0022-538X/93/084914-09$02.00/0
Copyright©)1993, American Society forMicrobiology
RNA
Structure
and Heterologous Recombination
in
the
Double-Stranded
RNA
Bacteriophage
4)6
SHIROH
ONODERA,l
XUEYINGQIAO,1
PAULGOTTLIEB,l
JEFFREYSTRASSMAN,l
MIKKO FRILANDER,2 ANDLEONARDMINDICH'*
DepartmentofMicrobiology, ThePublic Health Research Institute, New
York,
NewYork10016,1
andDepartment ofGenetics, UniversityofHelsinki, SF-00100,
Helsinki,
Finland2Received 1February 1993/Accepted 7 May 1993
Bacteriophage
+6
has a genome ofthree segments of double-stranded RNA, designated L, M, and S. A 1.2-kbp kanamycin resistance gene was inserted into segment M but was shown to begenetically
unstable because of a high recombination rate between segment M and the3' ends of segments S and L. The high rate ofrecombinationis due to complementary homopolymer tracts boundingthekan gene. Removalof onearmof thispotential hairpin stabilizes the insertion. The insertion ofa241- or427-bp WacZ' gene into segment M leads to astableLac+ phage. The insertion of thesamegenesbounded bycomplementaryhomopolymerarmsleads torecombinationalinstability.Astable derivative of this phage wasshown tohave lostoneof thehomopolymer arms. Several other conditions foster recombination. The truncation ofa genomic segment at the 3' end preventsreplication, but such adamagedmolecule canbe rescuedbyrecombination.Similarly,
insertion of the entire 3-kb lcZ gene preventsnormalformation ofvirus, but the viral genescanbe rescuedby recombination. Itappears that conditions leading to the retardation or absence of replicationofa particulargenomic segment facilitaterecombinational rescue.Bacteriophage 4)6 infects the plant pathogen Pseudo-monasphaseolicola (P.
syringae
pv. phaseolicola) HB1OY (HB1OY) (37). It hasa genome of three different pieces of double-stranded RNA(dsRNA) packaged within a polyhe-dral nucleocapsid (NC) (34). The NC contains a procapsid that has RNApolymerase activity (30).We havedevelopedaninvitrogenomic packagingsystem that utilizes single-stranded RNA (ssRNA) obtained from thein vitrotranscriptionofviral NCsorRNAtranscriptsof plasmids carrying cDNA copies of the viral genomic seg-ments. The RNA is packaged by procapsids isolated from
Escherichia coli cultures that are expressing the cloned genes coding for the four proteins of the
4)6
procapsids.Thesefilled particles are capable ofinfecting spheroplasts.
We have previously demonstrated that a transcriptof seg-ment Mthatcontainsaninsertion of the gene forkanamycin
resistance can be incorporated into the genome of viable phage (29). Virus that contains the kanamycin gene is genetically unstable, and virus preparations contain many particles that have lost part of themodifiedMsegment(29).
This losswasshown to be the result ofheterologous recom-bination between the genomic segments, resulting in the
replacementof the 3' end of segment Mwith that of either segment S or segment L (25). In the present report, we describeaninvestigationofsomeof the factors that promote heterologous recombination in
46.
Heterologous recombina-tion has been found in several ssRNAviruses, some with unit genomes and some with multipartite genomes(18, 31), mostly with positive-strand viruses but with negative-strand viruses aswell (4). This type of recombination appears to constitute a mechanism for the rescue of segments with damaged 3' ends. It also is a means of enlarging the genome of the virus.Several viruses isolated in nature appear to have been formed by heterologous recombination.*Correspondingauthor.
MATERIALSANDMETHODS
Bacterial strains, phage, and plasmids. P. phaseolicola
HB1OY (HB1OY) (37) was used as the host for 4)6 lysate preparationsand forphagetitration(Table 1).E. coli JM109
(39)wasused for the propagation of recombinantplasmids.
Construction of plasmids. Plasmids derived from pT3T7
191J
(Table 2) were used to prepare templates for theproductionof ssRNApackagingsubstratesfor the formation of 4)6 virus containing modified genomic segments (27).
PlasmidpLM672was theoriginalconstruction that had the kan gene ofpUC4KinacDNA copyofgenomicsegmentM
(29). Thekan gene is boundedbyaPstIsiteoneach sideas wellas 12 G'son the 5' side and 12 C'son the 3' side. This cartridgewasinserted into thePstIsite in the 3' noncoding
region of segment M(Fig. 1).
PlasmidspLM778 and pLM779were prepared bypartial cutting ofplasmid pLM672with PstI andbydigestingwith exonucleaseBAL31. Theresultingdigestswereligatedand tested for kanamycinresistance in E. coliJM109. Plasmids from resistant colonies were sequenced, and pLM778 and
pLM779werefoundto have deletions of 53 and 131bases, respectively, at the 3' ends of the kan gene. pLM778 is
missing 10 bases of thekan insert, and pLM779 ismissing onlythehomopolymerarm.Inneithercase wasthe 75-base
terminal replication region (25) impaired.
Polymerase chain reaction (PCR) was used to prepare
fragmentsof theE. coli lacZ gene boundedbyEcoRVsites.
lacGwassynthesizedonthetemplateofpUC8withprimers
OLM81 and OLM82 (Table 3). It contains the multiple cloningsites(MCS)ofpUC8butis trimmedtohaveonly241 nucleotides when it is cutwith EcoRV. ThelacZfragment has 60 amino acids.lacHwassynthesized on thetemplate of
pMC9(21)withprimersOLM81 andOLM102(Table 3).The cartridge contains 427 nucleotides when it is cut with EcoRV. ThelacHfragment has 133 amino acids.Although
bothcartridgesaremissingpartof thelac operator, theyare controlled by the lac repressor inplasmidsthat donothave anadditional lac operator.
