonal rabbit anti-p53 gave various but generally low amounts of large T by coimmunoprecipitation in comparison
with anti-phosphoserine-18 p53. Thus, while polyomavirus large T coprecipitates with p53 phosphorylated on serine 18, some form of interaction with unphosphorylated p53 or p53 modified at other sites is also possible. Neither normal IgG nor the mouse monoclonal antibody to p53 previously found to be negative (61) was found to coprecipitate polyomavirus large T. The E7 protein encoded by the high-risk HPVs, in addition to binding pRb, also interacts with p21. This interaction con-tributes to the ability to override p53-mediated G1arrest and
to promote continued cell division in differentiating keratino-cytes (27, 32). The possibility that a similar interaction occurs between polyomavirus large T and p21 was investigated using anti-p21 as the precipitating antibody and extracts of infected BMK cells. A substantial amount of large T was found to coprecipitate with anti-p21 (Fig. 9C). Thus, large T may act in a manner similar to that of HPV E7 in binding p21 and block-ing its cell cycle inhibitory functions.
DISCUSSION
Mice developing polyomavirus tumors show no evidence of effects attributable to the virus leading to a p53 block (18). Polyomavirus tumors fail to show stabilization of p53 in the manner of SV40-induced tumors, and the majority of primary polyomavirus tumor-derived cell lines retain p53 and show a normal p53 response to DNA damage (18). Thus, in the con-text of tumor cell growth, polyomavirus appears neither to elicit nor to block p53.
In contrast, results of the present study clearly show a p53 response elicited by polyomavirus during productive viral in-fection, both in cell culture and in the mouse. Lytic infection and virus amplification are essential steps leading to the induc-tion of tumors. These findings thus raise quesinduc-tions at two levels. First, what is the mechanism of p53 induction operating in productively infected cells but not in nonproductively in-fected (i.e., tumor) cells, and second, how is the virus able to circumvent the cell cycle arrest and proapoptotic functions of p53 during its own replication?
In polyomavirus-infected primary BMK cells, p53 begins to accumulate along with expression of the T Ags and prior to the onset of viral DNA replication. By 20 to 24 h postinfection, levels of p53 are 10- to 20-fold higher than those found in mock-infected cells. Coincident with its accumulation, p53 be-comes phosphorylated on serine 18, leading to its stabilization. p53 target genes are selectively induced. p21 is highly induced, BAX is induced to a lesser degree, and mdm2 remains essen-tially unchanged. This pattern of induction is similar to that seen in the same cells following Act D treatment. This suggests that lytic infection by polyomavirus in some manner triggers a DNA damage-like response. The latter typically involves acti-vation of ATM, ATR, DNA-PK, and the CHK1 and CHK2 kinases, which are known to phosphorylate p53 on N-terminal serine residues. Though the specific pathway leading to p53 phosphorylation has not been determined, some aspect of this pathway upstream of p53, or possibly the p53 response itself, may be important to the virus.
[image:5.587.110.214.73.358.2]Results suggest that the viral trigger for induction of a p53 response in a lytic infection may be viral DNA replication initiation complexes. These complexes, best characterized for
FIG. 9. Polyomavirus large T (LT) coprecipitates with p53 phos-phorylated on serine 18 and with p21. Confluent BMK cells were infected at an MOI of 5 to 10 and incubated for 24 h. (A) Immune complexes were prepared with rabbit polyclonal anti-p53 (Cell Signal-ing), rabbit polyclonal anti-phosphoserine-18 (Pser18) p53, or mouse monoclonal anti-p53 (Pab 421). (B) Immune complexes were prepared with rabbit polyclonal anti-phosphoserine-18 p53 preincubated with or without blocking peptide. (C) Immune complexes were prepared with rabbit polyclonal anti-p21 antibody. The corresponding normal IgGs were used as controls. Immunoblots were performed with rat poly-clonal anti-T Ag ascites (55).
on November 8, 2019 by guest
http://jvi.asm.org/
SV40 (56), consist of large T bound to the viral origin along with replication proteins encoded by the host. The viral DNA is partially unwound at the origin, mimicking cellular DNA origins that have fired (“theta” structures). Results from two experiments support this interpretation and at the same time indicate an important role for E2F-1. The large T mutant RB1, which is defective in binding pRb and in inducing cellular as well as viral DNA synthesis, is also defective in inducing a p53 response. Wild-type virus, on the other hand, is able to induce p53, even when blocked from inducing synthesis of viral and cellular DNA by addition of mimosine to infected cultures. Under these conditions, wild-type large T is expected to bind pRb leading to activation of E2F-1-responsive genes. The lat-ter include DNA polymerase ␣ and PCNA (17, 22), which function along with T antigen in viral DNA initiation and replication complexes (56). Thus, cells harboring free viral DNA with fired origins but unable to enter S phase may re-spond with a DNA damage response.
