Feline Herpesvirus 1 US3 Blocks the Type I Interferon Signal
Pathway by Targeting Interferon Regulatory Factor 3
Dimerization in a Kinase-Independent Manner
Jin Tian,
aYongxiang Liu,
aXiaoxiao Liu,
aXue Sun,
aJikai Zhang,
aLiandong Qu
aaDivision of Zoonosis of Natural Foci, State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary
Research Institute, Chinese Academy of Agricultural Sciences, Harbin, People's Republic of China
ABSTRACT
As a prevalent agent in cats, feline herpesvirus 1 (FHV-1) infection
con-tributes to feline respiratory disease and acute and chronic conjunctivitis. FHV-1 can
successfully evade the host innate immune response and persist for the lifetime of
the cat. Several mechanisms of immune evasion by human herpesviruses have been
elucidated, but the mechanism of immune evasion by FHV-1 remains unknown. In
this study, we screened for FHV-1 open reading frames (ORFs) responsible for
inhib-iting the type I interferon (IFN) pathway with an IFN-

promoter reporter and
analy-sis of IFN-

mRNA levels in HEK 293T cells and the Crandell-Reese feline kidney
(CRFK) cell line, and we identified the Ser/Thr kinase US3 as the most powerful
in-hibitor. Furthermore, we found that the anti-IFN activity of US3 depended on its N
terminus (amino acids 1 to 75) and was independent of its kinase activity.
Mechanis-tically, the ectopic expression of US3 selectively inhibited IFN regulatory factor 3
(IRF3) promoter activation. Furthermore, US3 bound to the IRF association domain
(IAD) of IRF3 and prevented IRF3 dimerization. Finally, US3-deleted recombinant
FHV-1 and US3-repaired recombinant FHV-1 (rFHV-dUS3 and rFHV-rUS3, respectively)
were constructed. Compared with wild-type FHV-1 and rFHV-rUS3, infection with
rFHV-dUS3 induced large amounts of IFN-

in vitro
and
in vivo
. More importantly,
US3 deletion significantly attenuated virulence, reduced virus shedding, and blocked
the invasion of trigeminal ganglia. These results indicate that FHV-1 US3 efficiently
inhibits IFN induction by using a novel immune evasion mechanism and that FHV-1
US3 is a potential regulator of neurovirulence.
IMPORTANCE
Despite widespread vaccination, the prevalence of FHV-1 remains high,
suggesting that it can successfully evade the host innate immune response and
in-fect cats. In this study, we screened viral proteins for inhibiting the IFN pathway and
identified the Ser/Thr kinase US3 as the most powerful inhibitor. In contrast to other
members of the alphaherpesviruses, FHV-1 US3 blocked the host type I IFN pathway
in a kinase-independent manner and via binding to the IRF3 IAD and preventing
IRF3 dimerization. More importantly, the depletion of US3 attenuated the anti-IFN
activity of FHV-1 and prevented efficient viral replication
in vitro
and
in vivo
. Also,
US3 deletion significantly attenuated virulence and blocked the invasion of
trigemi-nal ganglia. We believe that these findings not only will help us to better
under-stand the mechanism of how FHV-1 manipulates the host IFN response but also
highlight the potential role of US3 in the establishment of latent infection
in vivo
.
KEYWORDS
FHV-1, type I IFN, US3, inhibitors, IRF3
F
eline herpesvirus 1 (FHV-1), an agent that causes feline viral rhinotracheitis, was first
isolated in 1957 (1). Following entry, FHV-1 first replicates in the mucosae of both
the conjunctiva and the upper respiratory tract, resulting in multiple symptoms during
acute disease, including depression, fever, and ocular and nasal discharge (2). Chronic
Received9 January 2018Accepted26 March 2018
Accepted manuscript posted online4 April 2018
CitationTian J, Liu Y, Liu X, Sun X, Zhang J, Qu L. 2018. Feline herpesvirus 1 US3 blocks the type I interferon signal pathway by targeting interferon regulatory factor 3 dimerization in a kinase-independent manner. J Virol 92:e00047-18.https://doi.org/10.1128/JVI.00047-18.
EditorRozanne M. Sandri-Goldin, University of California, Irvine
Copyright© 2018 American Society for Microbiology.All Rights Reserved. Address correspondence to Jin Tian, tj6049345@126.com, or Liandong Qu, qld@hvri.ac.cn.
J.T. and Y.L. contributed equally to this work.
crossm
on November 6, 2019 by guest
http://jvi.asm.org/
FHV-1 infection can cause blindness in cats and occurs in combination with feline
calicivirus (FCV) infection and secondary bacterial infection (3). Severe nonsuppurative
meningoencephalitis caused by FHV-1 was reported recently (4), suggesting the
po-tential of this virus to cause central nervous system disease in cats. Latent chronic
infection is another hallmark of FHV-1 infection. To establish lifelong infection in cats,
the virus spreads through the sensory nerves to the trigeminal ganglia (3), which are
the main sites of latency. Latent infections in cats can be reactivated, resulting in virus
shedding into the environment. In addition to acutely infected cats, latently infected
cats are another primary mediator of virus shedding. However, the molecular
mecha-nism of FHV-1 latent infection is elusive.
The
Herpesviridae
family is classified into three subfamilies:
Alphaherpesvirinae
,
Betaherpesvirinae
, and
Gammaherpesvirinae
(5). Latent infection in the
Herpesviridae
family is a common feature. Unlike tumor viruses, which have multifunctional viral
oncogenes that control viral replication and oncogenesis, herpesviruses manipulate the
early innate immune response in the host in many different ways, and this feature is
important for the establishment of infection. Herpes simplex virus 1 (HSV-1) infection
induces a host type I interferon (IFN) response during early infection, but the
function-ality of this response is subsequently attenuated by viral proteins (6, 7), including ICP0
(8–10), UL11 (11), UL36 (12), UL42 (13), VP16 (14), VP24 (15), and US3 (16). Human
herpesvirus 6 (HHV-6), a member of the betaherpesvirus subfamily, has a worldwide
distribution. The HHV-6 immediate early 1 (IE1) protein prevents IFN-

gene expression
by preventing IFN regulatory factor 3 (IRF3) from binding efficiently to the IFN-

promoter sequence (17). Castleman’s disease, characterized by an atypical B cell
lymphoproliferative disorder, is caused by HHV-8 (Kaposi’s sarcoma-associated
herpes-virus [KSHV]) (18–20). Viral IRF1 (vIRF1) of HHV-8 can efficiently suppress the herpes-
virus-induced expression of endogenous IFN-

by inhibiting the formation of IRF3-CBP/p300
transcriptional complexes (coactivators of interferon gene transcription) (21). However,
few reports have revealed that these viral gene products are associated with the
establishment of virus latency.
Two recent reports have demonstrated that viral genes with anti-IFN activity affect the
establishment of viral latency or reactivation. Murine gammaherpesvirus 68 (MHV-68) open
reading frame 54 (ORF54) induces the degradation of the type I interferon receptor (22).
Moreover, ORF54 is required to establish latent infection, and this requirement is
based on its anti-IFN activity (22). Ma et al. demonstrated that cyclic GMP-AMP (cGAMP)
synthase (cGAS) and STING play important roles in regulating KSHV reactivation from
latency (23). Furthermore, those authors screened KSHV proteins for their ability to
inhibit the type I interferon pathway and found that KSHV vIRF1 targets STING and
disrupts the interaction of STING with TBK1, thereby inhibiting STING phosphorylation
and concomitant activation and consequently suppressing the interferon pathway (23).
These data indicate that the modulation of the type I interferon pathway is
indispens-able for efficient infection by and the lifelong persistence of herpesviruses.
Compared to the characteristics of infection with human herpesviruses, the
char-acteristics of FHV-1 infections are similar, but there are several differences. To date, no
study has been performed to investigate the effects of FHV-1 ORFs on the type I
interferon pathway. In the present study, we show that US3, an inhibitor, impedes the
interferon response by blocking the dimerization of IRF3. Moreover, we compared the
anti-IFN activity abilities of wild-type, US3-null mutant, and US3-repaired recombinant
FHV-1
in vitro
and
in vivo
. We show that US3-null mutant FHV-1 exhibits impaired
anti-IFN activity and that the restoration of US3 expression increases the anti-IFN
activity of the mutant. More importantly, a lack of US3 reduces virulence and blocks the
establishment of virus latency.
RESULTS
The FHV-1 US3 protein is the most potent IFN signaling antagonist.
To evaluate
the effect of FHV-1 infection on the IFN-