4914
on November 9, 2019 by guest
http://jvi.asm.org/
TABLE 1. Bacterialstrains, phages, andplasmids
Bacteriumorphage Description Referenceor source
Bacteria
P.phaseolicola HB1OY Hostof+6 37
E. coli JM109 recAl end41gyrA96 thi hsdRl7 supE44 reLA1 A-A(lac-proAB) [F' J.Messing traD36proABlacIqZAm15]
Phages
4)6 Wild-typephage 37
,06K1
4)6containing kan insegment M 294)1767 Recombinant ofMwith 3' end of Sresultingfromtransfection withtran- Thisstudy script of pLM655 truncatedatPstI site
4)1789 Recombinantof M with 3' endofSresultingfromtransfectionwithtran- Thisstudy scriptofpLM780 whichcontains entirelacZin the PstI site
,01791
RecombinantofMwith 3' end of Sresultingfromtransfection withtran- This study script of pLM780whichcontainsentire lacZinthePstIsite4)1797 Recombinant ofMwith 3' end of Lresultingfrom transfectionwithtran- This study script of pLM656 truncatedatthePstIsite
41798 Recombinant of Swith 3' end of Lresultingfromtransfectionwith tran- Thisstudy script ofpLM658 truncatedattheClaIsite
+1800 TransfectantcontainingtranscriptofpLM789; lacG in M withhomopoly- Thisstudy merarms; 18G's and27C's;unstable
4)1807
Mutantof+1800that is stableandmissingsequencefrom nucleotide This study 3498 toposition -2ofthelacopen readingframe41817 Transfectantcontaining transcriptofpLM847; lacH inMwithEcoRV Thisstudy cutligatedtobluntedPstIsite; stable
+1819 Transfectantcontaining transcriptofpLM844; lacHin M withhomopoly- This study merarms; 21 G'sand14C's; unstable
PlasmidspLM763andpLM765wereprepared by inserting and tailed with poly(dG) (32). After transformation of the EcoRV-cutPCRproductof lacG into either theClaI site JM109, Lac+ colonies were picked, and plasmids were ofpLM759, which contains the cDNA copy of segment S screened by sequencing for homopolymer arms ofvarious (see Fig. 1) and is missing thevector ClaI site, or into the lengths. PlasmidpLM789 has lacG in its PstIsitebounded PstI site of pLM656, which contains the cDNA copy of by 18G's atthe5' side and 27 C's atthe 3' side.
segmentM. In both cases, the sites were bluntedby treat- The derivatives of4o6containing lacG formedfluorescent mentwith T4 DNApolymerase (27). plaqueswhen platedon alawn of HB1OY carrying plasmid PlasmidpLM780was constructed by insertingthe 3-kbp pLM746 (LM1034) ina mediumcontaining
4-methylumbel-PstIcartridgeoflacZfrompMC1871(7)into the PstIsite of
liferyl-o-D-galactoside
(MUG). Phages carrying lacH pro-pLM765 andselectingfora Lac+ frameshift mutation. ducedplaqueswith about 10 timesthefluorescence of those Plasmid pLM789 was constructed by terminal deoxynu- carrying lacG and also resulted in blue plaques on platescleotidyltransferase tailingoflacGwithpoly(dC)and anneal- containing X-Gal
(5-bromo-4-chloro-3-indolyl-o-D-galacto-ingthismaterialtoplasmid pLM656whichwas cutwithPstI pyranoside; Fig. 2). Plasmid pLM746 is a derivative of
TABLE 2. Plasmids
Plasmid Description Referenceor source
pGHS1 Lactococcusplasmid containingE. coli lacZAm15 15
pT7T319U ampPT7 PT3lacZ' Pharmacia
pUC-4K amplacZ' kancartridge inPstI site 38
pMC1871 CartridgewithmostoflacZ 7
pLM254 amplacZ'withpUC8 MCS;vectorforpseudomonads 23
pLM655 ampPT7;exactcopyofsegment M inpT7T3; 19U's 29
pLM656 ampPT,;exactcopy ofsegmentM inpT7T3; 19U's 29
pLM658 ampPT,;exactcopy of segmentS 12
pLM672 ampP17; kangene in PstIsite ofpLM656 29
pLM746 InsertoflacflinpLM254; forlaccomplementation Thisstudy
pLM759 amp
P17;
exactcopy of segment S; vector lacksClaIsite ThisstudypLM763 ampPT,;lacG inClaI site ofS;nohomopolymerarms Thisstudy
pLM765 amp
Pm7;
lacGinPstI site ofM;nohomopolymerarms ThisstudypLM778 pLM672 with53-basedeletion of 3'homopolymerarm Thisstudy
pLM779 pLM672with 131-base deletion of 3'homopolymerarm Thisstudy
pLM780 ampPT7;complete lacZgene in PstI site of M Thisstudy
pLM789 ampPT7;lacG inPstIsite of M with 18 G'son5' side and 27 C'son3' side Thisstudy pLM844 amp
PT,;
lacHin PstIsite of M with 21 G's on5' side and 14 C'son3' side ThisstudypLM847 amp
PT7;
lacHinPstIsiteofM;nohomopolymerarms Thisstudyon November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.76.561.529.720.2]TABLE 3. Oligonucleotides for cloning and sequencing
Oligonucleotidename Sequence Description
OLM60 GATCTGCTGTCTCAACAGATC Forward from 2847 in M (before EcoRV)
OLM62 CCCTCTAGAGAGAGAGCCCCCGAAG Complementary to 3' termini withXbaIsite
OLM68 GCGAACTACGAAGGTAGAACG Forward from 2400 in S
OLM70 GAAAGCCCGGCATCGATGTCGTTTGCA Ssequence from 2673 to 2694
OLM71 AACGACATCGATGCCGGGCTTTCTGCA Complement ofOLM70
OLM81 GAGCGGATATCAATTTCACACAGG PCRprimer for making lacZ' constructions; contains anEcoRV site and starts in lac operator
OLM82 CCCGATATCAGGCGCCATTCGCCATTCAGG PCRprimer for3' end oflacG; containsanEcoRV site and ends at same place as pUC8 lacZ' OLM102 CCCGATATCAGCTTTCATCAACATTAAATGTGAGCGAG PCRprimerfor 3' end oflacH;contains an EcoRV
site andends at 133 amino acids oflacZ
M13 GTAAAACGACGGCCAGT ForsequencingpT7T3 19 U
RevM13 CAGGAAACAGCTATGAC For sequencing pT7T3 19 U
shuttlevectorpLM254 (23),into whichwehaveinsertedpart of the lacZAmJSgeneofplasmidpGSH1 (15)andpartof the lacZgeneofplasmid pMC1871 (7).Theplasmidhas the wide
hostrange of RSF1010 (1), the lacpromoterupmutation of
pLM254 (23), and the lacZAmJSgene,with the result thatit is a general lac complementation plasmid for many
gram-negative species (38).
In vitro synthesis ofpositive-sense transcripts by T7
poly-merase.Plasmidswerecutwith endonucleaseXbaI,and the resulting 5' overhangwasremovedwithmungbeannuclease
before transcription with T7 RNA polymerase (27). Trun-catedsegmentswereproduced by cuttingtheplasmidswith
endonuclease ClaI orPstI before thetranscription reaction (Fig. 1). RNAwaspurified by extractingonce withphenol:
chloroform:isoamyl alcohol and then with chloroform: isoamyl alcohol.
Plasmid pLM931 containsa27-baseinsert in the PstI site
of the cDNA copy ofsegment M, 5' to alacH insert. The
27-base insert is identical tothe sequence around the ClaI
site near the 3' end of segment S. pLM844 contains the cDNAcopyofsegmentMwith lacH inserted in the PstI site withhomopolymer tails of21G'sonthe 5' side and 14 C'son
the 3' side. Theplasmid preparationwas partiallycutwith PstI andreligatedafterbluntingwith T4 DNApolymeraseto
removethe 3' PstI site.Theresulting plasmid, pLM927,was
cutwith PstI andligatedin thepresenceofoligonucleotides OLM70andOLM71. PlasmidpLM931wasfoundtocontain the insert in the correct orientation. The orientation was
confirmedby sequencing.