Two other mechanisms of induction are possible but less likely. One is that the introduction of linear fragments of cel-lular DNA via pseudovirions (42) would mimic the generation of double-strand breaks by ionizing radiation and thereby in-duce a p53 response. This seems unlikely to be the only mech-anism, since the RB1 mutant is impaired in its ability to induce the response. A second possibility is that induction of p19Arf by E2F-1 (3) would lead to p53 stabilization by interference with mdm2-dependent p53 degradation (62). In an established line of rat fibroblasts, middle T can induce a p53-dependent cell cycle block via p19Arf, a block that large and/or small T can overcome (39). In the mouse system, however, stabilization of p53 occurs following polyomavirus infection of INK-4a⫺/⫺
MEF and is therefore not dependent solely on the p19Arf/ mdm2 pathway.
Induction of p53 during lytic infection potentially could lead to G1 arrest or to apoptosis. Either cellular response would
effectively block virus replication. However, several mecha-nisms are available to the virus to bypass or block p53 re-sponses. First, the binding of pRb by large T would be expected to act downstream of p21, bypassing the G1 arrest brought
about by inhibition of the G1cyclin–cyclin-dependent kinases
acting on pRb. Induction of p21, which can be brought about by the virus either via p53 or possibly E2F-1 (30), may con-ceivably serve a positive function for the virus by titrating the levels of free p21 so as to aid in the assembly and promote rather than inhibit the activity of these kinases (30, 36, 64).
Coprecipitation of large T with phosphorylated p53 may indicate direct or indirect association. Only a small fraction of large T appears to be involved. Interaction could be mediated by the CBP/p300 coactivators of p53 which also bind polyoma-virus large T (11, 43a). Alternatively, large T and p53 may be brought together on the viral DNA, which contains p53 and large T binding sites adjacent to the core origin (42a). The anti-p53 monoclonal antibody Pab 421 fails to bring down complexes with polyomavirus large T (reference 61 and present results). This antibody binds to p53 at the C terminus (57) and effectively brings down complexes with SV40 large T, which binds to the central DNA-binding domain of p53 (2, 58). This suggests that polyomavirus large T may interact differ-ently from SV40 large T by binding at the C terminus of p53 such that these complexes would not be recognized by the Pab
421 monoclonal antibody. Large T does not block the induc-tion of p21 by p53. However, the binding of large T to p21 provides a plausible mechanism for counterbalancing the cell growth arrest functions of p53 in a manner similar to that shown for HPV E7 (27, 32).
The proapoptotic functions of p53 mediated by BAX could well be overridden by middle T activation of the phosphatidyl-inositol 3-kinase/Akt pathway, which is known to protect against apoptosis in virus-infected cells (13, 41, 46). Akt can block apoptosis at several levels, including phosphorylation of BAD, caspases, and p21 (7, 14, 65). Both the hamster and mouse polyomaviruses encode middle T proteins that activate this antiapoptotic pathway (13, 48), distinguishing them from other DNA tumor viruses that lack a middle T and that target p53 more directly.
ACKNOWLEDGMENTS
D. Dey and J. Dahl contributed equally to this work.
We thank James DeCaprio for p53⫺/⫺fibroblasts, Ron DePinho
and Norman Sharpless for providing the INK-4a⫺/⫺MEF, and Walter
Eckhart and Carol Prives for helpful discussions. The technical assis-tance of John Carroll and Rebecca Dowgiert is also gratefully acknowl-edged.
This work has been supported by grants R35-44343, PO1 CA-50661, and RO1 CA-90992 from the National Cancer Institute.
