Heba H. Mostafa,
a*
Thornton W. Thompson,
a*
Adam J. Konen,
aSteve D. Haenchen,
a*
Joshua G. Hilliard,
aStuart J. Macdonald,
aLynda A. Morrison,
bDavid J. Davido
aaDepartment of Molecular Biosciences, University of Kansas, Lawrence, Kansas, USA
bDepartment of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, St. Louis,
Missouri, USA
ABSTRACT
In the process of generating herpes simplex virus 1 (HSV-1) mutations
in the viral regulatory gene encoding infected cell protein 0 (ICP0), we isolated a
vi-ral mutant, termed KOS-NA, that was severely impaired for acute replication in the
eyes and trigeminal ganglia (TG) of mice, defective in establishing a latent infection,
and reactivated poorly from explanted TG. To identify the secondary mutation(s)
re-sponsible for the impaired phenotypes of this mutant, we sequenced the KOS-NA
genome and noted that it contained two nonsynonymous mutations in
UL39
, which
encodes the large subunit of ribonucleotide reductase, ICP6. These mutations
re-sulted in lysine-to-proline (residue 393) and arginine-to-histidine (residue 950)
substi-tutions in ICP6. To determine whether alteration of these amino acids was
re-sponsible for the KOS-NA phenotypes
in vivo
, we recombined the wild-type UL39
gene into the KOS-NA genome and rescued its acute replication phenotypes in
mice. To further establish the role of
UL39
in KOS-NA’s decreased pathogenicity,
the
UL39
mutations were recombined into HSV-1 (generating UL39
mut), and this
mutant virus showed reduced ocular and TG replication in mice comparable to that of
KOS-NA. Interestingly, ICP6 protein levels were reduced in KOS-NA-infected cells relative
to the wild-type protein. Moreover, we observed that KOS-NA does not counteract
caspase 8-induced apoptosis, unlike wild-type strain KOS. Based on alignment studies
with other HSV-1 ICP6 homologs, our data suggest that amino acid 950 of ICP6 likely
plays an important role in ICP6 accumulation and inhibition of apoptosis, consequently
impairing HSV-1 pathogenesis in a mouse model of HSV-1 infection.
IMPORTANCE
HSV-1 is a major human pathogen that infects
⬃
80% of the human
population and can be life threatening to infected neonates or
immunocompro-mised individuals. Effective therapies for treatment of recurrent HSV-1 infections are
limited, which emphasizes a critical need to understand in greater detail the events
that modulate HSV-1 replication and pathogenesis. In the current study, we
identi-fied a neuroattenuated HSV-1 mutant (i.e., KOS-NA) that contains novel mutations in
the UL39 gene, which codes for the large subunit of ribonucleotide reductase (also
known as ICP6). This mutant form of ICP6 was responsible for the attenuation of
KOS-NA
in vivo
and resulted in diminished ICP6 protein levels and antiapoptotic
ef-fect. Thus, we have determined that subtle alteration of the UL39 gene regulates
ex-pression and functions of ICP6 and severely impacts HSV-1 pathogenesis, potentially
making KOS-NA a promising vaccine candidate against HSV-1.
KEYWORDS
HSV-1, ICP6, UL39, antiapoptosis, pathogenesis, ribonucleotide
reductase, viral mutant
Received18 October 2017Accepted3 January 2018
Accepted manuscript posted online10 January 2018
CitationMostafa HH, Thompson TW, Konen AJ, Haenchen SD, Hilliard JG, Macdonald SJ, Morrison LA, Davido DJ. 2018. Herpes simplex virus 1 mutant with point mutations inUL39is impaired for acute viral replication in mice, establishment of latency, and explant-induced reactivation. J Virol 92:e01654-17.https://doi .org/10.1128/JVI.01654-17.
EditorRichard M. Longnecker, Northwestern University
Copyright© 2018 American Society for Microbiology.All Rights Reserved. Address correspondence to David J. Davido, ddavido@ku.edu.
*Present address: Heba H. Mostafa, Clinical Microbiology Laboratories, University of Rochester Medical Center, Rochester, New York, USA; Thornton W. Thompson, Department of Molecular and Cell Biology, University of California, Berkeley, Berkeley, California, USA; Steve D. Haenchen, University of Arizona College of Public Health, Tucson, Arizona, USA.
on November 6, 2019 by guest
http://jvi.asm.org/
H
erpes simplex virus 1 (HSV-1) is the primary cause of infectious blindness in
western industrialized countries. The virus alternates between two phases during
its life cycle: lytic and latent (1). The lytic replication cycle, which occurs in epithelial
cells of the cornea or the mucocutaneous regions of the oral labial region, is
charac-terized by a temporal cascade of immediate early (IE), early (E), and late (L) gene
expression (2). Latency, on the other hand, is established in sensory neurons that
innervate the primary site of lytic infection. During latency, neither productive infection
nor significant viral gene expression occurs, with the exception of the
latency-associated transcripts (LATs) (3). Reactivation of the latent viral genome can be
trig-gered under various conditions of stress. IE genes are the first to be expressed, and
among these is the gene for the viral protein infected cell protein 0 (ICP0).
ICP0 is a multifunctional protein that has important functions in acute replication,
establishment of latency, and reactivation (4, 5). We examined how phosphorylation of
HSV-1 ICP0 in 3 specific regions of the protein affects its functions, and viral replication,
by creating the viral mutants Phos 1, 2, and 3 (6). When these mutants were
charac-terized using the mouse ocular model of HSV-1 latency and reactivation, Phos 1 and 2
had attenuated phenotypes
in vivo
, whereas Phos 3 behaved like wild-type (WT) HSV-1
in these assays (7). During the course of generating the Phos 3 mutant, we isolated a
Phos 3 variant (termed KOS-NA) that, in contrast, was severely impaired in its acute
replication in mouse eyes and trigeminal ganglia (TG), significantly reduced in its
capacity to establish latency, and reactivated poorly when latently infected TG were
explanted in culture. Given the severe attenuation of KOS-NA relative to Phos 3 in mice,
we hypothesized that KOS-NA contained a secondary mutation(s) in one or more key
viral genes that regulate HSV-1 pathogenesis. Furthermore, such secondary mutations
would provide insight into HSV-1 pathogenesis and could be useful in developing
therapeutic interventions against HSV-1 (e.g., vaccine).