Preparation of
+6
ssRNA. 46 ssRNAwas synthesized by aninvitrotranscriptionreactionessentially accordingtothe methoddescribedbyEmorietal. (9)withNCsprepared asdescribed by Mindich et al. (23). The resulting mixture of ssRNA anddsRNAwas fractionatedon acellulose column
with elutionbufferscontaining decreasingconcentrations of ethanolaccordingtothemethoddescribedbyFranklin(11). The ssRNA eluted at 20% ethanol was concentrated by
ethanol precipitationand dissolved inwater.
Preparationofprocapsids. 4)6 procapsidswerepreparedas
describedby Gottliebetal. (13), with the expression plasmid pLM450 (14) propagated in E. coli JM109. The procapsid preparation thatwasusedcontained 400 pug of proteinperml asdeterminedbytheBradfordassay(5). Purified procapsids
weredividedinaliquots and frozenat -70°C. Aliquotswere
thawedjustprior use.
Polymerase reaction conditions. The 4)6 RNApolymerase reactions contained 50 mM Tris-Cl, pH 8.2, 3 mM MgCl2, 100 mM NH40 acetate, 20 mM NaCl, 5 mM KCl, 5 mM
dithiothreitol, 0.1 mM Na2 EDTA, 1 mM(each)ATP, GTP, CTP, and UTP, 5% polyethylene glycol 4000, and 40 mg of Macaloid per ml(10).Toeach25-,ul reactionmixture,500to 750 ng of46ssRNA and 1.4 ,ug ofprocapsidprotein were added. Inthe experiments in which cDNA-derived M seg-ment RNA was used, the 4)6 ssRNA was derived from mutantsusSO7 (3),which hasanamber mutation in gene 6 of segment M.When cDNA-derived S segmentRNA wasused,
RNAfrom sus297,which has an amber mutation in gene 12 of segment
S,
was used (22). The amount of the synthetictranscriptwasSto10 ,ug.The reactionswereroutinelyrun at 28°C for 75 min.
Assembly
of coat protein. Purified protein P8was assem-bledonto thepackaged procapsids in 31.5-pl mixturescon-taining 10
RI
of the specific polymerase reaction mixtures andastandardamountof 2.2pugofpurifiedP8(28).Thefinalcomposition of the assemblymixture chosen (includingthe components carriedoverfrom thepolymerase reaction and from the P8preparation)was15.9mMTris-Cl(pH 8.2),0.95 mMMgCl2,0.21mMNa2EDTA, 6.3mMNaCl,97 mMKCl,
95 mM NH4Cl, 1.6 mM dithiothreitol, 7.0 mM potassium
phosphate (pH 7.4), 31.7mMNH4acetate, 0.32mM (each)
of ATP, GTP, CTP, and UTP, 1.6% polyethylene glycol
4000, and 0.71 mM CaCl2. The reaction mixture was incu-bated for 1 h at24°C.
TheNC infection. HB1OY cellswererendered competent for
4)6
NC infection as described previously(26). To each120-pul
aliquotofcells, 20 ,ul of eachcoat protein assemblymixture wasadded, and the infectionwasallowedtoproceed in
35-pul
dropletson Millipore VSWPO2500filterson Luria-Bertani(LB)plates overlaid withLBtopagar-3%lactose-20 mMpotassiumphosphate, pH 7.2,at room temperaturefor 60min.Thereafter,the cellswerewashedoncewith1mlof theLKSB buffer(26)andresuspendedin 100pul
of thesame buffer, and infectious center titers were determined on HB1OYlawns.Control transfections with ssRNAfrom wild-type4)6
gave several thousand plaques, RNA from amber mutantsgavefewor noplaques, and mixtures ofRNAfrom amber mutants and T7 polymerase transcripts of cDNA resultedin fewtoseveral hundredplaques,dependingonthe construction andthepreparation.Isolation of recombinational derivatives. Bacteriophage
4)6K1,
which contains the kan gene in segment M, was plated on a lawnofHB1OY. Mostplaques were small and turbid. These contained thekan insert. Large clear plaques werefoundtocontain recombinants. High-titer stockswere prepared by using HB1OYandthesloppy agar method(35).Thesepreparationswereextracted withphenol andanalyzed
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.66.550.83.222.2]1 2 3 4 5 6
C/_
>)I>
CO CO Cl)ILU: a.LCO CDOCEL C, C XL I
I SL5 ANK 51d~~~~~~~~
gene 2 gene 4
= - co >
0.c>,.'CQ, cu 8 = Q.>3a8E Q.>
~~~~~~~~~~~~~~~~~~~cu
> 8 ,>=,Co,LIXCDLQ cnW < Co aLW LX a.
segment
M I]I_
-K
I
i
gene 10 gene6 gene 3 gene13 HE Er{- - _- -_
= _
00coa > > > >0c >00~
m ci w cmLCL0 cn o-ci cj 5
\'
IIY
'flI
1
gene 12 gene5/11
gene8 gene9
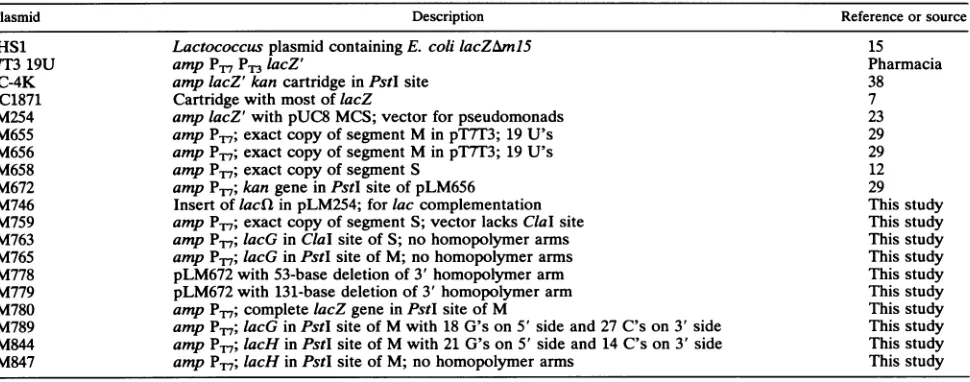
FIG. 1. Restrictionmapsof the cDNAcopiesof the threegenomicsegmentsof+6.ThePstI site in M and theClaIsite in S thatwereused
forgeneinsertionsareshown in boldface type.
for the size ofsegmentMbyagarosegel electrophoresis(29).
Virtually all such isolates showed unique migrationpatterns forsegmentM.Severalpreparations ofinterestwereusedto
prepare large-volume lysates (8), which were used for the preparation of ssRNA.