response, Crandell-Reese feline kidney (CRFK)
cells were infected with FHV-1, and IFN-

mRNA levels were then determined from 2 h to
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
12 h. The level of IFN-

mRNA was increased 2 h after infection and reached a peak at 4 h
but began to decline from 6 h (Fig. 1A). Next, we analyzed whether the cGAS-STING signal
pathway is important for IFN induction by FHV-1 infection. As shown in Fig. 1B and C,
knockdown of cGAS or STING reduced the induction of IFN-

mRNA, suggesting that the
cGAS-STING pathway is critical for IFN induction upon FHV-1 infection 4 h after infection. To
identify the viral genes/proteins responsible for modulating the IFN-

response, we cloned
a total of 70 FHV-1 ORFs and analyzed the effects of the ectopic expression of each ORF on
IFN-

promoter activity induced by cGAS-STING in HEK 293T cells. HEK 293T cells were
cotransfected with a plasmid-borne Flag-tagged virus ORF and an IFN-luciferase (Luc)
promoter reporter plasmid, together with the cGAS and STING plasmids. The cellular lysates
were subjected to dual-luciferase reporter (DLR) assays to measure IFN-

promoter activity
at 24 h posttransfection. As shown in Fig. 1D, we identified 11 inhibitors (UL30, ICP0, UL11,
UL55, UL1, UL45, UL27, UL3.5, UL48, UL4, and US3), and the ectopic expression of each
inhibitor resulted in at least a 50% reduction in IFN-

promoter activity. Among these
inhibitors, US3 was the most powerful antagonist and caused a 90% reduction in the IFN-

promoter activity. We also analyzed the levels of endogenous IFN-

mRNA by real-time
quantitative PCR (qPCR) during the ectopic expression of these inhibitors (Fig. 1E), and the
results confirmed that US3 was the inhibitor that most effectively antagonized the IFN
response.
Next, we analyzed the effects of the ectopic expression of the 11 inhibitors on
poly(dA·dT)-mediated IFN-

promoter activation in feline CRFK cells using reporter
assays and real-time qPCR analysis. Compared with vector transfection, the ectopic
expression of UL11, UL55, UL1, UL45, UL27, UL3.5, UL48, UL4, and US3 was able to
induce an
⬃
50% decrease in the IFN-

promoter activity (Fig. 1F). Additionally, US3 was
the most powerful antagonist and resulted in a 90% reduction (Fig. 1F). The efficient
anti-IFN activity of the US3 protein was further validated by measuring endogenous
IFN-

mRNA levels by real-time qPCR (Fig. 1G). Moreover, US3 expression significantly
inhibited the expression of the IFN-stimulated genes (ISGs) induced by poly(dA·dT)
(data not shown).
All data indicated that US3 was the most effective IFN signaling inhibitor.
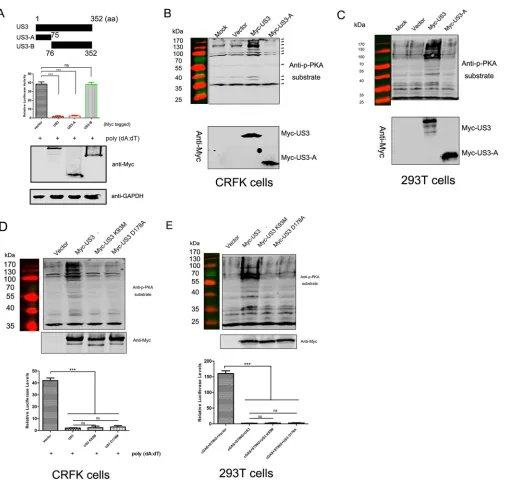
US3 kinase activity is not required for its anti-IFN function.
US3 was the most
powerful inhibitor of the IFN response. To analyze its minimal anti-IFN response
domain, we constructed a series of truncated US3 mutants. As shown in Fig. 2A, the
ability of US3-A, comprising residues 1 to 75, to inhibit poly(dA·dT)-mediated IFN-

promoter activation was comparable to that of full-length US3, but US3-B, comprising
residue 76 to the end of the protein, was unable to inhibit IFN-

promoter activation.
HSV-1 US3 has kinase activity similar to that of protein kinase A (PKA) (24). We
analyzed the Ser/Thr kinase activities of US3 and US3-A using an anti-phospho-Ser/Thr
PKA substrate antibody in CRFK and HEK 293T cells. Compared with mock-transfected
CRFK cells or those transfected with either the vector or US3-A, an increased number
of bands was detected in cells transfected with US3 (Fig. 2B). A similar result was
observed for HEK 293T cells (Fig. 2C). Both results indicated that US3 kinase activity may
be not required for its anti-IFN activity.
Furthermore, we constructed kinase-dead (KD) US3 mutants, namely, K93M and
D178A, and confirmed the lack of kinase activity in CRFK cells (Fig. 2D, top) and HEK
293T cells (Fig. 2E, top). To examine the role of US3 kinase activity in the inhibition of
IFN-

promoter activity in CRFK and HEK 293T cells, a luciferase reporter plasmid
(IFN-Luc) was cotransfected with the US3 K93M or US3 D178A plasmid, and IFN-

promoter activity was then measured. The ectopic expression of the K93M or D178A
mutant failed to affect the anti-IFN activity of US3 (Fig. 2D and E, bottom).
The results indicated that the anti-IFN activity of US3 was not dependent on its
kinase activity and that the N-terminal (residues 1 to 75) region of US3 was a potential
functional domain for counteracting type I IFN activation.
US3 inhibits IRF3-mediated IFN-

promoter activation.
cGAS is a cytosolic DNA
sensor that induces the type I IFN response mainly via the transcription factor IRF3 (25)
on November 6, 2019 by guest
http://jvi.asm.org/
FIG 1Screening of the viral proteins responsible for blocking the IFN-response. (A) CRFK cells (2⫻105) were infected with FHV-1 at an MOI of 1 for the indicated times, and the levels of IFN-mRNA from infected cells were analyzed by real-time qPCR. (B and C) CRFK cells (2⫻105) were transfected with 10 pmol of si-NC (negative control) or si-cGAS (B) or si-STING (ST) (C) for 24 h and then infected with FHV-1 at an MOI of 1 for 4 h. The levels of IFN-mRNA from infected cells were analyzed by real-time qPCR. The expression levels of cGAS and STING were determined by Western blotting. (D) HEK 293T cells (5⫻104) were transfected with 200 ng/well of the IFN-Luc plasmid and 20 ng/well of the pRL-TK plasmid, together with 50 ng/well of pFlag-cGAS, 5 ng/well of pFlag-STING, and 250 ng/well of an FHV-1 ORF expression plasmid. At 24 h posttransfection, cells were lysed, the firefly andRenillaluciferase activities in the
(Continued on next page)
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:4.585.43.547.65.667.2]and partially via the transcription factor NF-
B (26). To investigate which transcription
factor was inhibited by US3 in the process of activating IFN-