To understand the genetic basis for KOS-NA’s
in vivo
phenotypes, we sequenced its
genome and compared it to that of its wild-type parental virus, strain KOS (8, 9). We
discovered, besides ICP0’s phosphorylation mutations, two nonsynonymous mutations
in the ICP4 gene, one nonsynonymous mutation in the glycoprotein I (gI) gene, and two
nonsynonymous mutations (L393P and R950H) in the UL39 gene of the KOS-NA
genome. Given the parallels between the
in vivo
replication phenotypes of published
UL39
mutants and KOS-NA (10–12), we proposed that at least one of the mutated
codons in
UL39
of KOS-NA is responsible for its attenuated phenotypes.
UL39
encodes
the large subunit of ribonucleotide reductase, also known as ICP6. The ribonucleotide
reductase enzyme complex is essential for viral DNA replication because it converts
ribonucleotides to deoxyribonucleotides (13, 14). Other key functions of ICP6 have
been associated with inhibiting apoptosis (15) and activating or inhibiting necroptosis
(16, 17). Although this gene is not essential for viral growth and replication in dividing
cell lines, its function is required for viral replication in quiescent cells, such as neurons
(18, 19), and several studies have shown that
UL39
mutants are severely impaired for
replication, establishment of latency, and/or reactivation in murine models of HSV-1
infection (10, 11). Based on these observations, we wanted to test whether KOS-NA’s
attenuation is related to its
UL39
mutations.
To investigate this possibility, we rescued the mutated UL39 gene within the
KOS-NA genome with a wild-type copy of
UL39
(KOS-NAR), and we independently
introduced the mutated copy of the
UL39
from the KOS-NA genome into the wild-type
KOS genome (UL39
mut). The acute replication of both viruses in the eyes and TG of
mice, as well as establishment and reactivation from latency, was examined.
RESULTS
Mutations in KOS-NA impair acute viral replication in the eyes and TG.
We
initially examined acute replication of KOS-NA at the periphery in ocular epithelia. For
these experiments, CD-1 mice were infected with 2
⫻
10
5PFU of wild-type HSV-1 (KOS),
the attenuated ICP0 null mutant 7134, or KOS-NA per eye. 7134 was used as an
ICP0-null, attenuated control virus in these experiments (20). Eye swabs from mice were
on November 6, 2019 by guest
http://jvi.asm.org/
taken at days 1, 3, 5, 7, and 9 postinfection. As shown in Fig. 1A, the levels of replication
of KOS-NA on days 1 and 3 postinfection were 79-fold (
t
test,
P
⫽
0.015) and 25-fold (
t
test,
P
⫽
0.006) lower than that of KOS, respectively. On day 5 postinfection, no KOS-NA
infectious virus was detected, whereas KOS still replicated to levels
ⱖ
1,000-fold above
the limit of detection (
t
test,
P
⫽
3.9
⫻
10
⫺5) and continued to replicate through 7 days
postinfection. Interestingly, the replication of KOS-NA was more impaired than that of
7134 on days 3, 5, and 7 postinfection. When acute replication in neurons of the
trigeminal ganglia (TG) was examined, KOS-NA showed no detectable replication at all
time points examined. Relative to KOS titers, these represented decreases of up to
5,000-fold on days 3, 5, and 7 postinfection (Student’s
t
test,
P
⫽
1.4
⫻
10
⫺5to 3.5
⫻
10
⫺9). 7134, as expected (7), replicated in TG neurons of CD-1 mice, albeit poorly
(Fig. 1B).
Mutations in KOS-NA reduced the establishment of latency.
To quantify the
relative amount of viral DNA present in latently infected neurons, TG were collected at
days 28 to 30 postinfection and assayed for the presence of HSV-1 DNA using
quantitative real-time PCR. As shown in Fig. 2, the amounts of latent viral DNA present
in TG were significantly reduced for KOS-NA (53-fold; one-way analysis of variance
[ANOVA],
P
⬍
0.05) and 7134 (12-fold; one-way ANOVA,
P
⬍
0.05) relative to the level
for KOS. Decreases in the establishment of latency for KOS-NA are probably linked to
its impaired replication during acute infection in the TG.
FIG 1Acute replication of KOS-NA in mice. Groups of mice were infected with 2⫻105PFU per eye. Tear
film was collected on days 1, 3, 5, 7, and 9 postinfection, and TG were collected at the same time points from cohorts. Infectious virus in swab samples and homogenized TG was quantified by plaque assay. (A) Acute corneal replication of KOS-NA. (B) Acute replication of KOS-NA in TG. Results are geometric means⫾SEMs (n⫽6 samples per group per time point). The horizontal dotted lines represent the lower limit of detection.*, Student’sttest,P⬍0.05 compared with KOS. A portion of the control data was previously presented in reference 7.
FIG 2Viral genome loads in latent TG. Mice were infected with 2⫻105PFU per eye, and TG were
collected 28 to 30 days postinfection. DNA was extracted from latent TG, and the amount of HSV-1 DNA present was quantified by real-time PCR (n⫽4 to 10 TG per group). Results shown are the fold reduction compared to KOS. Control data were previously presented in reference 7.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:3.585.73.336.70.204.2] [image:3.585.137.269.573.698.2]The efficiency of KOS-NA reactivation is reduced.
To determine if the efficiency
and kinetics of reactivation from latency were altered for KOS-NA, TG of all viral groups
were collected on days 28 to 30 postinfection and subsequently cultured by
explan-tation. Samples were examined daily for cytopathic effect to monitor reactivation, and
the rates of reactivation and total percentage of reactivating samples were determined
for KOS-NA and 7134 mutants relative to that of KOS. KOS began to reactivate on day
3 postexplantation at 66% and reached 100% reactivation by day 5 postexplantation
(Fig. 3). KOS-NA reactivation peaked at day 4 postexplantation, maintaining a
reacti-vation efficiency of 8% throughout the study (Fisher’s exact test,
P
⬍
0.0001) (Fig. 3).
7134 reactivated on day 10 postexplantation, and after heat shock it reached its highest
levels (73%) on day 16 postexplantation (Fig. 3). In contrast, heat shock did not alter the
percentage of TG reactivating KOS-NA. It is very likely that KOS-NA’s diminished
capacity to establish a latent infection (Fig. 2) is directly associated with its inefficient
reactivation phenotype.
Whole-genome sequencing of the KOS-NA mutant.