REFERENCES
1.Appella, E., and C. W. Anderson.2001. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem.268:2764–2772. 2.Bargonetti, J., I. Reynisdottir, P. N. Friedman, and C. Prives.1992.
Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev.6:1886–1898.
3.Bates, S., A. C. Phillips, P. A. Clark, F. Stott, G. Peters, R. L. Ludwig, and K. H. Vousden.1998. p14ARFlinks the tumour suppressors RB and p53. Nature395:124–125.
4.Bauer, P. H., R. T. Bronson, S. C. Fung, R. Freund, T. Stehle, S. C. Harrison, and T. L. Benjamin.1995. Genetic and structural analysis of a virulence determinant in polyomavirus VP1. J. Virol.69:7925–7931.
5.Bean, L. J. H., and G. R. Stark.2001. Phosphorylation of serines 15 and 37 is necessary for efficient accumulation of p53 following irradiation with UV. Oncogene20:1076–1084.
6.Bolen, J. B., S. E. Fisher, K. Chowdhury, T. C. Shan, J. E. Williams, C. J. Dawe, and M. A. Israel.1985. A determinant of polyomavirus virulence enhances virus growth in cells of renal origin. J. Virol.53:335–339. 7.Cardone, M. H., N. Roy, H. R. Stennicke, G. S. Salvesen, T. F. Franke, E.
Stanbridge, S. Frisch, and J. C. Reed.1998. Regulation of cell death pro-tease caspase-9 by phosphorylation. Science282:1318–1321.
8.Chang, T. H., F. A. Ray, D. A. Thompson, and R. Schlegel.1997. Disregu-lation of mitotic checkpoints and regulatory proteins following acute expres-sion of SV40 large T antigen in diploid human cells. Oncogene14:2383– 2393.
9.Chao, C., S. Saito, C. W. Anderson, E. Appella, and Y. Xu.2000. Phosphor-ylation of murine p53 at ser-18 regulates the p53 responses to DNA damage. Proc. Natl. Acad. Sci. USA97:11936–11941.
10.Chehab, N. H., A. Malikzay, E. S. Stavridi, and T. D. Halazonetis.1999. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA96:13777–13782.
11.Cho, S., Y. Tian, and T. L. Benjamin.2001. Binding of p300/CBP co-activator by polyoma large T antigen. J. Biol. Chem.276:33533–33539.
12.Craig, A. L., L. Burch, B. Vojtesek, J. Mikutowska, A. Thompson, and T. R. Hupp.1999. Novel phosphorylation sites of human tumour suppressor pro-tein p53 at Ser20 and Thr18 that disrupt the binding of mdm2 (mouse double minute 2) protein are modified in human cancers. Biochem. J.342:133–141. 13.Dahl, J., A. Jurczak, L. A. Cheng, D. C. Baker, and T. L. Benjamin.1998. Evidence of a role for phosphatidylinositol 3-kinase activation in the block-ing of apoptosis by polyomavirus middle T antigen. J. Virol.72:3221–3226. 14.Datta, S. R., A. Brunet, and M. E. Greenberg.1999. Cellular survival: a play
in three Akts. Genes Dev.13:2905–2927.
15.Dawe, C. J., R. Freund, G. Mandel, K. Ballmer-Hofer, D. A. Talmage, and T. L. Benjamin.1987. Variations in polyoma virus genotype in relation to tumor induction in mice. Characterization of wild type strains with widely differing tumor profiles. Am. J. Pathol.127:243–261.
16.Debbas, M., and E. White.1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev.7:546–554.
on November 8, 2019 by guest
http://jvi.asm.org/
17.DeGregori, J., T. Kowalik, and J. Nevins.1995. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol.15:4215–4224.
18.Dey, D. C., R. P. Bronson, J. Dahl, J. P. Carroll, and T. L. Benjamin.2000. Accelerated development of polyoma tumors and embryonic lethality: dif-ferent effects of p53 loss on related mouse backgrounds. Cell Growth Differ. 11:231–237.
19.Doherty, J., and R. Freund.1999. Middle T antigen activation of signal transduction pathways does not overcome p53-mediated growth arrest. J. Vi-rol.73:7882–7885.
20.Doherty, J., and R. Freund.1997. Polyomavirus large T antigen overcomes p53 dependent growth arrest. Oncogene14:1923–1931.