signaling, the NF-
B–Luc
or IRF3-Luc luciferase reporter plasmid was cotransfected with pMyc-US3, together with
the cGAS and STING plasmids, into HEK 293T cells for 24 h. Compared to the empty
vector control, US3 expression significantly decreased IRF3-mediated transactivation by
7- to 70-fold in a dose-dependent manner (Fig. 3A) but did not significantly affect
NF-
B-mediated transactivation (Fig. 3B).
To determine which adaptor protein in the IRF3 pathway was blocked by US3, CRFK
cells were cotransfected with the US3 plasmid and plasmids expressing the STING
adaptor protein, TBK1, and the active form of IRF3 (IRF3/5D). US3 was able to inhibit
IRF3 promoter activity driven by all expression constructs (Fig. 3C to E). The activation
induced by STING and TBK1 was inhibited by more than 90% (Fig. 3C and D), and the
activation driven by IRF3/5D was inhibited by 80% to 90% (Fig. 3E). Collectively, these
results suggested that US3 inhibited the IFN antiviral response at the IRF3 level or
downstream.
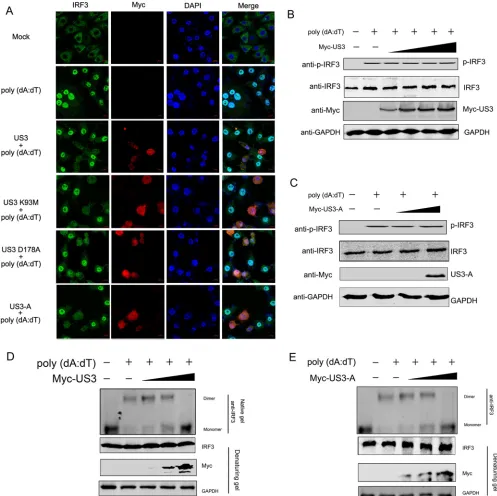
US3 inhibits the dimerization of IRF3.
With the activation of interferon signaling,
the IRF3 dimer subsequently translocates to the nucleus, where it binds to the IFN-

promoter. First, we confirmed whether US3 was able to reduce the entry of activated
IRF3 into the nucleus. CRFK cells were transfected with Myc-US3 or its mutants for 12
h and then transfected with poly(dA·dT). The levels of IRF3 in the nucleus after
stimulation were evaluated by confocal microscopy. As shown in Fig. 4A, transfection
with poly(dA·dT) increased the level of IRF3 in the nucleus. However, the ectopic
expression of US3 or its mutants inhibited IRF3 translocation into the nucleus,
suggest-ing that US3 inhibited the IFN antiviral response by targetsuggest-ing IRF3.
Furthermore, we performed Western blot analysis to determine whether US3
blocked IRF3 entry into the nucleus by reducing the level of phosphorylated IRF3
(p-IRF3). We found that poly(dA·dT) treatment increased the level of p-IRF3. However,
in the presence of increasing amounts of US3, the level of p-IRF3 was not reduced (Fig.
4B). Moreover, the ability of the truncated US3-A construct to inhibit the IFN-

pathway
remained comparable to that of full-length US3, and US3-A expression also did not
affect the level of p-IRF3 (Fig. 4C).
Phosphorylated IRF3 forms an IRF3 homodimer, which then enters the nucleus and
binds to the IFN-