As our ICP0 phosphorylation
study showed (7), the ICP0 phosphorylation site mutant Phos 3 was not impaired for
acute replication in eyes or TG of mice, the establishment of latency, or reactivation. The
differences in the pathogenesis between Phos 3 and KOS-NA strongly suggested that
KOS-NA contained secondary mutations in its viral genome. To identify the secondary
mutations in KOS-NA responsible for its highly attenuated phenotypes, we sequenced
its viral genome in the same manner as for KOS (8). Subsequently, the genomes of
KOS-NA and KOS (8, 9) were aligned using fast statistical alignment (FSA) to identify
nucleotide polymorphisms between these two viruses (21). The alignment of these
genomes revealed that KOS-NA contained 5 nonsynonymous mutations in three genes:
UL39
, which encodes ICP6,
US7
, which encodes gI, and
RS1
, which encodes the viral
transcriptional regulator ICP4.
Construction of KOS-NAR, UL39
mut, and HrR3 R viruses.
Because the acute
replication, latency, and explant reactivation capabilities of KOS-NA were remarkably
similar to those of ICP6 mutant viruses in mice (10), we hypothesized that the L393P
(lysine to proline) and/or R950H (arginine to histidine) amino acid substitution encoded
in the open reading frame of ICP6 was responsible, at least in part, for KOS-NA’s
attenuated pathogenesis. To address this possibility, we rescued the UL39 gene of
KOS-NA with a wild-type copy of
UL39
, and we introduced the KOS-NA mutated UL39
gene into the genome of the HrR3 virus, an ICP6::
lacZ
insertion mutant of KOS (18). The
latter HSV-1 mutant we called UL39
mut. As a control, we also generated an HrR3 rescue
(R) virus in which we rescued the ICP6::
lacZ
cassette with a wild-type copy of
UL39
.
FIG 3Explant-induced reactivation of KOS-NA. Mice were infected with 2⫻105PFU per eye. On days 28 to
30 postinfection, TG were collected and explanted onto Vero cells. The time required for reactivation to occur from the latent TG was determined by assaying the culture medium daily for the presence of infectious virus. Each time point represents the cumulative percentage of samples that showed reactivation (n⫽19 to 20 TG per group). The arrow at the top of the graph indicates that at day 10, samples were heat shocked at 43°C for 3 h. Control data were previously presented in reference 7.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:4.585.101.307.68.226.2]UL39
mutations are responsible for the diminished acute replication
pheno-type of KOS-NA.
To establish whether the mutations identified in the UL39 gene of
KOS-NA resulted in its attenuated pathogenesis, we tested the acute replication
potential of KOS-NAR and UL39
mutin the eyes and TG of mice. As shown in Fig. 4A,
UL39
mutreplicated to levels comparable to those of KOS-NA and was reduced up to
500-fold relative to KOS in the eyes of mice at days 1, 3, and 5 postinfection (
t
test,
P
ⱕ
1.2
⫻
10
⫺7). In contrast, KOS-NAR and HrR3 R replicated to levels similar to those
of KOS (Fig. 4A).
We then analyzed acute replication in the TG of our viral groups on day 5
postin-fection, as this time point had shown the maximum fold reduction between KOS-NA
and KOS (Fig. 1B). Replication of UL39
mutin TG at day 5 postinfection could not be
detected (
ⱖ
1,412-fold reduction relative to KOS;
t
test,
P
⫽
6.7
⫻
10
⫺15) (Fig. 4B). Acute
replication of KOS-NAR and HrR3 R was comparable to that of KOS (Fig. 4B). The lack of
detectable viral replication for UL39
mutwas also apparent in the TG on day 3
postin-fection (H. H. Mostafa and D. J. Davido, unpublished data).
KOS-NA is reduced for ICP6 protein accumulation without appreciably
affect-ing the interaction between the two subunits of the ribonucleotide reductase.
To
understand the mechanism by which mutations within the UL39 gene impact ICP6, we first
examined the accumulation of ICP6 protein after KOS-NA infection versus KOS. As shown
in Fig. 5A, the ICP6 protein levels after KOS-NA infection were reduced by 50% relative to
FIG 4Mutations in the UL39 gene are responsible for the reduced acute replication phenotype of KOS-NA. (A) Acute replication of KOS-NAR, UL39mut, and HrR3 R in mouse eyes. Mice were infected with
2⫻ 105PFU per eye, and tear film was collected from each eye after 4 h and days 1, 3, and 5
postinfection. The amount of infectious virus collected in each sample was determined by plaque assay. (B) Acute replication of KOS-NAR, UL39mut, and HrR3 R in mouse TG at day 5 postinfection. Mice were
infected as described above, and TG were collected. TG were then homogenized, and the amount of infectious virus present in each sample was determined by plaque assay. Results shown are geometric means⫾SEMs (n⫽8 samples per group per time point). The horizontal dotted line represents the lower limit of detection.*, Student’sttest,P⬍0.05.
FIG 5KOS-NA ICP6 protein accumulation and interaction with pUL40. (A) Vero cells were infected at an MOI of 2 with KOS or KOS-NA for 24 h. Infected monolayers were harvested, and the indicated proteins were examined by Western blot analyses. ICP0 was used as a loading control for infection. (B) Vero cells were mock infected (M) or infected at MOIs of 2 for KOS and 5 for KOS-NA for 24 h. UL40 was immunoprecipitated (IP), and the levels of ICP6 and UL40 were analyzed by Western blotting.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:5.585.49.363.72.208.2] [image:5.585.71.340.585.687.2]those produced by KOS. On the other hand, the UL40 (small subunit of ribonucleotide
reductase) protein levels produced by the two viruses were comparable (Fig. 5A). This
indicates that one or both substitutions in ICP6 reduce its protein levels. We then examined
the interaction between UL40 and ICP6 produced by KOS-NA versus KOS by
immunopre-cipitating pUL40; however, in these experiments we were unable to pull down detectable
amounts of pUL40 in KOS-NA-infected cells. We also infected cells with KOS-NA at a
mulitiplicity of infection (MOI) higher than for KOS, trying to express more KOS-NA ICP6 and
UL40 proteins. Even under these conditions we could, at best, detect only a very faint UL40
band after immunoprecipitating pUL40 (Fig. 5B). Conversely, a robust amount of pUL40
could be pulled down from KOS-infected cells, and consequently a stronger ICP6 band (Fig.
5B). This result suggests that the conformation of pUL40 may depend on its interaction with
ICP6, and thus, reduced ICP6 levels expressed by KOS-NA might make it difficult to pull
down pUL40 with our antibody. On the other hand, the fact that we were able to detect
some ICP6 when immunoprecipitating pUL40 from KOS-NA-infected cells (Fig. 5B) argues
against the premise that the amino acid substitutions in ICP6 from KOS-NA substantially
affect its interaction with UL40.