Phagescarrying the kan insert with only one homopoly-mer armformedplaquesof normal size. Plaquesof all sizes werepicked, and phage stockswereprepared asdescribed
above.Allcontainedkan inserts.
Phages carrying lac inserts were screened by plating on
LM1034in the presence of MUG (50 ,ug/ml) orX-Gal (200
ug/ml).
Nonfluorescent orwhite plaques were picked, andstockswereprepared.
PCRamplification of cDNA. Twomethodswere used for PCRamplification. In the first,in vitrotranscriptsofphages
were incubated with oligonucleotide primer OLM62(Table 3) and avianmyeloblastosisvirusreversetranscriptase(24).
OLM62iscomplementarytothe 3'end of all of thegenomic segmentsand containsatail with anXbaI site. The product
was extracted with phenol, precipitated with ethanol, and
dissolved in DNA buffer. This DNA was then used as
template for the PCR with oligonucleotide primer OLM60 (Table 3) along withOLM62. The amplifiedDNAwasthen
cut at the XbaI site and atEcoRV, which is centered at nucleotides 2978 to 2979 in gene 3 of segment M (Fig. 1). ThisDNAwasthencloned intoplasmid pT7T3 19U (Phar-macia) that had beencutwith XbaIandSmaI. Phage 41798 transcriptswereamplifiedwithOLM62andOLM68,andthe
productwascloned intopT7T3 19UwhichwascutwithSall andXbaI.
In the second method, a small preparation of phage
containing about 10"l to 1012 PFU was extracted with
phenol, the RNAwas precipitated with ethanol, dissolved,
and electrophoresed in 1% agarose, and the bands were
electroeluted, precipitated, and dissolved in water. The RNAwas then used as template for cDNA synthesis with
either avianmyeloblastosis virusreversetranscriptaseorthe
Pharmacia kit with cloned murine leukemia virus reverse
transcriptase. The resulting DNA was cut and ligated to pT7T3 19U.
DNAsequenceanalysis. Sequence analysiswasperformed
by the dideoxy chain termination method (33) with bacterio-phage T7 DNA polymerase (Sequenase; U.S. Biochemi-cals). Thesynthetic deoxyoligonucleotides used asprimers
were the standard M13 sequencing primer and the reverse
M13sequencing primer (Table 3). In allcases,the sizeof the recombinationproductascalculated for the sequenceatthe
crossoversite agreed with the size of the genomicsegment
asdetermined by gelelectrophoresis.
RESULTS
Stability of kan insegmentM. Wehave prepareda
deriv-ative ofbacteriophage 46,designated 46K1, in whichagene
for resistancetokanamycinhas been insertednearthe3' end ofgenomicsegmentM (Fig. 1)(29). Thiswasaccomplished
by inserting a kanamycin resistance gene cartridge of
pUC-4K(Table 2)into the PstI site ofacDNAcopyofthe genomic segmentthat had been cloned intoa T7 promoter vectorcalledpT7T3 19U (Table 2). The cartridge is 1.2 kbp and isboundedby 12G's and aPstI site onthe5' side and
12 C's and a PstI site on the 3' side. A transcript of the scale (kbp)
-i >
Ecom
c cnC)W cgene 7
U
gene1
segment L 6374 bp
4061 bp
segment S 2948 bp
on November 9, 2019 by guest
http://jvi.asm.org/
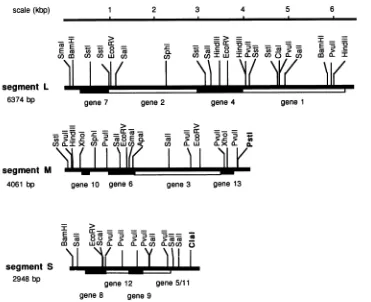
[image:4.612.123.494.63.365.2]FIG. 2. Plaques of
01817,
which has lacH stably inserted into the PstI site of segment M, on LM1034 in LB agar with X-Gal (A); plaques ofo01819,
which isgenetically unstable duetohomopolymerarms attheposition of the lacH insertion (B).resulting plasmid, pLM672,was packaged along with puri-fied ssRNA segments s and 1 into procapsids which were
then coated with protein P8andintroduced into spheroplasts
ofthe host bacterium. The resulting bacteriophage,
46K1,
formed small turbid plaques. Large clear plaques were
observed at a frequency of 0.1 to 10%. Whereas
46K1
containednormal-size segments S and Landa larger-than-normalsegmentM, the clear plaque formshad M segments withsizesvaryingfrom a few hundredbase pairslarger than
normal to several hundredbasepairs smaller than normal. These M segments were shown to be the products of heterologous recombination in which the 3' end ofM was
replacedby the 3' end ofsegmentSorL(25). Although the plaques formed by thephages containingthekan insertwere
smaller than normal, thephage yieldwas at normal levels, indicatingthatthe high proportionofrecombinantswas not
dueto the lowproductivity of the insert-bearing phage.
To test thepossibility that theinstabilityof thekan gene was due to the
homopolymer
arms that bounded it, weprepared plasmids inwhich one ofthe homopolymer arms was removed bytreatmentwith exonuclease BAL 31. Two
plasmids (pLM778 and pLM779) were constructed. They
haddeletions atthe 3' endsofthekangene that includeda
few bases in the kan insert and either 34 or 123 bases in segment M.Transcriptswereprepared,packaged, andused to transfect
HB10Y.
Plaques wereisolated,
phage were prepared, and RNA was analyzedandshown to contain thekangene.Whereastranscripts of theparentplasmid pLM672 resulted in genetically unstable phage, both of these plas-midsresultedinphagethat werecompletelystable. Whereas
46K1
producedmainly
small turbid plaques that containedthekan insert insegment M andlarge clear plaquesthat had lost the insert, theplaques formed byphages thathad lost one homopolymer arm were larger and clearer than the
406K1
plaques,andphagein all of theplaques, whether theywerelargeorsmall, containedthekan insert.
Stability
of lac in segment M. We constructed a Laccomplementation system for pseudomonadsby
preparing
a wide host range plasmid containing the w fragment of thelacZgene. Thisplasmidwas designated pLM746
(Table
2).Wethenpreparedplasmidswith cDNAcopiesofsegment M
containing inserts of the a portion of the lacZgene. One
insert,which is derived from the pUC8 (38) MCS, is
desig-nated lacG. Itis 241baseslong. Anotherinsert, designated lacH, is 427 bases long. Transcripts from these plasmids
were used in transfection experiments to produce 46
con-tainingpartsof lacZ.PhagewithlacHproducedblueplaques
onHB1OYcontaining pLM746 in thepresenceofX-Gal(Fig. 2A). Phage with lacG formed plaques thatwerefluorescent with MUG but were not blue with X-Gal. The level of
P-galactosidase
activityinHB10Yisdependentonthelength of the afragment. lacG has 60 amino acids, while lacH has133amino acids.