21.Dumaz, N., and D. W. Meek.1999. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J.18:7002–7010.
22.Dyson, N.1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245–2262.
23.el-Deiry, W. S., T. Tokino, V. E. Velculescu, D. B. Levy, R. Parsons, J. M. Trent, D. Lin, W. E. Mercer, K. W. Kinzler, and B. Vogelstein.1993. WAF1, a potential mediator of p53 tumor suppression. Cell75:817–825. 24.Feunteun, J., L. Sompayrac, M. Fluck, and T. L. Benjamin.1976.
Localiza-tion of gene funcLocaliza-tions in polyoma virus DNA. Proc. Natl. Acad. Sci. USA 73:4169–4173.
25.Freund, R., P. H. Bauer, H. A. Crissman, E. M. Bradbury, and T. L. Ben-jamin.1994. Host range and cell cycle activation properties of polyomavirus large T-antigen mutants defective in pRB binding. J. Virol.68:7227–7234. 26.Freund, R., R. T. Bronson, and T. L. Benjamin.1992. Separation of
immor-talization from tumor induction with polyoma large T mutants that fail to bind the retinoblastoma gene product. Oncogene7:1979–1987.
27.Funk, J. O., S. Waga, J. B. Harry, E. Espling, B. Stillman, and D. A. Galloway. 1997. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev.11:2090–2100.
28.Herzig, M., M. Novatchkova, and G. Christofori.1999. An unexpected role for p53 in augmenting SV40 large T antigen-mediated tumorigenesis. Biol. Chem.380:203–211.
29.Hirao, A., K. Y. Y., S. Matsuoka, A. Wakeham, J. Ruland, H. Yoshida, D. Liu, S. J. Elledge, and T. W. Mak.2000. DNA damage-induced activation of p53 by the checkpoint kinase chk2. Science287:1824–1827.
30.Hiyama, H., A. Iavarone, and S. A. Reeves.1998. Regulation of the cdk inhibitor p21 gene during cell cycle progression is under the control of the transcription factor E2F. Oncogene16:1513–1523.
31.Hollstein, M., D. Sidransky, B. Vogelstein, and C. C. Harris.1991. p53 mutations in human cancers. Science253:49–53.
32.Jones, D. L., R. M. Alani, and K. Munger.1997. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev.11:2101–2111.
33.Kapoor, M., and G. Lozano.1998. Functional activation of p53 via phos-phorylation following DNA damage by UV but not gamma radiation. Proc. Natl. Acad. Sci. USA95:2834–2837.
34.Ko, L. J., and C. Prives.1996. p53: puzzle and paradigm. Genes Dev. 10:1054–1072.
35.Krude, T.1999. Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res.247:148–159. 36.LaBaer, J., M. D. Garrett, L. F. Stevenson, J. M. Slingerland, C. Sandhu, H. S. Chou, A. Fattaey, and E. Harlow.1997. New functional activities for the p21 family of CDK inhibitors. Genes Dev.11:847–862.
37.Lees-Miller, S. P., K. Sakaguchi, S. J. Ullrich, E. Appella, and C. W. Ander-son.1992. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell. Biol.12:5041–5049.
38.Li, Y., C. W. Jenkins, M. A. Nichols, and Y. Xiong.1994. Cell cycle expres-sion and p53 regulation of the cyclin-dependent kinase inhibitor p21. Onco-gene9:2261–2268.
39.Lomax, M., and M. Fried.2001. Polyoma virus disrupts ARF signaling to p53. Oncogene20:4951–4960.
40.Lu, H., Y. Taya, M. Ikeda, and A. J. Levine.1998. Ultraviolet radiation, but not gamma radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc. Natl. Acad. Sci. USA95:6399–6402.
41.Meili, R., P. Cron, B. A. Hemmings, and K. Ballmer-Hofer.1998. Protein kinase B/Akt is activated by polyomavirus middle-T antigen via a phospha-tidylinositol 3-kinase-dependent mechanism. Oncogene16:903–907. 42.Michel, M. R., B. Hirt, and R. Weil.1967. Mouse cellular DNA enclosed in
polyoma viral capsids (pseudovirions). Proc. Natl. Acad. Sci. USA58:1381– 1388.