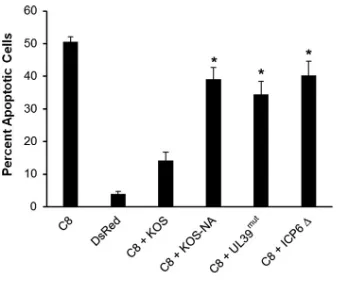
Mutation in
UL39
reduces ICP6-mediated inhibition of caspase 8-dependent
apoptosis.
It has been shown that HSV-1 ICP6 binds to caspase 8, inhibiting apoptosis
(15). To test whether the UL39 mutations impaired the antiapoptotic activity of ICP6,
which might be an underlying mechanism of KOS-NA
in vivo
attenuation, we
trans-fected Vero cells with a plasmid expressing human caspase 8 and then intrans-fected these
cells with KOS, KOS-NA, UL39
mut, or ΔICP6, an ICP6-null mutant. Apoptotic cells were
then counted after staining with Hoechst dye. Our data in Fig. 6 indicate that KOS
impaired caspase 8-induced apoptosis compared to that in KOS-NA-, UL39
mut-, and
ΔICP6-infected samples. This result indicates that one or both codon changes reduce
the antiapoptotic activity of ICP6.
DISCUSSION
We have isolated a virus, KOS-NA, with two nonsynonymous mutations in the UL39
gene, which encodes the large subunit of ribonucleotide reductase. These mutations
result in amino acid substitutions L393P and R950H. KOS-NA containing these
muta-tions is significantly reduced in its acute replication in mouse corneas and TG, is greatly
impaired in its capacity to establish latency, and poorly reactivates from a latent
infection after explant-induced reactivation.
The HSV-1 ribonucleotide reductase proteins encoded by
UL39
and
UL40
belong to
class I ribonucleotide reductases that have a heterodimeric structure which is active in
the
␣
2

2 configuration (22–26) and is similar to that in mammalian and bacterial
FIG 6UL39mutations from KOS-NA reduce ICP6-mediated inhibition of caspase 8-dependent apoptosis. Vero cells were transfected with a plasmid that expresses either CMV-DsRed (DsRed) or human caspase 8 (C8). Five hours later, Vero cells were treated with acyclovir and after 1 h, cells were infected with each indicated virus at an MOI of 2.5 for 18 h. Cells were stained with Hoechst dye, and apoptotic cells were counted by fluorescence microscopy. Data are the averages from two independent experiments (n⫽6 samples per group). Error bars represent SEMs.*, Student’sttest,P⬍0.05.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:6.585.119.290.69.210.2]ribonucleotide reductases. The
UL39
-encoded large subunit contains the catalytic and
allosteric sites which confer substrate specificity. The smaller subunit is responsible for
generation of free radicals that are required for substrate activation (27). Viruses
mutated in the UL39 gene replicate normally in dividing cells at 37°C, where the
reductase activity is thought to be compensated by the cellular homolog. The viral
ribonucleotide reductase activity becomes important, however, for viral DNA
replica-tion in neurons and nondividing cells or at elevated temperatures (e.g., 41°C) (18). Of
relevance to our study, the activity of this viral enzyme complex is essential for efficient
acute replication
in vivo
, which impacts the establishment of latency and reactivation
(10, 28). ICP6 is expressed early, before its small-subunit partner, during viral infection
in cell culture (29–32) and contains an extra N-terminal domain that is not required for
HSV ribonucleotide reductase activity (33). This N-terminal domain possesses
antiapo-ptotic, anti- or pronecroptotic (15–17, 34), chaperone-like (35), and protein kinase (36)
activities. Additionally, ICP6 is a target of HSV-directed cytotoxic T lymphocytes in mice,
a function that is largely conferred by a single amino acid (37).
Previous studies showed that the N-terminal region of ICP6 is required for
interdo-main linking (38), but the interaction of ICP6 with the smaller subunit of ribonucleotide
reductase is conferred by its C-terminal region (39). Amino acid sequence alignment
studies indicate that L393 is not conserved between strain KOS and other HSV strains,
syn17
⫹
(HSV-1), McKrae (HSV-1) HG52 (HSV-2), and 4 other sequenced strains of HSV-1
(Fig. 7A), which, like KOS-NA, encode a proline at this site. This conservation in other
HSV strains suggests that the L393P mutation would not negatively impact ICP6
activities involving this residue and strongly supports the concept that the L393P
substitution in ICP6 of KOS-NA does not contribute to its attenuated phenotype. Amino
acid sequence alignments indicate that R950 is conserved between all HSV-1 and HSV-2
strains we examined (i.e., KOS, syn17
⫹
, McKrae, and the HSV-2 HG52 strain) (Fig. 7B) as
well as varicella-zoster virus (VZV) (40), suggesting that the R950H mutation in KOS-NA
is unique. Moreover, a single mutation at amino acid 961 of HSV-1 ICP6 (41) has been
reported to reduce the ribonucleotide reductase activity of the enzyme (42). Although
this site is important for interactions of the two ribonucleotide reductase subunits, it
was previously concluded that this effect might be indirect (41). Position 950 is close to
amino acid 961, which is within a region identified as “block 10” (residues 960 to 963)
FIG 7Sequence alignments around residue 393 (A) in ICP6 of KOS-NA and various HSV strains and around residue 950 (B) in ICP6 orthologs of KOS-NA, various HSV strains, VZV, and HCMV.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.120.288.70.304.2]that is highly conserved between viral ribonucleotide reductases of herpesviruses (40).
Although the region encompassing amino acids 950 and 961 lies in a region highly
conserved between HSV strains, as shown by our sequence alignment (Fig. 7B), this
region was not as highly conserved when we aligned ICP6 to the large ribonucleotide
reductase subunits from human cytomegalovirus (HCMV) and VZV (40) (Fig. 7B) and
other eukaryotic species (data not shown). Nonetheless, the R950 residue itself was
conserved between alpha- and betaherpesviruses we examined and the eukaryotic
large subunits (data not shown). This highlights the functional importance of the R950
site and indicates that it is likely the cause of KOS-NA phenotypes.