Phages with either of the twolac insertsweregenetically
stable. White or nonfluorescent plaques appeared with a
frequency of about 0.1%. These plaques were found to containphages withmutations to Lac-rather than deletions of the lacinsert. Plasmidswerethenprepared with inserts of lacG bounded by arms of poly(G) and poly(C). Phage producedby these transcriptswere foundtobegenetically unstable.Theinstability increasedaccordingtothelength of
thehomopolymerarms.Phage derivedfromthetranscript of pLM789, which contained lacG bounded by 18 G's and27 C's in addition to the PstI
sites,
were so unstable that all fluorescent plaques containedmixedpopulations and could not be purified because progeny fluorescent plaquescon-tainedover50% recombinants.Asimilar resultwasobtained
with phages containing thelacH insert (Fig. 2B), in which
homopolymerarmsof21G's and14C'sresultedinthe same
degreeofinstability.Ingeneral, thelargeLac-plaqueshad
lostthe lac insert, and analysis ofthe RNAbygel
electro-phoresisshowedthatthesizeof segment M wasaltered
(Fig.
3).Inallcases, the alteration was due torecombination.The
phages that contained inserts bounded by homopolymer
armswereabletoreproducewellenoughtoyield stocksthat had normal titers. Thehighproportionofrecombinantswas notduetotherelativeefficiencies of
production
between the insert-bearing strains and the recombinants, although the recombinants would eventually overwhelm the parental phages if stocks were propagated without plaquepurifica-tion.
Occasionally,
alarge
Lac' plaque
formedby
theunstableon November 9, 2019 by guest
http://jvi.asm.org/
L -1 M
S-mI
4- L
4- S
a b c d e f g h i
j
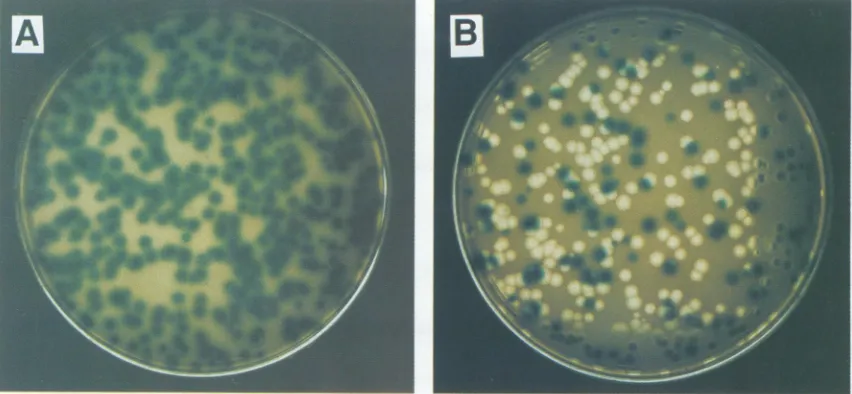
FIG. 3. Agarose gel electrophoresis of dsRNA extracted from
phage samples derived from phage ji1800 that contains lacG
bounded by homopolymer arms. RNAs in lanes a andj are from
normal
46;
lane b, d, and f RNAs derive from Lac' plaques,whereas lanec,e,g,h,andi RNAs derive from Lac-plaques.Note
that the Lac' phage have M segments with the same size (4,340
kbp), while the Msegmentsof the recombinantsvaryin size.
phagewasfoundtobe stable.One of these, 4i1807,wasused
astemplate for cDNAsynthesis, PCR, cloning,and
sequenc-ing. Itwas found that a deletion had been formed at the 5' end of the lac insert that hadremovedoneof the
homopoly-mer arms. In this case, therewasno evidence of
heterolo-gous recombination but rather an internal deletion from
position 3501 in segment M (which is after the Shine-Dalgarno sequence forgene 13) and the -2 position of the
lacG open reading frame. This results in an intact open
reading frame with an adequately placed Shine-Dalgarno
sequence.
Therefore, it appeared that the homopolymer arms
con-tributed to heterologous recombination and that inserts of 241, 427, and 1,200baseswithout homopolymer arms were
stable in the PstI site of segment M. We then prepared transcriptscontaining the entire lacZgenein thesame site.
Transfection experiments yielded very few plaques. The
yield was reduced by about 100-fold relative to that for normalsegmentM.The plaquesthatwereobtainedwerenot
Lac'onHB10Y, butsomewereLac'on LM1034 (HB1OY
withpLM746).RNAprepared from the phages showedthat theywere recombinants with sizes ofsegment M ranging fromlessthannormaltoseveral kilobaseslarger than normal (Fig. 4). Sequencingofseveral recombinants confirmed that theyweretheproducts of heterologous recombination(Fig.
L -_ M
-_w
S -0.4-- L
4- M
4- S
[image:6.612.82.272.69.178.2]a b c d e f g h
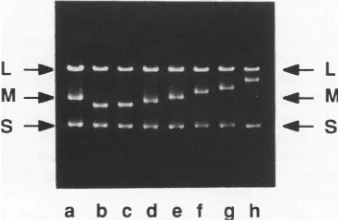
FIG. 4. Agarose gel electrophoresis of dsRNA extracted from
phage samples derived from plaquesproduced by transfectionwith RNAderived frompLM780, which contains the entire lacZgenein segmentM.RNA inlaneais fromnormal 06. RNAs in lanesb,c,
d,e,f,g,and harefromrecombinantplaques. NoneareLac'alone;
however,thephagesused for laneg wereLac'onlawns ofLM1034. Thesequenceof thecrossoverjunction is shown in Fig. 5 (1791).
5). It appears that the 3-kb size of the entire lacZ gene is too big to add to the 4 kb in segment M, but recombination does rescue a small percentage of the M segments.
The effect of 3' truncation. We have shown that the 5' end of each genomic segment carries the information for pack-aging specificity and that segments that are missing their 3' ends can be packaged but do not serve as templates for minus-strand synthesis (12). Transcripts of S copies that had been truncated at the ClaI site or M copies that had been truncated at the PstI site were prepared (Fig. 1). The transcripts of the truncated segments were packaged with normal heterologous segments and used to transfect host cells. Plaques were obtained at a small percentage of the frequency found withnormaltranscripts. Phage stocks were prepared, and RNA was analyzed by gel electrophoresis. It was found that the plaques were all recombinants. Sequenc-ing of cDNA copies of the RNA showed that they were all products of heterologous recombination. The crossover pointswere usually 5' to the truncation site, although in one case (Fig. 5)the crossover point was at the presumptive end formed by the ClaI cut of the copy of segment S.