42a.Miller, S. D., G. Farmer, and C. Prives.1995. p53 inhibits DNA replication in vitro in a DNA-binding-dependent manner. Mol. Cell. Biol.15:6554–6560. 43.Mor, O., M. Read, and M. Fried.1997. p53 in polyoma virus transformed
REF52 cells. Oncogene15:3113–3119.
43a.Nemethova, M., and E. Wintersberger.1999. Polyomavirus large T antigen binds the transcriptional coactivator protein p300. J. Virol.73:1734–1739. 44.Pipas, J. M., and A. J. Levine.2001. Role of T antigen interactions with p53
in tumorigenesis. Semin. Cancer Biol.11:23–30.
45.Prives, C., and P. A. Hall.1999. The p53 pathway. J. Pathol.187:112–126. 46.Qian, W., and K. G. Wiman.2000. Polyoma virus middle T and small t
antigens cooperate to antagonize p53-induced cell cycle arrest and apoptosis. Cell Growth Differ.11:31–39.
47.Scheffner, M., B. A. Werness, J. M. Huibregtse, A. J. Levine, and P. M. Howley.1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell63:1129–1136.
48.Scherneck, S., R. Ulrich, and J. Feunteun.2001. The hamster polyomavi-rus—a brief review of recent knowledge. Virus Genes22:93–101. 49.Schuchner, S., and E. Wintersberger.1999. Binding of polyomavirus small T
antigen to protein phosphatase 2A is required for elimination of p27 and support of S-phase induction in concert with large T antigen. J. Virol. 73:9266–9273.
50.She, Q.-B., N. Chen, and Z. Dong.2000. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem.275: 20444–20449.
51.Shieh, S. Y., J. Ahn, K. Tamai, Y. Taya, and C. Prives.2000. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev.14:289–300.
52.Shieh, S. Y., M. Ikeda, Y. Taya, and C. Prives.1997. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell91:325–334. 53.Shieh, S. Y., Y. Taya, and C. Prives.1999. DNA damage-inducible
phos-phorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J.18:1815–1823.
54.Siliciano, J. D., C. E. Canman, Y. Taya, K. Sakaguchi, E. Appella, and M. B. Kastan.1997. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev.11:3471–3481.
55.Silver, J. B., B. S. Schaffhausen, and T. L. Benjamin.1978. Tumor antigens induced by nontransforming mutants of polyoma virus. Cell15:485–496. 56.Simmons, D. T.2000. SV40 large T antigen functions in DNA replication
and transformation. Adv. Virus Res.55:75–134.
57.Stephen, C. W., P. Helminen, and D. P. Lane.1995. Characterization of epitopes on human p53 using phase-displayed peptide libraries: insights into antibody-peptide interactions. J. Mol. Biol.248:58–78.
58.Tan, T.-H., J. Wallis, and A. J. Levine.1986. Identification of the p53 protein domain involved in formation of the simian virus 40 large T-antigen–p53 protein complex. J. Virol.59:574–583.
59.Tibbetts, R. S., K. M. Brumbaugh, J. M. Williams, J. N. Sarkaria, W. A. Cliby, S. Y. Shieh, Y. Taya, C. Prives, and R. T. Abraham.1999. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13:153–157.
60.Unger, T., R. V. Sionov, E. Moallem, C. L. Yee, P. M. Howley, M. Oren, and Y. Haupt.1999. Mutations in serines 15 and 20 of human p53 impair its apoptotic activity. Oncogene18:3205–3212.
61.Wang, E. H., P. N. Friedman, and C. Prives.1989. The murine p53 protein blocks replication of SV40 DNA in vitro by inhibiting the initiation functions of SV40 large T antigen. Cell57:379–392.
62.Weber, J. D., L. J. Taylor, M. F. Roussel, C. J. Sherr, and D. Bar-Sagi.1999. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol.1:20–26. 63.Winocour, E.1963. Purification of polyoma virus. Virology19:158–168. 64.Zhang, H., G. J. Hannon, and D. Beach.1994. p21-containing cyclin kinases
exist in both active and inactive states. Genes Dev.8:1750–1758. 65.Zhou, B. P., Y. Liao, W. Xia, B. Spohn, M. H. Lee, and M. C. Hung.2001.
Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol.3:245–252.