promoter along with other cotranscription factors. Since the level of
phosphorylated IRF3 was not decreased by US3 expression, the dimerization of
phos-phorylated IRF3 may have been blocked. To confirm this hypothesis, we first analyzed
the effect of US3 expression on the IRF3 dimerization induced by poly(dA·dT). CRFK
cells were transfected with increasing amounts of Myc-US3 or the empty vector for 12
h and then transfected with poly(dA·dT). The lysates were resolved by native gel
electrophoresis, and both the monomeric and dimeric forms of IRF3 were detected. As
shown in Fig. 4D, an increase in the level of dimeric IRF3 was detected in cells
stimulated with poly(dA·dT), whereas in the presence of US3, the dimerization of IRF3
was decreased in a dose-dependent manner. Additionally, the inhibitory effect of US3-A
on IRF3 dimerization was also detected in a dose-dependent manner (Fig. 4E).
These data indicated that targeting IRF3 dimerization served as a strategy for US3 to
block the IFN response.
US3 targets IRF3 by binding to the IRF association domain.
After activation with
a virus or stimulator, the formation of the IRF3 homodimer depends on the IRF
FIG 1Legend (Continued)
total cell lysates were measured, and the relative luciferase activity was determined. The results are shown as a heat map. (E) HEK 293T cells (5⫻104) were transfected with 50 ng/well of pFlag-cGAS, 5 ng/well of pFlag-STING, and 250 ng/well of an FHV-1 ORF expression plasmid. At 20 h posttransfection, the mRNA levels of endogenous IFN-in the cells were measured by real-time qPCR. (F) CRFK cells (5⫻104) were transfected with 200 ng/well of the IFN-Luc plasmid and 20 ng/well of the pRL-TK plasmid, together with 250 ng/well of an FHV-1 ORF expression plasmid. At 12 h posttransfection, 2g/ml poly(dA·dT) was transfected into the cells for 24 h, and the relative luciferase activities were then determined. (G) CRFK cells (5⫻104) were transfected with 250 ng/well of an FHV-1 ORF expression plasmid. At 12 h posttransfection, 2g/ml poly(dA·dT) was transfected into the cells for 24 h, and the mRNA levels of endogenous IFN- in the cells were then measured by real-time qPCR. The data shown represent the means⫾SD, and all experiments were repeated three times. Differences (*,P⬍0.05;**,P⬍0.01;***,P⬍0.001) between the experimental and control groups are noted.
on November 6, 2019 by guest
http://jvi.asm.org/
association domain (IAD) region. Since the ectopic expression of US3 inhibits IRF3
dimerization, we speculated that US3 binds to the IAD of IRF3, which may disrupt the
dimerization of IRF3.
To confirm this hypothesis, we first examined the interaction between US3 and IRF3
by performing coimmunoprecipitation (co-IP) analyses. The association of US3 with
nonactive or activated IRF3 was evaluated by co-IP. The results indicated that Myc-US3
could coprecipitate with Flag-IRF3 or Flag-IRF3/5D (Fig. 5A). Moreover, the association
between US3 and endogenous IRF3, including phosphorylated IRF3, was also
demon-FIG 2Identification of the US3 domain required for its anti-IFN function. (A) CRFK cells (5⫻104) were transfected with 200 ng/well of the IFN-Luc plasmid and 20 ng/well of the pRL-TK plasmid, together with 250 ng/well of the full-length US3 or truncated US3 plasmids, as indicated. At 12 h posttransfection, 2g/ml poly(dA·dT) was transfected into the cells for 24 h, and the relative luciferase activities were then determined. aa, amino acids. (B to E) CRFK cells (B and D) or HEK 293T cells (C and E) (2⫻105) were transfected with 1g/well of the US3, truncated US3-A, or US3 mutant plasmid or the empty vector. At 24 h posttransfection, cells were lysed and subjected to Western blot analysis using the anti-phospho-Ser/Thr PKA substrate and anti-Myc antibodies for the detection of phospho-serine/threonine peptides and US3, US3-A, or US3 mutants, respectively. The method for evaluating the effect of the ectopic expression of the US3 mutant on IFN-promoter activity (D and E, bottom) is described above. The data shown represent the means⫾SD, and all experiments were repeated three times. Differences (***,P⬍0.001) between the experimental and control groups are shown. ns, nonsignificant.
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:6.585.45.551.65.547.2]strated (Fig. 5B). Furthermore, a pulldown assay was performed with the His-tagged
US3 protein and the Flag-tagged IRF3 protein. The results showed that His-US3
interacted with Flag-IRF3 (Fig. 5C). Since US3-A could antagonize the function of
IRF3/5D, we found that Myc–US3-A could also coprecipitate with IRF3 and
Flag-IRF3/5D (Fig. 5D).
To determine the critical US3-binding domain of IRF3, a series of IRF3 deletion
mutants was constructed (Fig. 5E) and tested by co-IP. The results indicated that the
IRF3 mutant with the deletion of the IAD could not interact with US3 (Fig. 5F) and that
FIG 3US3 inhibits IRF3-mediated type I IFN signaling. (A and B) The Myc-US3 plasmid at increasing amounts was cotransfected into HEK 293T cells with 250 ng/well of the IRF3-Luc (A) or NF-B–Luc (B) plasmid and 20 ng/well of the pRL-TK plasmid, together with 50 ng/well of pFlag-cGAS and 5 ng/well of pFlag-STING. (C to E) A total of 200 ng/well of the Myc-US3 plasmid was cotransfected into CRFK cells with 250 ng/well of the IRF3-Luc plasmid and 20 ng/well of the pRL-TK plasmid, together with 250 ng/well of pFlag-STING, (C) pFlag-TBK1 (D), or pFlag-IRF3/5D (E). Cells were harvested at 24 h posttransfection and subjected to the relative luciferase activity analysis. The data shown represent the means⫾SD, and all experiments were repeated three times. Differences (***,P⬍0.001) between the experimental and control groups are shown.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.42.403.67.549.2]the green fluorescent protein (GFP)-IAD fusion protein could coprecipitate with US3-A
(Fig. 5G), suggesting that the IAD region mediated the IRF3-US3 association.
These results demonstrated that US3 directly targeted the IAD of IRF3.
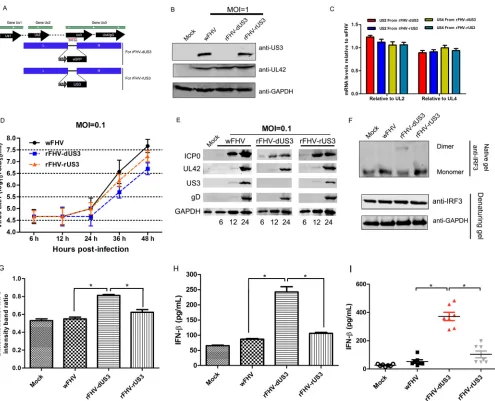
US3 deletion increases the production of IFNs
in vitro
and
in vivo
.
To analyze the
function of US3 during FHV-1 infection, we generated US3-deleted recombinant FHV-1
FIG 4US3 inhibits the formation of the IRF3 dimerization complex. (A) CRFK cells (5⫻104) were transfected with poly(dA·dT) (2g/ml) following transfection with the Myc-US3 or mutated US3 plasmids for 12 h. Cellular localization analysis of IRF3 was performed by observing fluorescence 18 h after stimulation with poly(dA·dT). US3 and its mutants were detected with an anti-Myc polyclonal antibody and visualized with Alexa Fluor 647 (red). IRF3 was detected with an anti-IRF3 antibody and visualized with Alexa Fluor 488 (green). (B to E) CRFK cells transfected with the empty vector or increasing levels of Myc-US3 (B and D) or Myc–US3-A (C and E) for 24 h were transfected with poly(dA·dT) (2g/ml) for 12 h, and the lysates were then subjected to immunoblotting with anti-phospho-IRF3, anti-IRF3, anti-Myc, and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies (B and C) or subjected to native polyacryl-amide gel electrophoresis to detect both the monomeric and dimeric forms of IRF3 using an anti-IRF3 antibody (D and E). All experiments were repeated three times.
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.41.538.72.568.2](rFHV-dUS3) and US3-repaired recombinant FHV-1 (rFHV-rUS3) (Fig. 6A), and the
re-combinant viruses were verified by Western blotting (Fig. 6B). Compared with wild-type
FHV-1 (wFHV), the deletion or recovery of US3 did not affect the expression of the
upstream gene US2 or the downstream gene US4 (Fig. 6C), as demonstrated by
real-time qPCR. Next, we found that rFHV-dUS3 showed significantly attenuated growth
kinetics in cells compared to those of wFHV and rFHV-rUS3 (Fig. 6D). To confirm the
different replication of these viruses, we analyzed the expression of ICP0, UL42, US3,
and gD. The results showed that the deletion of US3 reduced the expression of ICP0,
UL42, and gD, confirming the results of the replication kinetics assay (Fig. 6E).
Afterwards, IRF3 dimerization after infection by wFHV and recombinant viruses was
evaluated by native PAGE. Compared with the dimer levels in wFHV and rFHV-rUS3
infections, a high IRF3 dimer level was detected after infection with rFHV-dUS3 (Fig. 6F
and G). To further examine the anti-IFN activity of US3 upon infection, the level of IFN-

secretion in the cell supernatant was measured by an enzyme-linked immunosorbent
assay (ELISA). The results indicated that rFHV-dUS3 infection induced significantly
higher levels of IFN-

secretion than did wFHV and rFHV-rUS3 infections (Fig. 6H). In
addition, domestic cats were infected with 10
650% tissue culture infective doses
(TCID
50) of wFHV, rFHV-dUS3, and rFHV-rUS3 for 24 h, and the serum IFN-

level was
measured. Compared with wFHV and rFHV-rUS3 infections, rFHV-dUS3 infection
in-duced a significantly large amount of IFN-

(Fig. 6I).
These data revealed that US3 plays an important role in inhibiting IFN-