Chabaud et al. have shown that there is a constitutive interaction between HSV-1
ICP6 or its HSV-2 homolog, ICP10, and caspase 8 and proposed that this interaction is a
protective mechanism against HSV-induced apoptosis (43). Interestingly, the C-terminal
domain was shown to be essential to inhibit cytokine-induced apoptosis, which is a
conserved activity of ICP10 in HSV-2 (43). In this study, we showed that one amino acid
at the C terminus of HSV-1 ICP6 is responsible for its antiapoptotic effect and its
mutation has deleterious effects on acute viral replication, both in the mouse cornea
and in the TG. The same domain was recently demonstrated to be responsible for ICP6’s
antiapoptotic activity of blocking virus- or cytokine-induced necroptosis (16). Given the
previous studies regarding ICP6 and apoptosis, our data in Fig. 6 strongly suggest that
the H950 residue in ICP6 of KOS-NA impairs caspase 8-induced apoptosis, contributing
to its attenuated phenotype in mice (Fig. 1 to 3). It is not clear if the R950H substitution
in ICP6 of KOS-NA impairs its ribonucleotide reductase activity, although the R950H
substitution does not directly alter conserved motifs known to be required for this
catalytic activity (44).
Deletion of
US7
, which encodes glycoprotein I (gI), can also attenuate the acute viral
replication in sensory ganglia and the central nervous system (CNS) in mice without
having a pronounced effect in cell culture (45, 46). Consequently, we tested the effect
of introducing the KOS-NA
US7
mutation into the genome of ICP0 phosphorylation
mutant Phos 3. Our results showed that this mutation in the background of Phos 3 virus
was able to efficiently replicate in the eyes and TG of mice at levels comparable to those
of the wild-type strain KOS (Mostafa and Davido, unpublished). This result and the
restoration of wild-type replication by KOS-NAR indicate that the
US7
mutation from
KOS-NA is not responsible for the neuroattenuated phenotype of KOS-NA.
The profound attenuation of KOS-NA in the nervous system suggests that this virus
might be useful in safely stimulating immunity to HSV-1. The severe impairment of
KOS-NA in establishing an efficient latent infection and reactivation from latency is
likely related to its diminished acute replication phenotype in the eyes and TG of mice,
as has been observed with other HSV-1 mutants (28, 47). However, KOS-NA injected
directly into the brains of mice also replicates to barely detectable levels (L. A. Morrison
and D. J. Davido, unpublished data). Indeed, we have data from an initial vaccine study
showing that KOS-NA has very good efficacy as a potential prophylactic vaccine against
HSV-1 (D. J. Davido, E. M. Tu, H. Wong, M. Korom, H. H. Mostafa, B. Combs, S. D.
Haenchen, and L. A. Morrison, unpublished data). The efficacy of KOS-NA as a vaccine
may be due, in part, to its ability to express a mutant form of ICP6. Notably, published
studies indicate that expression of HSV-1 ICP6 and its orthologs can generate
ICP6-specific antibodies and cytotoxic T lymphocytes in HSV-infected or vaccinated animals
and individuals (37, 48, 49). These host immune responses would not occur if an
ICP6-null mutant was used as a vaccine.
MATERIALS AND METHODS
Cell lines and viruses.Vero cells and L7 (ICP0-containing Vero) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 100 g/ml of penicillin, 100 U/ml of streptomycin, and 2 mML-glutamine. The wild-type HSV-1 strain KOS, 7134 (an ICP0-null mutant virus) (20), KOS-NA, KOS-NA marker rescue (MR), UL39mut, HrR3 (a virus that contains a
lacZinsertion inUL39[18]) and ΔICP6 (originally termed ICP6delta [10]), both kindly provided by Sandra Weller, and HrR3 R viruses were propagated and viral titers were determined as previously described (50, 51).
on November 6, 2019 by guest
http://jvi.asm.org/
assembled against the reference HSV-1 strain 17 syn⫹genome using Seqman pro (DNASTAR, Inc.). The final KOS-NA genome is 152,011 bp, has 13 short gaps totaling 1,582 bp exclusively at variable number tandem repeat (VNTR) regions, and was sequenced to an average per-base-pair coverage of 3,104⫻. We transferred annotation from the reference genome strain 17 syn⫹using RATT (56) and confirmed the final KOS-NA annotation manually. To identify and characterize nucleotide differences between KOS and KOS-NA, we aligned the genomes using FSA (21) and applied a custom R script (http://www.r-project .org). We identified 34 single nucleotide polymorphisms (SNPs) discriminating the strains, including 17 nonsynonymous changes residing in six genes (10 of which are the introduced phosphorylation mutations in both copies of ICP0, 2 in the UL39 gene, 4 in both copies of the ICP4 gene, and 1 in the gI gene), and 11 short (1 to 4bp) insertion-deletion events, all present in intergenic regions.
Construction of UL39 mutant and rescue viruses.KOS-NA contains mutations in ICP0 phosphor-ylation sites and was constructed as previously described (7, 51, 52). To generate UL39mut, the UL39 gene
was cloned out of the KOS-NA viral genome using BglII and KpnI sites and ligated into pSP72 using the same sites (pSP72:UL39). Vero cells were plated on 60-mm dishes at 4⫻105per plate. Twenty-four hours
postplating, cells were cotransfected with 1g of viral DNA from HrR3 and 2.5g of pSP72:UL39 plasmid digested with BglII and KpnI. Transfections were performed using Fugene HD (Roche) at a ratio of 3:1 (microliters of transfection reagent to microgram of DNA) based on manufacturer recommendations. Mutants were identified by blue/white selection in the presence of 5-bromo-4-chloro-3-indolyl--D -galactopyranoside (X-Gal). White plaques were isolated, and viral isolates containing the two mutated sites were screened with the PCR amplification refractory mutation system (ARMS) technique using the following primers for the codon 950 site: WT (R950) primer (5=-CGTGTTTCATCATGCTCTAGC-3=), H950 mutant primer (5=-CGTGTTTCATCATGCTCTAGT-3=), and common primer (5=-TGCACACGGCCTGCCTGAA GCT-3=). Primers for the codon 393 site were WT (L393) primer (5=-CTGGACGTTCCTCCGGTACT-3=), P393 mutant primer (5=-CTGGACGTTCCTCCGGTACC-3=), and common primer (5=-TGGAAGACGGACTCCATGTA G-3=). Candidates were confirmed by DNA sequencing. Correct insertion of the gene into the viral genome was confirmed by XhoI digestion followed by Southern blotting (Mostafa and Davido, unpub-lished). The KOS-NAR virus was generated by cotransfection of 1g of KOS-NA viral DNA and 2.5g of pKHF plasmid (18), which contains WT UL39, digested with EcoRI and XbaI. Plaques were picked randomly and screened with the PCR ARMS technique as described above. Confirmation of the mutation rescue was performed by PCR, followed by sequencing and Southern blot analyses using the restriction enzyme BamHI (Mostafa and Davido, unpublished). The HrR3 R (rescue) virus was constructed by cotransfecting Vero cells with 1g of HrR3 viral DNA and 2.5g of pKHF plasmid digested with EcoRI and XbaI. Rescuants were identified by blue/white selection and confirmed by Southern blotting by digesting the viral DNA with XhoI (Mostafa and Davido, unpublished).