The effect of sequence identity on recombination. Although wefind that inabout half of the cases, there are only one or two overlapping bases at the crossover point, in the other half we find six or more identical bases. This suggests that the heterologous recombination system does in fact prefer regions with similar sequences. A test was devised to see whether sequence identity would stimulate recombination. We prepared a 27-nucleotide insert that is identical to the sequence around the ClaI site of segment S (Table 3). This insert wasplaced 5' to a lacH insert bounded by homopoly-mer arms (21 G's and 14 C's) at thePstI site of segment M in pLM844. The new plasmid was called pLM931. We knew that the lac construction is extremely unstable, and we expected that recombinants whose crossover sites utilized theidentical sequences would all have a particular size. The phagecontaining lac and the duplication of theClaI site was unstable, and white plaques appeared at a frequency of more than 50% inpassaged small blue plaques. Small blue plaques were picked andreplated. Eighteen white-plaque derivatives were picked, phage stocks were prepared, and RNA was analyzed by electrophoresis. It was found that most of the recombinant phages showed segment M sizes incompatible with the sizes expected for crossovers at the identical sequence. Several preparations looked as if they might be products of such an event on the basis of probing with oligonucleotides. However, sequence analysis showed that none of the crossovers occurred in the 27-base sequence. The absence of crossovers in the 27-nucleotide identity region suggests that the initiation of the crossover is not determined by sequence matching; instead, it appears that the matchinginfluences the location of the crossover on the receptor strand.
DISCUSSION
Homologous recombination in several positive-strand RNAviruses, particularly in poliovirus (16, 17) and corona-virus (18, 19), has been demonstrated and studied. In addi-tion, theproducts ofheterologous recombination have been observed in virus isolates from the wild (see reference 18) or in experiments in which defective segments of segmented genome virus could be rescued (6). Although we have not explored the mechanism of the recombinational event, our results are consistent with the currently held copy choice model (17).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.93.262.538.648.2]Junction Segment size MtruncatedatPstl (3830)
M GCGTTCACCAAAGGCACGATCGTGATCTGCCTGGTGGTGGTCGTCCTC 1767 GCGTTCACCAAAGGCACGATCGTGATCCTGGACGTCGTCAAGGCCCCT S AGTGAACGCTCCACCGGCACCCAGATCCTGGACGTCGTCAAGGCCCCT
Mtruncatedat Pstl (3830)
M
1797 GCAACCGCTTGT'T TTGGAATCCLCGCTl"ACGTACuCCAATTTCT L GCCTACGTGTACCGCGTTGGTCGCACCGCTACGTACCCCAATTTCT
S truncatedatClal (2686)
S CATAGGCTTGGAAAGCCCGGCATCGATGTCGTTCCATGTCGCGCCA
1798 CATAGGCTTGGAAAGCCCGGCATCGCGCATCGTGGATCAGATGGCC L GTCTGCCGTCCACCTCGCGCAGTCGCGCATCGTGGATCAGATGGCC
LacZin MPstl site(3835)
LacZ TCGCTCACATTTAATGTTGATGAAAGCTGGCTACAGGAAGGCCAGA 1 791 TCGCTCACATTTATGTTGATGAAAGCTGATCGGAAGCTATGAAAG
S TTCATGCATCTCAGATTTGCGTAAAGCTGATCGGAAGCTATGAAAG
LacZinMPstl site(3835)
M~ ~~ ~
M TCACCAAAGGCACGATCGTGATCTGCCTGGTGGTGGTCGTCCTC M 1789 TCACCAAAGGCACGATCGTGATCTCGTTCCATGTCGCGCCAGACGC
S GCTTGGAAAGCCCGGCATCGATGTCGTTCCATGTCGCGCCAGACGC
5'1 3' M3465
M3569 S2686
LacZ401
M3466
S1422 L4480
L6039
S2601 S2691
4992
5464 3022 4583
3726
FIG. 5. Base sequences ofrecombination products around the crossoverregions of representative recombinants. The sequenceofthe originalsegmentthat donates the5'end isunderlined, and that of the original L or S segment that donatesthe 3' end is shown below. The extentof sequenceidentityatthe crossoverpointis indicatedby the dark horizontal lines. Phages41767 and+1797weretheproducts of transfections with truncated M segments. +1798 was the product of a truncated S segment. +1791 and +1789 were the products of transfections with the entire lacZgeneinserted into thePstI site of segment M. 41791 has a crossover in thelacZgene, andenough of the generemainssothat thisphageisLac'onw-complementingstrains.+1789has a crossoverwithinthe M sequenceandhas therefore lostthe entirelacsequence.
The experiments that we have described in the present reportshow that the frequency ofrecombinantsin a
popu-lation of
+6
can be increased more than 1,000-fold by the incorporation of a sequence bounded by homopolymer G-C arms. Using the lac screening system, we find that the normalrecombinant frequency is less than 1 in 1,000. We do notknow whether the increase in recombination is due to theformationof ahairpin or to some other consequence of the homopolymer tracts; however, it seems likely that hairpins would beformed. The hairpins could promote recombination
by providingabarrier tosynthesis of minus-strand RNA, or
they might prevent plus-strand genomic precursors from being completely packaged. The normal 3' termini of the segments contain double-stranded structures (25), but they are probably much smaller than those formed by the
ho-mopolymertracts.
Although it is difficult to precisely quantitate the relative
frequencies of transfection, the level of rescue recombina-tion thatwefindfor truncatedgenomicprecursorsis about 1 to10%of theexpectednumber ofplaqueswithintact RNA. This would imply thatrecombination is being facilitated in these cases, since we found that the frequency of recombi-nantswas less than 1per 1,000 in the absence structures that promote recombination. It is worth emphasizing that the recombinational rescue events that we report here take place
during the first round of replication of the virus. The fre-quency is not amplified by selection during successive rounds of viralreplication.
We have shown in other studies that the synthesis of minus strandsdoes not take place until all three plus strands have beenpackaged by a procapsid (12). However,synthesis ofminus strands on intact templates takes place even if the other packaged molecules are truncated and cannot repli-cate.Thisregulatory program might serve to coordinate the
replication of the genomic segments so that their numbers
remain equal; however, it may also serve to set up condi-tions that would favor recombinational rescue ofgenomic
segmentsthat arepackagedwith damaged3' ends.
Thefinding that the truncated genomic precursors can be rescuedbyrecombination implies that the recombination is taking place during the synthesis of minus-strand RNA, since in the absence of minus-strand synthesis there is no
transcription. Additionally, we have shown that the trun-catedsegments donotsupportminus-strandsynthesis(12).
If the recombination is taking place during minus-strand
synthesis,itisnotclear howthe recombinationalcomplexes
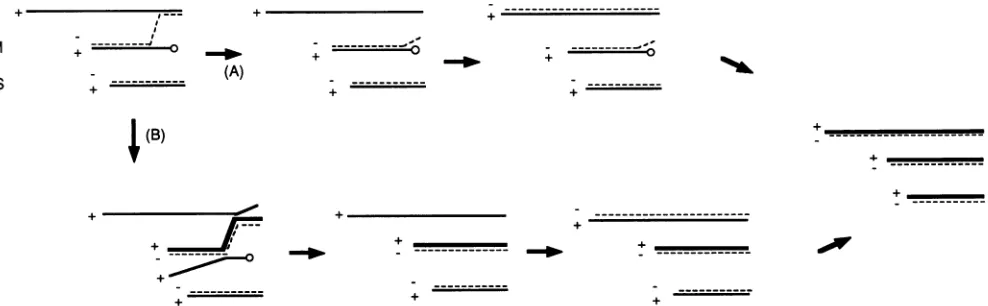
areresolved. Figure6shows therecombinant minusstrand
bindingtoboth the donor and therecipient plusstrands. Two
possibilitiesare(Fig. 6, pathway A)that thechimericminus strand leaves itsoriginal3'template spontaneouslyand(Fig.