production
during FHV-1 infection.
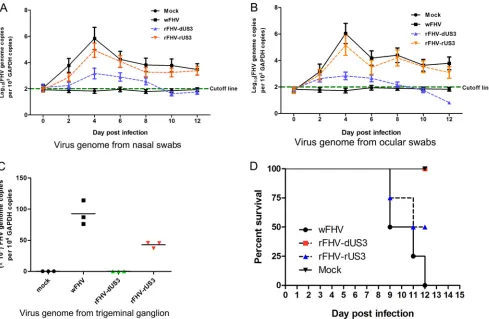
US3 is required for virus shedding and invasion of the trigeminal ganglion.
Some reports have indicated that US3 is a multifunctional protein and affects virus
virulence. We examined the effect of US3 on the virulence of FHV-1. Virus shedding in
nasal and ocular swabs, the viral DNA load in the trigeminal ganglion, and mortality
were analyzed by infecting domestic cats with the viruses. As shown in Fig. 7A and B,
FIG 5US3 binds to the IAD of IRF3. (A) CRFK cells (106) were cotransfected with the Flag-IRF3 or Flag-IRF3/5D and Myc-US3 expression plasmids, followed by co-IP for Flag-IRF3 or Flag-IRF3/5D using an anti-Flag antibody or for Myc-US3 using an anti-Myc antibody. (B) Following transfection with Myc-US3 or the empty vector (vec) for 24 h, the cells were transfected with poly(dA·dT) (2g/ml) for 12 h. Cell lysates were prepared and then subjected to co-IP with an anti-Myc antibody, followed by immunoblotting with anti-Myc, anti-IRF3, and anti-p-IRF3 antibodies. (C) CRFK cells (106) were transfected with the Flag-IRF3 plasmid. His pulldown assays were then conducted, followed by immunoblotting with anti-Flag and anti-His antibodies. (D) Analysis of the association of US3-A with IRF3 or IRF3/5D by co-IP. (E) Schematic representation of the Flag-IRF3 constructs with deletions of the DNA-binding domain (DBD), the IRF association domain (IAD), or the autoinhibition element (AIE). (F) Following cotransfection with the Myc-US3 and Flag-IRF3 mutants for 24 h, cell lysates were prepared and then subjected to co-IP with an anti-Myc antibody, followed by immunoblotting with anti-Myc and anti-Flag antibodies. (G) CRFK cells were transfected with the indicated GFP-IAD constructs and Flag–US3-A plasmids. Co-IP was then conducted, followed by immunoblotting with anti-Flag and anti-GFP antibodies. All experiments were repeated three times.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:9.585.53.540.68.292.2]the deletion of US3 from FHV-1 remarkably decreased virus discharge in the nasal and
ocular swabs compared to the virus discharge in the wFHV and rFHV-rUS3 infection
groups. On day 4 postinfection, the viral genome copy numbers in the nasal swabs and
ocular swabs in the wFHV and rFHV-rUS3 infection groups were more than 100-fold
higher than those in the rFHV-dUS3 infection group, suggesting the determinant role
of US3 in viral replication in peripheral tissues. More importantly, on day 6
postinfec-tion, higher viral DNA loads were detected in the trigeminal ganglia from
wFHV-infected and rFHV-rUS3-wFHV-infected cats, but no viral DNA was detected in the trigeminal
ganglia in rFHV-dUS3-infected cats (Fig. 7C), suggesting that a lack of US3 blocked virus
entry into the trigeminal ganglion.
As important virulence determinants for US3 upon FHV-1 infection, wFHV and
rUS3 challenges resulted in 100% and 75% mortalities, respectively, but
rFHV-FIG 6US3-null mutant FHV-1 induces more IFN-production than wild-type FHV-1 and US3-repaired recombinant FHV-1. (A) Schematic diagram of the transfer vectors for constructing recombinant FHVs. (B) Expression of US3 and UL42 in the US3-deleted (rFHV-dUS3) and US3-repaired (rFHV-dUS3) recombinant FHV-1 was verified by Western blotting using anti-US3 and anti-UL42 antibodies, respectively. (C) The mRNA expression levels of US2 and US4 relative to those of UL2 or UL4 in rFHV-dUS3 and rFHV-dUS3 12 h after infection. (D) Analysis of the growth kinetics among the wFHV, rFHV-dUS3, and rFHV-rUS3 groups. (E) CRFK cells were infected with the indicated viruses at an MOI of 0.1. Protein expression assays of ICP0, UL42, US3, and gD were conducted at the indicated time points. (F and G) CRFK cells were infected with the indicated viruses at an MOI of 1 for 12 h. (F) Cellular lysates were subjected to native polyacrylamide gel electrophoresis to detect both the monomeric and dimeric forms of IRF3 using an anti-IRF3 antibody. (G) Band intensity ratios of the dimers/monomers from three independent experiments, expressed as means⫾SD. (H) CRFK cells were infected with wFHV, rFHV-dUS3, or rFHV-rUS3 at an MOI of 1 for 24 h. The amounts of IFN-in the cellular supernatants from infected cells were analyzed by an ELISA. (I) Cats were infected with 106TCID
50of wFHV, rFHV-dUS3, or rFHV-rUS3 or inoculated with 100l of DMEM (mock). At 24 h postinoculation, sera were collected and subjected to an ELISA to measure IFN-production. The data shown represent the means⫾SD. Differences (*,P⬍0.05) between the experimental and control groups are shown.
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
[image:10.585.46.541.68.469.2]dUS3 challenge was not lethal to the cats (Fig. 7D). These results revealed that the US3
deletion decreased the pathogenicity of FHV-1 in cats and appeared to be involved in
the establishment of a latent viral infection in the trigeminal ganglion.
DISCUSSION
Evading the host early innate immune response may be key for the establishment
of latent and persistent infections by herpesviruses. In the present study, we identified
the FHV-1 US3 protein as an efficient inhibitor of the IFN pathway, and the mechanism
of the anti-IFN activity of US3 was probed. We found that the kinase activity of US3 was
not essential for its anti-IFN activity. Furthermore, US3 interacted with the IAD region
of IRF3, which blocked IRF3/IRF3 dimerization.
In vivo
infection analysis demonstrated
that infection with the US3-null mutant increased IFN-

secretion in the serum, reduced
pathogenicity, and blocked the establishment of latent viral infection in the trigeminal
ganglion. These data constitute the first evidence that US3 is involved in the
estab-lishment of latent viral infection in the trigeminal ganglion.
Hosts can utilize the innate immune system to detect immunostimulatory
mole-cules. In the cytoplasm, RNA viruses are detected by RIG-I-like receptors (RLRs), and
DNA viruses are sensed by cGAS (27, 28), IFI16 (gamma interferon-inducible protein 16)
(29), DAI (DNA-dependent activator of IFN regulatory factors) (30), AIM2 (absent in
melanoma 2), and RNA polymerase III (31). However, viruses have evolved multiple
strategies, such as antagonizing the IFN response, to evade the host antiviral response.
Human herpesviruses can manipulate the early innate immune response of the host in
many ways. Many anti-IFN response effectors encoded by herpesviruses have been
identified. In this study, we found that 11 inhibitors (UL30, ICP0, UL11, UL55, UL1, UL45,
FIG 7US3 deletion decreases virus shedding, blocks invasion into the trigeminal ganglion, and attenuates virulence in cats. Cats were infected with 106TCID 50 of wFHV, rFHV-dUS3, or rFHV-rUS3 or inoculated with DMEM (mock), as described in Materials and Methods. The virus genome copy numbers relative to GAPDH values in nasal (A) and ocular (B) swabs or trigeminal ganglia (C) and mortality (D) were monitored at the indicated time points. The data shown represent the means⫾SD, and all experiments were repeated two times.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:11.585.45.534.70.389.2]UL27, UL3.5, UL48, UL4, and US3) in FHV-1 were potential antagonists that suppressed
the host innate immune response. These proteins may function cooperatively to
contribute to immune evasion upon FHV-1 infection.
HSV-1 US3 is reportedly a multifunctional protein that can regulate viral replication
by phosphorylating a number of viral and cellular substrates (32). Although the US3
gene is conserved among all alphaherpesviruses (33), the function of this gene may
vary among different species of alphaherpesviruses. HSV-1 US3 interacts with IRF3, and
the kinase activity of US3 phosphorylates IRF3 at Ser175 to inhibit IRF3 dimerization,
which prevents the activation of the IFN antiviral response (16). However, we first
discovered that FHV-1 US3 competitively bound to the IAD of IRF3 in a kinase
activity-independent manner, which hindered IRF3 dimerization, revealing a novel
mechanism of the anti-IFN response by alphaherpesvirus US3. IRF3 is an important
transcription factor that functions during the activation of type I IFN signaling in
response to pathogenic invasion. Inhibition of the formation of the transcriptional
activator IRF3 and other coactivator complexes or inhibition of the binding of IRF3 to the
IFN-

promoter is a mechanism used by viruses. HHV-8 vIRF1 interacts with the CBP/p300
coactivators and efficiently inhibits the formation of transcriptionally competent IRF3-CBP/
p300 complexes (34). The HHV-6 IE1 protein prevents IFN-