Ocular infection of mice.CD-1 outbred female mice (6 to 7 weeks old) were obtained from Charles River Laboratories (Shrewsbury, MA), cared for according toGuide for the Care and Use of Laboratory Animals(57), and infected as previously described (7). Mice were housed according to institutional and federal guidelines and were used in a protocol approved by the University of Kansas. Briefly, mice were anesthetized by intraperitoneal injection of ketamine (75 to 100 mg/kg of body weight) and xylazine (10 mg/kg of body weight). Corneas were scarified with a 26-gauge needle and were infected with KOS, 7134, KOS-NA, KOS-NAR, UL39mut, or HrR3 R at 2⫻105PFU of virus per eye in 3 to 5l of medium.
Determination of viral titers in eyes and TG.Four hours and 1, 3, 5, 7, and/or 9 days postinfection, eye swabs and/or TG samples were collected. For eye swabs, tear film was collected by swabbing the eye with cotton-tipped swabs and placed in microcentrifuge tubes containing 500 l of 5% FBS-supplemented growth medium. For TG samples, the mice were euthanized by CO2asphyxiation, and the
TG were removed and placed in microcentrifuge tubes with 500l of growth medium and 100l of 1-mm beads. These samples were homogenized using a Mini-Beadbeater 8 (BioSpec, Bartlesville, OK). In all cases, titers of the wild-type, UL39mut, and HrR3 R viruses were determined on Vero cells, and titers
of KOS-NA and KOS-NAR viruses were determined on L7 cells. Statistical analyses were performed using Student’sttest.
Latent viral genome loads in TG.At 28 to 30 days postinfection, latently infected TG were collected, and DNA was isolated from each TG as previously reported (26). PCR primers for the HSV-1 UL50 gene and the mouse adipsin gene were used to amplify viral DNA and as a loading control for cellular DNA, respectively (7). Real-time PCRs were performed in a total volume of 25l containing FastStart SYBR green Master (Rox) (Roche, Indianapolis, IN) and primers (300 nM) in an ABI Prism 7500 real-time PCR system (Applied Biosystems, Foster City, CA). UL50 PCR samples contained 125 ng of DNA per reaction
on November 6, 2019 by guest
http://jvi.asm.org/
mixture, adipsin PCR samples contained 10 ng of DNA per reaction mixture, and all samples were analyzed in duplicate or triplicate. Standard curves for each PCR condition were determined as described before (7) to quantify the amount of viral DNA present in each sample relative to the adipsin gene using the threshold cycle (2⫺ΔΔCT) method. Statistical analyses were performed using the one-way ANOVA test. Viral explant-induced reactivation studies.At days 28 to 30 postinfection, latently infected mice were euthanized and each TG was collected, cut into 8 pieces, and cultured on Vero cells in a well of a 24-well plate containing 1.5 ml of growth medium. The cultures were sampled daily for up to 16 days for the presence of infectious virus by cytopathic effect on Vero cells for KOS-infected samples and on L7 cells for KOS-NA- and 7134-infected samples. At day 10 postexplantation, cultures were heat shocked at 43°C for 3 h as an additional stimulus for reactivation. Statistical analyses were performed using Fisher’s exact test.
Western blotting.To examine ICP6 and UL40 protein levels, 1⫻105Vero cells were plated per well
of a 12-well plate. Twenty four hours postplating, cells were infected at an MOI of 2 for each virus. Samples were harvested 24 h postinfection in 50l of 1⫻Laemmli buffer (100°C) supplemented with 1⫻ protease inhibitors (leupeptin at 1g/ml, aprotinin at 1g/ml, and phenylmethylsulfonyl fluoride [PMSF] at 1 mM). Samples were heated at 95°C for 5 min, vortexed, centrifuged, loaded onto a 4 to 12% gradient gel (Invitrogen), and run at 120 V for 1 h. Proteins were transferred to nitrocellulose membranes using a semidry transfer unit (GE Healthcare; catalog no. TE77). Each membrane was blocked in 5% bovine serum albumin in Tris-buffered saline (TBS) and 0.1% Tween 20 (TBS-T) for 1 h at room temperature. Primary antibodies were incubated with membranes overnight at 4°C, then the membranes were washed 3 times in TBS-T, and secondary antibodies were added at room temperature for 1 h. Membranes were washed 3 times in TBS-T and developed using SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Pictures were captured with a Kodak 4000R image station.
Immunoprecipitations.Vero cells were plated in 60-mm dishes at 5⫻105per plate. Twenty four
hours later, cells were mock infected or infected with KOS at an MOI of 2 or KOS-NA at MOI of 5 for 24 h. Cells were harvested in 100l of a buffer containing 100 mM Tris-HCl (pH 8), 50 mM NaCl, 10% glycerol, 20 mM-mercaptoethanol, and 1% Nonidet P-40 with protease inhibitors as described above. Samples were sonicated at 100 W for 30 s, incubated on ice for 30 min, and centrifuged at 15,000 rpm for 10 min at 4°C. After aspiration of the buffer, 50l of protein G Dynabeads (Invitrogen) was incubated with 200
l of phosphate-buffered saline (PBS) with 0.05% Tween 20 and 5l of an anti-UL40 mouse monoclonal antibody (kindly provided by Roger Everett), by rotating for 1 h at room temperature. Beads bound to the antibody were washed with PBS and 0.05% Tween 20 and incubated with the sample lysates, with rocking at 4°C overnight. The beads were then washed twice with PBS-Tween and transferred to new tubes. Fifty microliters of 1⫻Laemmli buffer (100°C) with protease inhibitors was added to each sample, and samples were boiled for 5 min, vortexed, boiled again, and vortexed twice for 1 min each. Western blot analyses were performed as described in the previous section.