6, pathway B) that transcription of new plus strands dis-places the chimeric minus strand from itsoriginal template,
allowing the original template to reinitiate minus-strand
synthesisthat wouldnowbe normal. There is evidence that minus-strand synthesis must occur on L before any tran-scription can take place (12). If it must becompleted, then model B would not stand, since L donates 3' ends to recombinants as well as S does. In such a case, the minus strand of L would not be complete until after resolution. However, model B would stand if minus-strandsynthesison Ldoesnothavetobecompletedbeforetranscriptionof the othersegmentscanbegin.
Many of the crossover points show limited sequence identity between the two parental strands. This region is often 6 bases or more inlength (Fig. 5).When weplaced an insert of 27 bases ofidentity 5' to the destabilizing region, we found noevidence ofpreferredcrossingoverin thisregion.
Our tentative conclusion is that the launching region is determinedbysome condition in the donor strand and that this donor strand then seeksout asimilar sequence on which
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.134.485.72.285.2]L +
I---M +
(A)
+
(B)
+_S
+
[image:8.612.63.557.70.223.2]_ o -o
FIG. 6. Models for the resolution ofrecombinational intermediates. The diagram illustratesasituation in which the plus strand ofMis
truncatedatthe3' endandcannotserveasthetemplate for minus-strand synthesis. Minus-strand synthesisoccursnormallyonS and begins on L. The nascent minus strand on L leaves its original template to copy segment M. In pathway A, the new recombinant strand spontaneously leaves its L template, and Lcan serve asthe template for its normal complement. In pathway B,transcription (boldface line) ofthe recombinantstructureleadstothedisplacement of the originaltemplate strands. The L templatewould thenbe freetoserveasthe
template for normal L minus-strand synthesis.
toland. Therewould be amuchgreaterprobability that the exploring strand wouldlandbackonitspropertemplate, but if another plus strand is available, itmightbe chosen. The
sequenceidentityisnotnecessarybut ispreferred. Since the frequency ofrecombination isverylow whenthesegments
areintact andnormal,theremustbesomething special about thecasesthatpromoterecombination that is differentfroma
situationinwhichonestrand issimply replicatingfaster than
another. It is possible that all three segments synthesize minus strands simultaneously in normal particles. Under thesecircumstances,thesystemmightbeverysensitivetoa
delay or absence of minus-strand synthesis on one of the segments.
Therearemanypossibilitiesformatchingsequencesofsix
nucleotides in two heterologous strands. If a 6-base
se-quenceattheend of the nascentchain hadtomatch witha
sequence within a region of a few hundred bases, the probabilitywould be of the order 10% that it would find a
matching site; however, ifthe matching sequence could be chosen fromsomepartof theexploringchain(not
necessar-ilythevery end ofit), theprobabilityofa matchwould be much higher. One might ordinarily think that the nascent minus strand would be tightly bonded to the template. However, thesituationmaybesimilarto thatproposedfor nascent RNA that is formed by E. coli DNA-dependent RNApolymerase, whereinitappearsthatthenewest partof the chain is not tightly associated with the template but rather isassociatedwith thepolymerase (36).
4)6is theonlydsRNA virus thathas shownintermolecular recombination. Deletions andduplications havebeen found in rotavirus (20), and themechanism ofproduction of these intramolecular rearrangements may be similar to that for heterologous recombination; however, this remains
un-demonstrated.An internal duplicationthatwasreportedfor
segment 10 of human rotavirus (2) shows a 7-base direct
repeatthatseemsreminiscentof thesequence relationships reportedinthepresentpaperand inourprevious description
of the 4)6 recombination system (25). It seems likely that onceit becomespossibletoscreenforintermolecular
recom-binants, theywill be found in the other segmenteddsRNA virus systems.
ACKNOWLEDGMENTS
This workwas supported by Public HealthService grant GM-31709 awardedtoL.Mindich. MikkoFrilanderwassupported bya
grantfrom theAcademy ofFinlandawardedtoDennisBamford. We thank David Dubnau, Alex Goldfarb, and Issar Smith for valuable discussions.
REFERENCES
1. Bagdasarian, M., R. Lurz, B. Ruckert, F. C. H. Franklin, M. M. Bagdasarian, J. Frey, and K. N. Timmis. 1981. Specific-purpose plasmid cloningvectors. II.Broad hostrange,highcopy
num-ber, RSF1010-derived vectors, and a host-vector system for genecloninginPseudomonas. Gene 16:237-247.
2. Ballard, A.,M. A.McCrae, and U. Desselberger. 1992.
Nucle-otidesequencesofnormal andrearrangedRNA segment 10 of
human rotaviruses.J. Gen. Virol.73:633-638.
3. Bamford,D.H.,M.Romantschuk,andP.J. Somerharju. 1987.
Membrane fusion inprocaryotes:bacteriophage 46membrane fuses with thePseudomonassyringaeoutermembrane.EMBO
J. 6:1467-1473.
4. Bergmann, M.,A.Garcia-Sastre, and P. Palese.1992.
Transfec-tion-mediated recombination of influenza A virus. J. Virol.
66:7576-7580.
5. Bradford, M. M. 1976. A rapid and sensitive method for the
quantitation of microgram quantities of protein utilizing the
principle ofprotein-dye binding. Anal.Biochem. 72:248-254.
6. Bujarski,J. J., and P. Kaesberg.1986. Geneticrecombination between RNAcomponentsofamultipartite plantvirus. Nature
(London)321:528-531.
7. Casadaban, M. J., A. Martinez-Arias, S. K. Shapira, and J. Chou. 1983. ,B-Galactosidase gene fusions for analyzing gene
expression in Escherichia coli and yeast. Methods Enzymol. 100:293-308.
8. Day, L. A., and L. Mindich. 1980. The molecularweight of
bacteriophage 4)6and itsnucleocapsid. Virology 103:376-385.
9. Emori, Y.,H.lba,and Y. Okada. 1983.Transcriptional
regula-tion of three double-stranded RNA segments ofbacteriophage +6in vitro. J.Virol. 46:196-203.
10. Fraenkel-Conrat, H.,B. Singer,and A.Tsugita. 1961. Purifica-tion of viral RNAbymeansofbentonite.Virology14:54-58. 11. Franklin,R. M. 1966. Purification andpropertiesof the
replica-tive intermediate of the RNAbacteriophage R17. Proc. Natl.