gene expression by preventing
IRF3 from binding efficiently to the IFN-

promoter sequence (17).
Alphaherpesviruses establish latency in the sensory neurons of the trigeminal
ganglia. The establishment of latency requires a series of complex molecular events
involving cellular and viral factors (35). The virus initially infects the mucosal epithelium,
and virions then enter the peripheral neurons via retrograde axonal transport and
establish a latent viral infection. In this study, we found that after primary infections in
cats, viral DNA of the US3-null mutant was not detected in the trigeminal ganglia,
suggesting that a lack of US3 blocked the establishment of a latent viral infection in the
trigeminal ganglion. However, whether this lack of US3 impairs retrograde axonal
transport or the ability of viruses to infect neurons needs further investigation.
In summary, we have identified multiple anti-IFN proteins of FHV-1, and among
these proteins, US3 is an efficient inhibitor of the IFN response. The binding of US3 to the
IAD of IRF3 impairs the dimerization of IRF3, which inhibits the production of type I
interferon. Moreover, US3 is a key determinant of virus virulence for FHV-1 infection in cats,
and US3 depletion in the context of FHV-1 infection prevents efficient viral replication
in
vitro
and
in vivo
and blocks the entry of virions into the trigeminal ganglion.
MATERIALS AND METHODS
Virus, cells, and antibodies.The FHV-1 strain (HR-1) used in this study was isolated from a cat displaying respiratory symptoms. Compared with the ORF sequences of the FHV-1 C-27 strain submitted to the GenBank database (accession no.FJ478159.2), most ORFs of HR-1 are conserved, and only the UL2, UL39, UL50, and V1 ORFs display certain mutations.
CRFK cells and HEK 293T cells (ATCC) were grown in Dulbecco’s modified Eagle medium (DMEM; Gibco) containing 8% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were incubated at 37°C in a 5% CO2humidified cabinet.
Antibodies, namely, rabbit anti-Flag (catalogue no. ab1162), rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GADPH) (catalogue no. ab22555), rabbit anti-IRF3 (catalogue no. ab68481), rabbit anti-IRF3 (phospho-S386) (catalogue no. ab76493), rabbit anti-Myc (catalogue no. ab9106), rabbit anti-cGAS (catalogue no. ab176177), mouse anti-Myc (catalogue no. ab62928), and rabbit anti-His (catalogue no. ab9108), were purchased from Abcam. The anti-phospho-Ser/Thr PKA substrate antibody (catalogue no. 9621) was pur-chased from Cell Signaling Technology. The mouse anti-US3, anti-UL42, and anti-ICP0 polyclonal antibodies and the mouse anti-gD monoclonal antibody were prepared by our laboratory. Poly(dA·dT) was purchased from InvivoGen.
Plasmid construction.The feline IFN-promoter luciferase reporter plasmid (pIFN-Luc) was de-scribed previously (36). A pRL-TK plasmid (Promega) expressing theRenillaluciferase protein was used as a control. The PRD II and PRD III plasmids expressed luciferase driven by an NF-B-positive regulatory domain (PRD) motif and an IRF3-PRD motif, respectively (36). Flag-US3, Myc-US3 and Myc-US3 mutants, and His-US3 were generated by cloning the ORF of US3 or mutated US3 into the p3⫻Flag-CMV10, pCMV-Myc, and pET-32a vectors (Clontech), respectively. The pFlag-IRF3 plasmid and its mutants were constructed by using standard molecular biology techniques. Feline IRF3/5D, which was constitutively active, and dominant negative IRF3 forms were constructed according to methods described previously by Lin et al. (37). The pFlag-STING plasmid encoded feline STING tagged with Flag, as described previously (38). The pFlag-cGAS plasmid encoded feline cGAS tagged with Flag.
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
Dual-luciferase reporter assay.Cells (104) were cotransfected with the IFN-Luc, NF-B–Luc, or IRF3-Luc reporter plasmid at 0.25g/well, together with 0.02g/well of the pRL-TK plasmid (control), using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, the plasmids and 0.5l of Lipofectamine 2000 were diluted with 12.5l of Opti-MEM (Gibco), respectively, and then mixed and incubated for 5 min. A DNA-lipid complex was added to cells. At 24 h posttransfection, luciferase assays were performed. The Promega luciferase assay system was used, according to the manu-facturer’s directions, to measure the activities of firefly andRenillaluciferase in the total cell lysates.
Real-time quantitative PCR.Total RNA was extracted from cells by using an Axygen multisource total RNA miniprep kit according to the manufacturer’s instructions. cDNA was obtained by using FastKing-RT SuperMix containing DNase (Tiangen, China).
Real-time qPCR was performed by using LightCycler 480 SYBR green I master (Roche, Switzerland) according to the manufacturer’s instructions. The following primer pairs were used: fe-IFN--forward (5=-GAAGGAGGAAGCCATATTGGT-3=) and fe-IFN--reverse (5=-CTCCATGATTTCCTCCAGGAT-3=), fe-IFITM1-forward (5=-CACCACCGTGATCAACATCCA-3=) and fe-IFITM1-reverse (5=-GACTTCACGGAGTAGGCA AAG-3=), fe-ISG15-forward (5=-TCCTGGTGAGGAACCACAAGGG-3=) and fe-ISG15-reverse (5=-TTCAGCCAGA ACAGGTCGTC-3=), fe-Viperin-forward (5=-CATGACCGGGGCGAGTACCTG-3=) and fe-Viperin-reverse (5= -GCAAGGATGTCCAAATATTCACC-3=), hu-IFN--forward (5=-AAATTGCTCTCCTGTTGTGCT-3=) and
hu-IFN--reverse (5=-CTGTCCTTGAGGCAGTATTCA-3=), US2-forward (5=-ATGAGTCCATATAAATCTGTG-3=) and US2-reverse (5=-ATCTATTGACGATATCCCAGT-3=), US4-forward (5=-TTCGATTCCGGGAGTTTCCTG-3=) and US4-reverse (5=-TACGATGATTCTTATCATTCG-3=), UL2-forward (5=-TCCTACCTAAAATACAAGATA-3=) and UL2-reverse (5=-GTAGTGCCGCGTAGATGTTTC-3=), and UL4-forward (5=-CCACTTACCACTTCAGCG GGA-3=) and UL4-reverse (5=-TGTTGGGGTGGTCGGGCTATC-3=).
For the determination of the viral DNA copy numbers, quantitative PCR was conducted. The following primer pairs were used: UL49.5-forward (5=-ATGGATCGTTTATCCGTAACA-3=) and UL49.5-reverse (5=-TGA TATTCACACCCCTTGCCG-3=) and GAPDH-forward (5=-TCCAGGTCTACATGTTCCAGT-3=) and GAPDH-reverse (5=-AAGGTTGTCAGCCCATCCTAG-3=). Both PCR products were cloned into the pJET1.2 T vector to produce a standard curve.
RNA interference (RNAi).The following small interfering RNA (siRNA) sequences were used: TCGG GACCAAATTGACAAA (si-feline cGAS) and GCCGAATACTTGAAGACAT (si-feline STING). The siRNAs were produced by RiboBIO (Guangzhou, China). According to the manufacturer’s instructions, CRFK cells were transfected with 10 pmol of siRNA diluted in 100l of Opti-MEM (Gibco) containing 3l of transfection reagent for 24 h. The knockdown efficiency was determined by Western blot analysis.
FHV-1 ORF construction.The FHV-1 ORF library was assembled from DNA from FHV-1. Total DNA was extracted from the supernatant of virus-infected cell cultures by using an AxyPrep body fluid viral DNA/RNA miniprep kit (Axygen) according to the manufacturer’s instructions. ORF primers were de-signed based on sequences from GenBank. PCR products were ligated into the p3⫻Flag-CMV10 plasmid. All constructs were verified by sequencing and immunoblotting using an anti-Flag tag antibody.
Immunoblotting and coimmunoprecipitation.Briefly, the cells were lysed by using radioimmu-noprecipitation assay (RIPA) lysis buffer (Beyotime, China) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), a protease inhibitor. Lysates were then collected and centrifuged. A total of 25g of each protein sample was separated by using 12% SDS-PAGE and then transferred to nitrocellulose membranes (Thermo) and subjected to immunoblotting.