Cell viability assay.Vero cells were plated in a 24-well glass plate at 5⫻104per well. Twenty-four
hours later, cells were transfected with 1.25g of a plasmid that expresses either CMV-DsRed (gift from Yoshi Azuma) or human caspase 8 (pcDNA3-Casp8) (Addgene; plasmid 11817) (58). Transfections were performed using Opti-MEM (Invitrogen) and Fugene HD (Promega). Five hours after transfection, medium was removed and Vero cell medium containing acyclovir (ACV; 44g/ml) was added to inhibit viral DNA replication. Six hours after transfection, cells were infected with each virus at an MOI of 2.5 for 1 h in the presence of ACV and washed three times with PBS plus ACV; Vero cell medium containing ACV was then added to the cells. Twenty-four hours after transfection, cells were stained with Hoechst 33342 dye (Invitrogen) by adding 0.5l of 10-mg/ml aqueous dye stock solution to 0.5 ml of phenol red-free DMEM (Thermo Scientific) in each well. Apoptotic cells were identified as having condensed chromatin/ fragmented nuclei compared to the total number of cells per field of vision by fluorescence microscopy. Three independent sets of images were taken for each sample. Statistical analyses were performed using Student’s t test (GraphPad Prism) for virally infected samples compared to caspase 8-transfected, KOS-infected cells.
ACKNOWLEDGMENTS
This work was supported by University of Kansas (D.J.D.) and National Institutes of
Health (NIH) grants R21EY019739 (L.A.M. and D.J.D.) and R01OD010974 (S.J.M.). The
Genome Technology Access Center is partially supported by NCI Cancer Center Support
grant no. P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA grant no.
UL1RR024992 from the National Center for Research Resources (NCRR), a component of
the NIH, and by the NIH Roadmap for Medical Research.
D.J.D. and L.A.M. are coinventors on U.S. patent 9616119, which describes using
KOS-NA as a vaccine against HSV-1 infections.
We thank Sandra Weller, Howard Marsden, and Roger Everett for providing reagents
related to this study.
The content of this article is solely the responsibility of the authors and does not
necessarily represent the official views of the NIH.
on November 6, 2019 by guest
http://jvi.asm.org/
herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J Virol 67:7501–7512.
5. Harris RA, Everett RD, Zhu XX, Silverstein S, Preston CM. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J Virol 63: 3513–3515.
6. Davido DJ, von Zagorski WF, Lane WS, Schaffer PA. 2005. Phosphoryla-tion site mutaPhosphoryla-tions affect herpes simplex virus type 1 ICP0 funcPhosphoryla-tion. J Virol 79:1232–1243.https://doi.org/10.1128/JVI.79.2.1232-1243.2005. 7. Mostafa HH, Thompson TW, Kushnir AS, Haenchen SD, Bayless AM,
Hilliard JG, Link MA, Pitcher LA, Loveday E, Schaffer PA, Davido DJ. 2011. Herpes simplex virus 1 ICP0 phosphorylation site mutants are attenu-ated for viral replication and impaired for explant-induced reactivation. J Virol 85:12631–12637.https://doi.org/10.1128/JVI.05661-11.
8. Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. 2012. Genome sequence of herpes simplex virus 1 strain KOS. J Virol 86:6371– 6372.
https://doi.org/10.1128/JVI.00646-12.
9. Colgrove RC, Liu X, Griffiths A, Raja P, Deluca NA, Newman RM, Coen DM, Knipe DM. 2016. History and genomic sequence analysis of the herpes simplex virus 1 KOS and KOS1.1 sub-strains. Virology 487:215–221.
https://doi.org/10.1016/j.virol.2015.09.026.
10. Jacobson JG, Leib DA, Goldstein DJ, Bogard CL, Schaffer PA, Weller SK, Coen DM. 1989. A herpes simplex virus ribonucleotide reductase dele-tion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology 173: 276 –283.https://doi.org/10.1016/0042-6822(89)90244-4.
11. Brandt CR, Kintner RL, Pumfery AM, Visalli RJ, Grau DR. 1991. The herpes simplex virus ribonucleotide reductase is required for ocular virulence. J Gen Virol 72(Part 9):2043–2049.
12. Yamada Y, Kimura H, Morishima T, Daikoku T, Maeno K, Nishiyama Y. 1991. The pathogenicity of ribonucleotide reductase-null mutants of herpes simplex virus type 1 in mice. J Infect Dis 164:1091–1097.https:// doi.org/10.1093/infdis/164.6.1091.
13. Averett DR, Lubbers C, Elion GB, Spector T. 1983. Ribonucleotide reduc-tase induced by herpes simplex type 1 virus. Characterization of a distinct enzyme. J Biol Chem 258:9831–9838.
14. Ponce de Leon M, Eisenberg RJ, Cohen GH. 1977. Ribonucleotide reduc-tase from herpes simplex virus (types 1 and 2) infected and uninfected KB cells: properties of the partially purified enzymes. J Gen Virol 36: 163–173.https://doi.org/10.1099/0022-1317-36-1-163.
15. Dufour F, Bertrand L, Pearson A, Grandvaux N, Langelier Y. 2011. The ribonucleotide reductase R1 subunits of herpes simplex virus 1 and 2 protect cells against poly(I·C)-induced apoptosis. J Virol 85:8689 – 8701.
https://doi.org/10.1128/JVI.00362-11.
16. Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. 2015. Herpes simplex virus suppresses necroptosis in hu-man cells. Cell Host Microbe 17:243–251.https://doi.org/10.1016/j.chom .2015.01.003.
17. Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, Zhong CQ, Xia W, Zhou R, Zheng C, Han J. 2015. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17:229 –242.https://doi.org/10.1016/j.chom.2015.01 .002.
18. Goldstein DJ, Weller SK. 1988. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J Virol 62:196 –205.
19. Goldstein DJ, Weller SK. 1988. Factor(s) present in herpes simplex virus type
591–593.
23. Cohen GH. 1972. Ribonucleotide reductase activity of synchronized KB cells infected with herpes simplex virus. J Virol 9:408 – 418.
24. Frame MC, Marsden HS, Dutia BM. 1985. The ribonucleotide reductase induced by herpes simplex virus type 1 involves minimally a complex of two polypeptides (136K and 38K). J Gen Virol 66(Part 7):1581–1587. 25. Ingemarson R, Lankinen H. 1987. The herpes simplex virus type 1
ribonucleotide reductase is a tight complex of the type alpha 2 beta 2 composed of 40K and 140K proteins, of which the latter shows multiple forms due to proteolysis. Virology 156:417– 422.https://doi.org/10.1016/ 0042-6822(87)90422-3.
26. Swain MA, Galloway DA. 1986. Herpes simplex virus specifies two sub-units of ribonucleotide reductase encoded by 3=-coterminal transcripts. J Virol 57:802– 808.