Acad. Sci. USA 55:1504-1511.
12. Frilander, M.,P.Gottlieb, J. Strassman,D. H.Bamford,andL.
Mindich. 1992. Dependence of minus strand synthesis upon
+
S
+~~~~~
O
+
+~~~~~~
o+
+~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
+--+
+- - l
+
on November 9, 2019 by guest
http://jvi.asm.org/
complete genomic packaging in the dsRNA bacteriophage 4)6R. J.Virol. 66:5013-5017.
13. Gottlieb, P., J. Strassman, D. H. Bamford, and L. Mindich.
1988. Production of a polyhedral particle in Eschenichia coli
froma cDNAcopyof the large genomic segmentof bacterio-phage 46. J. Virol. 62:181-187.
14. Gottlieb, P., J. Strassman, X. Qiao, A. Frucht, and L. Mindich. 1990. In vitro replication, packaging and transcription of the segmented dsRNA genomeof bacteriophage 46: studies with procapsids assembled from plasmid encoded proteins. J. Bac-teriol. 172:5774-5782.
15. Haima, P., D. van Sinderen, H. Schotting, S. Brown, and G. Venema.1990. Development ofa 3-galactosidase a-complemen-tation system for molecular cloning inBacillus subtilis. Gene 86:63-69.
16. Hirst, G. K. 1962. Genetic recombination with Newcastle
dis-ease virus, polioviruses and influenza. Cold Spring Harbor
Symp. Quant. Biol. 27:303-308.
17. Kirkegaard, K., and D. Baltimore. 1986. The mechanism of RNArecombination in poliovirus. Cell 47:433-443.
18. Lai, M. M. C. 1992. RNA recombination in animal and plant viruses. Microbiol. Rev. 56:61-79.
19. Lai, M. M. C., R. S. Baric, S. Makino,J. G.Keck, J. Egbert,
J. L. Leibowitz, and S. A. Stohlman. 1985. Recombination between nonsegmented RNA genomes of murine
coronavi-ruses. J. Virol. 56:449-456.
20. McIntyre,M., V. Rosenbaum, W. Rappold, M. Desselberger, D. Wood,andU.Desselberger. 1987. Biophysical characterization of rotavirus particles containing rearranged genomes. J. Gen. Virol.68:2961-2966.
21. Miller, J. H., J. S. Lebkowski, K. S. Greisen, and M. P. Calos. 1984. Specificity of mutations induced in transfected DNA by mammalian cells. EMBO J. 3:3117-3124.
22. Mindich, L., and J. Lehman. 1983. Characterization of +6
mutants that are temperature sensitive in the morphogenetic protein P12. Virology 127:438-445.
23. Mindich, L., G. MacKenzie, J. Strassman, T. McGraw, S. Metzger, M. Romantschuk, and D. Bamford. 1985. cDNA clon-ing of portions of the bacteriophage 4)6 genome.J. Bacteriol.
162:992-999.
24. Mindich, L., I. Nemhauser, P. Gottlieb, M. Romantschuk, J. Carton, S. Frucht, J. Strassman, D. H. Bamford, and N. Kalk-kinen. 1988. Nucleotidesequence of thelarge double-stranded RNA segment ofbacteriophage 4)6: the genes specifying the viralreplicase and transcriptase. J. Virol. 62:1180-1185. 25. Mindich, L., X. Qiao, S. Onodera, P. Gottlieb, and J. Strassman.
1992.Heterologous recombination in the double-stranded RNA bacteriophage 4)6. J. Virol. 66:2605-2610.
26. Ojala, P. M., M. Romantschuk, and D. H. Bamford. 1990. Purified4)6nucleocapsidsarecapable of productive infection of
host cells with partially disrupted outer membrane. Virology 178:364-372.
27. Olkkonen, V. M., P. Gottlieb, J. Strassman, X. Qiao, D. H. Bamford, and L. Mindich. 1990. In vitroassembly ofinfectious nucleocapsids ofbacteriophage4)6:formation ofarecombinant double-stranded RNA virus. Proc. Natl. Acad. Sci. USA 87: 9173-9177.
28. Olkkonen, V. M., P.Ojala,and D. H.Bamford. 1991. Genera-tionof infectiousnucleocapsids byinvitroassembly of the shell protein onto thepolymerase complex of the dsRNA bacterio-phage4)6.J. Mol. Biol. 218:569-581.
29. Onodera, S., V. M. Olkkonen, P. Gottlieb, J. Strassman, X. Qiao,D. H.Bamford,andL. Mindich.1992. Construction ofa transducing virus from double-strandedRNAbacteriophage 4)6: establishment of carrierstatesin host cells. J. Virol.66:190-196. 30. Partridge, J. E., J. L. Van Etten, D. E. Burbank, and A. K. Vidaver.1979. RNApolymerase activity associated with bacte-riophage4)6 nucleocapsid.J. Gen.Virol. 43:299-307.
31. Rott,M. E., J. H. Tremaine, and D. M. Rochon. 1991. Compar-isonof the 5'and3'termini oftomatoringspot virusRNA1and RNA2: evidence for RNA recombination. Virology 185:468-472.
32. Roychoudhury,R., E. Jay, and R. Wu.1976. Terminallabeling and addition ofhomopolymertractstoduplexDNAfragments by terminal deoxynucleotidyl transferase. Nucleic AcidsRes. 3:101-116.
33. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminatsequenc-ing inhibitors. Proc. Natl. Acad. Sci. USA74:5463-5467.
34. Semancik, J. S., A. K. Vidaver, and J. L. Van Etten. 1973. Characterizationofasegmented double-helicalRNAfrom bac-teriophage+6.J.Mol.Biol. 78:617-625.
35. Sinclair,J. F., J.Cohen,and L. Mindich.1976. The isolation of suppressible nonsense mutants ofbacteriophage +6. Virology 75:198-208.
36. Surratt, C. K., S. C. Milan, and M. J. Chamberlin. 1991. Spontaneouscleavage ofRNAin ternarycomplexes of Esche-richiacoliRNApolymerase and itssignificance for the mecha-nism of transcription. Proc. Natl. Acad. Sci. USA 88:7983-7987.
37. Vidaver, A. K., R. K. Koski, and J. L. Van Etten. 1973. Bacteriophage +6: a lipid-containing virus ofPseudomonas phaseolicola.J. Virol. 11:799-805.
38. Vieira, J., and J. Messing. 1982. The pUC plasmids, an
M13mp7-derivedsystemfor insertionmutagenesis and sequenc-ingwithsynthetic universal primers. Gene19:269-276. 39. Yanisch-Perron, C.,J. Vieira, and J. Messing. 1985. Improved
M13 phage cloningvectors and host strains: nucleotide se-quencesoftheM13mpl8 and pUC19vectors.Gene 33:103-119.