For co-IP, expression plasmids were transfected into HEK 293T cells for 24 h, and the cells were lysed in ice-cold RIPA lysis buffer containing 1 mM PMSF. The lysates were then collected and centrifuged, followed by protein isolation via incubation with the appropriate amount of anti-Flag M2 magnetic beads or anti-c-Myc agarose beads (Sigma) overnight at 4°C. After washing three times with phosphate-buffered saline (PBS) supplemented with Tween (PBST), the beads were boiled in 5⫻sample buffer before analysis by SDS-PAGE.
For His pulldown assays, His-tagged US3 was expressed in Escherichia coliBL21(DE3) cells. After sonication and centrifugation, the supernatant was incubated with the Ni-nitrilotriacetic acid (NTA) His-Bind resin (catalogue no. 70666; Novagen) for 12 h at 4°C. After washing six times with PBS, the resin was incubated at 4°C for 12 h with the lysates of CRFK cells transfected with pFlag-IRF3. After washing, the bound proteins in the resin were analyzed by SDS-PAGE and immunoblot analyses.
IRF3 dimerization assay.Monomeric and dimerized IRF3 were analyzed as previously described (16). Briefly, the cells were lysed with RIPA lysis buffer containing a protease inhibitor and phosphatase inhibitor cocktail. Native gels (8%) were prerun in Tris-glycine native running buffer (25 mM Tris [pH 8.3], 192 mM glycine, 0.1% deoxycholate [DOC]) for 30 min at 66 V, and the lysates were then resolved for 150 min at 70 V. The monomeric and dimerized IRF3 molecules were detected by immunoblot analysis using an anti-IRF3 antibody.
Subcellular localization.Subcellular localization was examined as previously described (38, 39). Briefly, at 24 h posttransfection, the cells were washed three times with PBST and then fixed with 4% paraformaldehyde for 30 min. Following permeabilization with 0.2% Triton X-100 for 20 min at room temperature, the cells were blocked with PBS containing 5% bovine serum albumin (BSA) for 1 h at room temperature. Primary antibodies, namely, mouse anti-Myc and rabbit anti-IRF3, were diluted in PBS and incubated with cells for 2 h. The cells were then washed three times with PBS and incubated with secondary antibodies [Alexa Fluor 488 goat anti-mouse IgG(H⫹L) (catalogue no. ab150077; Abcam) and Alexa Fluor 647 goat anti-rabbit IgG(H⫹L) (catalogue no. ab150119; Abcam)] for 1 h. The cells were then washed three times with PBS and stained by using 1M 4=,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). A confocal laser scanning microscope (Leica) was used to detect fluorescence.
on November 6, 2019 by guest
http://jvi.asm.org/
Generation of US3-deleted or US3-repaired recombinant FHV-1.To construct the recombinant transfer vector for the generation of US3-deleted recombinant FHV-1, two segments flanking the US3 gene, namely, US3-L and US3-R, were used as the homologous recombination arms. Both homologous arms US3-L and US3-R were amplified from FHV-1 strain HR-1 genomic DNA by using primer pair US3-L-forward (5=-attggatccacagcgtcgccgtctcgttgt-3=) and US3-L-reverse (5=-attgcggccgcctttcatcatcacta tcacta-3=) and primer pair US3-R-forward (5=-attgcggccgcgcgaccattgtctttagcttc-3=) and US3-R-reverse (5=-aattctagatagacttatatttaccaaggt-3=). A linker containing the BamHI, NotI, and XbaI restriction sites was inserted into a pcDNA3.1(⫹) vector that was cut by using BglII and ApaI, and the US3-L and US3-R fragments were then cloned into the vector to create pcDNA-LR. Enhanced GFP (EGFP) was cloned into pAAV-MCS (Stratagene) at the HindIII and BglII restriction sites, and the fragment containing the expression cassette under the control of the cytomegalovirus (CMV) promoter and the human growth hormone (hGH) poly(A) signal was then cut by using NotI and cloned into the NotI site of pcDNA-LR to construct the transfer vector pcDNA-LR-eGFP.
To generate US3-deleted recombinant FHV-1, CRFK cells (106) were transfected with 2g of the pcDNA-LR-EGFP transfer vector and then inoculated with FHV-1 at a multiplicity of infection (MOI) of 0.1 at 12 h posttransfection. Three days later, the cells were harvested, exposed to two to three freeze-thaw cycles, and inoculated into CRFK cells to screen the recombinant virus based on green fluorescence using the plaque method (23).
For the construction of US3-repaired recombinant FHV-1, EGFP from the pcDNA-LR-eGFP transfer vector was cut by restriction enzyme digestion, and the ORF of US3 was cloned to generate the CMV-US3 expression cassette. This recombinant virus was produced according to the methods described above, and the recombinant virus was screened by using the plaque method based on the disappearance of green fluorescence. US3-deleted recombinant FHV-1 and US3-repaired recombinant FHV-1 were verified by Western blotting using anti-US3 and anti-UL42 polyclonal antibodies.
Growth curves.The growth kinetics of wFHV and rFHV-dUS3 were assessed as described previously (36).
ELISA for IFN-.Secreted IFN-in the cell supernatant or serum was measured by using a cat interferon beta ELISA kit (catalogue no. MBS047371; MyBioSource) according to the manufacturer’s instructions.
Animal experimental design.The animal experimental protocols in this study were approved by the Harbin Veterinary Research Institutional Animal Care Committee.
The experimental cats tested negative for feline calicivirus, parvovirus, herpesvirus, and infectious peritonitis virus by reverse transcription-PCR (RT-PCR) or PCR. Five-month-old healthy domestic cats weighing between 2.0 and 2.5 kg were randomly divided into four groups (7/group) living in a single animal house (3 m by 3 m) for each group. The cats were anesthetized subcutaneously with Quan Mian Bao (10 mg/kg of body weight), which included lidocaine, ketamine, and haloperidol (40), according to American Animal Hospital Association (AAHA) anesthesia guidelines for dogs and cats (41). Cats were inoculated with 0.5 ml (0.2 ml for each nasal passage and 0.05 ml for each eye) of 106TCID
50of wild-type and recombinant FHV-1 via intranasal and ocular routes, respectively. The control group was mock challenged with 0.5 ml DMEM. The serum samples before challenge and after challenge for 24 h were collected for the analysis of the amount of IFN-. Nasal and ocular swabs were harvested to determine virus discharge. On day 6, three animals from each group were humanely euthanized with 20% sodium pentobarbital (0.3 ml/kg) intravenously (i.v.) according to protocols suggested by the World Society for the Protection of Animals for the euthanasia of dogs and cats (42). The trigeminal ganglia were isolated for the analysis of viral DNA copy numbers. During the experiment, infected cats were humanely euthanized when they began hunching in pain, were barely able to move or breathe, and lost their ability to eat and drink (40).
Statistics.The data are presented as the means⫾standard deviations (SD). Statistical significance was determined by using unpairedttests and one-way analysis of variance (ANOVA) with Prism 5.0 software (GraphPad Software). For all tests, aPvalue of⬍0.05 was considered to indicate a significant difference.
Accession number(s).All the ORF sequences have been submitted to the GenBank database under accession no.MH027306toMH027379.
ACKNOWLEDGMENTS
This work was funded by the National Natural Science Foundation of China (grant
no. 31770172).
We thank Chunfu Zheng’s laboratory for help with IRF3 dimerization assays.
REFERENCES
1. Crandell RA, Maurer FD. 1958. Isolation of a feline virus associated with intranuclear inclusion bodies. Proc Soc Exp Biol Med 97:487– 490. https://doi.org/10.3181/00379727-97-23783.
2. Tai SH, Niikura M, Cheng HH, Kruger JM, Wise AG, Maes RK. 2010. Complete genomic sequence and an infectious BAC clone of feline herpesvirus-1 (FHV-1). Virology 401:215–227.https://doi.org/10.1016/j.virol.2010.02.021. 3. Thiry E, Addie D, Belak S, Boucraut-Baralon C, Egberink H, Frymus T,