27. Nordlund P, Reichard P. 2006. Ribonucleotide reductases. Annu Rev Biochem 75:681–706. https://doi.org/10.1146/annurev.biochem.75.103004 .142443.
28. Katz JP, Bodin ET, Coen DM. 1990. Quantitative polymerase chain reac-tion analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J Virol 64:4288 – 4295. 29. Honess RW, Roizman B. 1973. Proteins specified by herpes simplex virus.
XI. Identification and relative molar rates of synthesis of structural and nonstructural herpes virus polypeptides in the infected cell. J Virol 12:1347–1365.
30. Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 14:8 –19.
31. Watson RJ, Preston CM, Clements JB. 1979. Separation and character-ization of herpes simplex virus type 1 immediate-early mRNA’s. J Virol 31:42–52.
32. Wymer JP, Chung TD, Chang YN, Hayward GS, Aurelian L. 1989. Identi-fication of immediate-early-type cis-response elements in the promoter for the ribonucleotide reductase large subunit from herpes simplex virus type 2. J Virol 63:2773–2784.
33. Conner J, Macfarlane J, Lankinen H, Marsden H. 1992. The unique N terminus of the herpes simplex virus type 1 large subunit is not required for ribonucleotide reductase activity. J Gen Virol 73(Part 1):103–112. 34. Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y.
2011. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apop-tosis by interacting with caspase-8. Apopapop-tosis 16:256 –271.https://doi .org/10.1007/s10495-010-0560-2.
35. Chabaud S, Lambert H, Sasseville AM, Lavoie H, Guilbault C, Massie B, Landry J, Langelier Y. 2003. The R1 subunit of herpes simplex virus ribonucleotide reductase has chaperone-like activity similar to Hsp27. FEBS Lett 545:213–218.https://doi.org/10.1016/S0014-5793(03)00547-7. 36. Cooper J, Conner J, Clements JB. 1995. Characterization of the novel protein kinase activity present in the R1 subunit of herpes simplex virus ribonucleotide reductase. J Virol 69:4979 – 4985.
37. Salvucci LA, Bonneau RH, Tevethia SS. 1995. Polymorphism within the herpes simplex virus (HSV) ribonucleotide reductase large subunit (ICP6) confers type specificity for recognition by HSV type 1-specific cytotoxic T lymphocytes. J Virol 69:1122–1131.
38. Conner J, Cross A, Murray J, Marsden H. 1994. Identification of structural domains within the large subunit of herpes simplex virus ribonucleotide reductase. J Gen Virol 75(Part 12):3327–3335.
39. Conner J, Furlong J, Murray J, Meighan M, Cross A, Marsden H, Clements JB. 1993. Herpes simplex virus type 1 ribonucleotide reductase large
on November 6, 2019 by guest
http://jvi.asm.org/
subunit: regions of the protein essential for subunit interaction and dimerization. Biochemistry 32:13673–13680.
40. Nikas I, McLauchlan J, Davison AJ, Taylor WR, Clements JB. 1986. Struc-tural features of ribonucleotide reductase. Proteins 1:376 –384. 41. Nikas I, Darling AJ, Lankinen HM, Cross AM, Marsden HS, Clements JB.
1990. A single amino acid substitution in the large subunit of herpes simplex virus type 1 ribonucleotide reductase which prevents subunit association. J Gen Virol 71(Part 10):2369 –2376.
42. Preston VG, Palfreyman JW, Dutia BM. 1984. Identification of a herpes simplex virus type 1 polypeptide which is a component of the virus-induced ribonucleotide reductase. J Gen Virol 65(Part 9):1457–1466. 43. Chabaud S, Sasseville AM, Elahi SM, Caron A, Dufour F, Massie B,
Langelier Y. 2007. The ribonucleotide reductase domain of the R1 sub-unit of herpes simplex virus type 2 ribonucleotide reductase is essential for R1 antiapoptotic function. J Gen Virol 88:384 –394.https://doi.org/ 10.1099/vir.0.82383-0.
44. Lembo D, Brune W. 2009. Tinkering with a viral ribonucleotide re-ductase. Trends Biochem Sci 34:25–32.https://doi.org/10.1016/j.tibs .2008.09.008.
45. Balan P, Davis-Poynter N, Bell S, Atkinson H, Browne H, Minson T. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J Gen Virol 75(Part 6):1245–1258.
46. Dingwell KS, Doering LC, Johnson DC. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J Virol 69: 7087–7098.
47. Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol 63:759 –768.
48. van Velzen M, Jing L, Osterhaus AD, Sette A, Koelle DM, Verjans GM. 2013. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently
infected human trigeminal ganglia. PLoS Pathog 9:e1003547.https://doi .org/10.1371/journal.ppat.1003547.
49. Geltz JJ, Gershburg E, Halford WP. 2015. Herpes simplex virus 2 (HSV-2) infected cell proteins are among the most dominant antigens of a live-attenuated HSV-2 vaccine. PLoS One 10:e0116091.https://doi.org/ 10.1371/journal.pone.0116091.
50. Cai W, Schaffer PA. 1991. A cellular function can enhance gene expression and plating efficiency of a mutant defective in the gene for ICP0, a trans-activating protein of herpes simplex virus type 1. J Virol 65:4078 – 4090. 51. Boutell C, Everett R, Hilliard J, Schaffer P, Orr A, Davido D. 2008. Herpes
simplex virus type 1 ICP0 phosphorylation mutants impair the E3 ubiq-uitin ligase activity of ICP0 in a cell type-dependent manner. J Virol 82:10647–10656.https://doi.org/10.1128/JVI.01063-08.
52. Mostafa HH, Thompson TW, Davido DJ. 2013. N-terminal phosphoryla-tion sites of herpes simplex virus 1 ICP0 differentially regulate its activ-ities and enhance viral replication. J Virol 87:2109 –2119.https://doi.org/ 10.1128/JVI.02588-12.
53. Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioin-formatics 11:485.https://doi.org/10.1186/1471-2105-11-485.
54. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25.https://doi.org/10.1186/gb-2009-10-3-r25. 55. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821– 829.https://doi .org/10.1101/gr.074492.107.
56. Otto TD, Dillon GP, Degrave WS, Berriman M. 2011. RATT: Rapid Anno-tation Transfer Tool. Nucleic Acids Res 39:e57.https://doi.org/10.1093/ nar/gkq1268.
57. National Reseach Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC.
58. Stennicke HR, Salvesen GS. 1997. Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem 272:25719 –25723.
on November 6, 2019 by guest
http://jvi.asm.org/