Gruffydd-Jones T, Hartmann K, Hosie MJ, Lloret A, Lutz H, Marsilio F, Pennisi MG, Radford AD, Truyen U, Horzinek MC. 2009. Feline herpesvirus infection.
ABCD guidelines on prevention and management. J Feline Med Surg 11:547–555.https://doi.org/10.1016/j.jfms.2009.05.003.
4. Hora AS, Tonietti PO, Guerra JM, Leme MC, Pena HFJ, Maiorka PC, Brandao PE. 2013. Felid herpesvirus 1 as a causative agent of severe nonsuppurative meningoencephalitis in a domestic cat. J Clin Microbiol 51:676 – 679.https://doi.org/10.1128/JCM.02462-12.
5. Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. 2009. The order Herpesvirales. Arch Virol 154:171–177.https://doi.org/10.1007/s00705-008-0278-4.
Tian et al. Journal of Virology
on November 6, 2019 by guest
http://jvi.asm.org/
6. Melroe GT, DeLuca NA, Knipe DM. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J Virol 78: 8411– 8420.https://doi.org/10.1128/JVI.78.16.8411-8420.2004.
7. Zheng C. 3 January 2018. Evasion of cytosolic DNA-stimulated innate im-mune responses by HSV-1. J Virolhttps://doi.org/10.1128/JVI.00099-17. 8. Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. 2004. The herpes
simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol 78:1675–1684.https:// doi.org/10.1128/JVI.78.4.1675-1684.2004.
9. Mossman K. 2005. Analysis of anti-interferon properties of the herpes simplex virus type I ICP0 protein. Methods Mol Med 116:195–205. 10. Zhang J, Wang K, Wang S, Zheng C. 2013. Herpes simplex virus 1 E3
ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J Virol 87:12935–12948.https://doi.org/10.1128/JVI.01952-13.
11. Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol 86:3528 –3540. https://doi.org/10.1128/JVI.06713-11.
12. Wang S, Wang K, Li J, Zheng C. 2013. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquiti-nating TRAF3. J Virol 87:11851–11860.https://doi.org/10.1128/JVI.01211-13. 13. Zhang J, Wang S, Wang K, Zheng C. 2013. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. Med Micro-biol Immunol 202:313–325.https://doi.org/10.1007/s00430-013-0295-0. 14. Xing J, Ni L, Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus
1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol 87:9788 –9801. https://doi.org/10.1128/JVI.01440-13.
15. Zhang D, Su C, Zheng C. 2016. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J Virol 90:5824 –5829.https://doi.org/ 10.1128/JVI.00186-16.
16. Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1 serine/ threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta inter-feron production. J Virol 87:12814 –12827.https://doi.org/10.1128/JVI .02355-13.
17. Jaworska J, Gravel A, Fink K, Grandvaux N, Flamand L. 2007. Inhibition of transcription of the beta interferon gene by the human herpesvirus 6 immediate-early 1 protein. J Virol 81:5737–5748. https://doi.org/10 .1128/JVI.02443-06.
18. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266:1865–1869.https://doi.org/10 .1126/science.7997879.
19. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86:1276 –1280.
20. Soulier J, Grollet L, Oksenhendler E, Miclea JM, Cacoub P, Baruchel A, Brice P, Clauvel JP, d’Agay MF, Raphael M, Sigaux F. 1995. Molecular analysis of clonality in Castleman’s disease. Blood 86:1131–1138. 21. Burysek L, Yeow WS, Lubyova B, Kellum M, Schafer SL, Huang YQ, Pitha
PM. 1999. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J Virol 73:7334 –7342.
22. Leang RS, Wu TT, Hwang S, Liang LT, Tong L, Truong JT, Sun R. 2011. The anti-interferon activity of conserved viral dUTPase ORF54 is essential for an effective MHV-68 infection. PLoS Pathog 7:e1002292.https://doi.org/ 10.1371/journal.ppat.1002292.
23. Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B. 2015. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A 112:E4306 –E4315.https://doi.org/10.1073/pnas.1503831112. 24. Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3
activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 101:9411–9416.https://doi.org/10.1073/pnas .0403160101.
25. Sun LJ, Wu JX, Du FH, Chen X, Chen ZJJ. 2013. Cyclic GMP-AMP synthase
is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786 –791.https://doi.org/10.1126/science.1232458. 26. Xu H, Su C, Pearson A, Mody CH, Zheng C. 2017. Herpes simplex virus
1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-kappaB activation. J Virol 91:e00025-17.https://doi.org/10.1128/ JVI.00025-17.
27. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730 –737.https://doi.org/10.1038/ni1087.
28. Ouyang SY, Song XQ, Wang YY, Ru H, Shaw N, Jiang Y, Niu FF, Zhu YP, Qiu WC, Parvatiyar K, Li Y, Zhang RG, Cheng GH, Liu ZJ. 2012. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36:1073–1086. https://doi.org/10.1016/j.immuni.2012.03.019.
29. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin TC, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Im-munol 11:997–1004.https://doi.org/10.1038/ni.1932.
30. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505.https://doi.org/10.1038/nature06013.
31. Chiu YH, MacMillan JB, Chen ZJJ. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576 –591.https://doi.org/10.1016/j.cell.2009.06.015.
32. Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85:9599 –9613.https://doi.org/10 .1128/JVI.00845-11.
33. Calton CM, Randall JA, Adkins MW, Banfield BW. 2004. The pseudorabies virus serine/threonine kinase Us3 contains mitochondrial, nuclear and membrane localization signals. Virus Genes 29:131–145.https://doi.org/ 10.1023/B:VIRU.0000032796.27878.7f.
34. Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ, Jr, Barber GN, Hiscott J. 2001. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. On-cogene 20:800 – 811.https://doi.org/10.1038/sj.onc.1204163.
35. Maroui MA, Calle A, Cohen C, Streichenberger N, Texier P, Takissian J, Rousseau A, Poccardi N, Welsch J, Corpet A, Schaeffer L, Labetoulle M, Lomonte P. 2016. Latency entry of herpes simplex virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog 12:e1005834.https://doi.org/10.1371/journal.ppat.1005834.
36. Tian J, Zhang X, Wu H, Liu C, Liu J, Hu X, Qu L. 2015. Assessment of the IFN-beta response to four feline caliciviruses: infection in CRFK cells. Infect Genet Evol 34:352–360.https://doi.org/10.1016/j.meegid.2015.06.003. 37. Lin R, Mamane Y, Hiscott J. 1999. Structural and functional analysis of
interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol 19:2465–2474.https://doi.org/10 .1128/MCB.19.4.2465.
38. Zhang X, Wu H, Liu C, Sun X, Zu S, Tian J, Qu L, Li S. 2016. The function of feline stimulator of interferon gene (STING) is evolutionarily con-served. Vet Immunol Immunopathol 169:54 – 62. https://doi.org/10 .1016/j.vetimm.2015.12.005.
39. Wu H, Zhang X, Liu C, Liu D, Liu J, Wang G, Tian J, Qu L. 15 August 2015. Molecular cloning and functional characterization of feline MAVS. Im-munol Reshttps://doi.org/10.1007/s12026-015-8682-9.
40. Tian J, Liu D, Liu Y, Wu H, Jiang Y, Zu S, Liu C, Sun X, Liu J, Qu L. 2016. Molecular characterization of a feline calicivirus isolated from tiger and its pathogenesis in cats. Vet Microbiol 192:110 –117.https://doi.org/10 .1016/j.vetmic.2016.07.005.
41. Bednarski R, Grimm K, Harvey R, Lukasik VM, Penn WS, Sargent B, Spelts K, American Animal Hospital Association. 2011. AAHA anesthesia guide-lines for dogs and cats. J Am Anim Hosp Assoc 47:377–385.https://doi .org/10.5326/JAAHA-MS-5846.
42. Anonymous. 2008. World Society for the Protection of Animals methods for the euthanasia of dogs and cats: comparison and recommendations. World Society for the Protection of Animals, London, United Kingdom. http://www.icam-coalition.org/downloads/Methods%20for%20the %20euthanasia%20of%20dogs%20and%20cats-%20English.pdf.
on November 6, 2019 by guest
http://jvi.asm.org/