0022-538X/06/$08.00
⫹
0
doi:10.1128/JVI.00322-06
Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Hypersusceptibility to Substrate Analogs Conferred by Mutations in
Human Immunodeficiency Virus Type 1 Reverse Transcriptase
Robert A. Smith,* Donovan J. Anderson, and Bradley D. Preston
Department of Pathology, University of Washington, Seattle, Washington 98195
Received 14 February 2006/Accepted 29 April 2006
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) contains four structural motifs
(A, B, C, and D) that are conserved in polymerases from diverse organisms. Motif B interacts with the incoming
nucleotide, the template strand, and key active-site residues from other motifs, suggesting that motif B is an
important determinant of substrate specificity. To examine the functional role of this region, we performed
“random scanning mutagenesis” of 11 motif B residues and screened replication-competent mutants for
altered substrate analog sensitivity in culture. Single amino acid replacements throughout the targeted region
conferred resistance to lamivudine and/or hypersusceptibility to zidovudine (AZT). Substitutions at residue
Q151 increased the sensitivity of HIV-1 to multiple nucleoside analogs, and a subset of these Q151 variants was
also hypersusceptible to the pyrophosphate analog phosphonoformic acid (PFA). Other AZT-hypersusceptible
mutants were resistant to PFA and are therefore phenotypically similar to PFA-resistant variants selected in
vitro and in infected patients. Collectively, these data show that specific amino acid replacements in motif B
confer broad-spectrum hypersusceptibility to substrate analog inhibitors. Our results suggest that motif B
influences RT-deoxynucleoside triphosphate interactions at multiple steps in the catalytic cycle of
polymerization.
Conversion of viral RNA to double-stranded DNA by
re-verse transcriptase (RT) is a defining step in the retroviral life
cycle (8) and a key target of therapy for human
immunodefi-ciency virus type 1 (HIV-1) infection (17). The 66-kilodalton
subunit of HIV-1 RT contains four motifs (A, B, C, and D)
that are similarly arranged in all known structures of
replica-tive DNA and RNA polymerases (21). Three additional
struc-tural elements, motifs E and F and premotif A, are also
conserved among RTs and viral RNA-dependent RNA
poly-merases (4, 21, 45, 76). Together, motifs A, B, C, and F and
premotif A form a closely packed protein framework that
po-sitions the templating nucleotide, the primer terminus, and
incoming deoxynucleoside triphosphate (dNTP) at the RT
ac-tive site (Fig. 1A). Amino acid substitutions within this
con-served core can affect dNTP insertion fidelity, susceptibility to
nucleoside analogs, and/or discrimination against
ribonucleo-side triphosphates (rNTPs) during DNA synthesis (40, 49, 64,
72). Thus, these motifs influence the stringency and specificity
of substrate incorporation by RT.
Motif B is of particular interest because of its central
posi-tion in the RT core structure (Fig. 1) (12, 24). Motif B contacts
the template strand, the incoming dNTP, and each of the other
motifs in the core structure (premotif A and motifs A, C, and
F) (Fig. 1A), including residues within these other motifs that
are known to affect dNTP substrate recognition (Fig. 1B) (12,
24). The importance of motif B in substrate selection is evident
from studies of HIV-1 mutants resistant to nucleoside analogs
(49, 72). Two amino acid substitutions in motif B are
associ-ated with resistance to chain-terminating inhibitors: Q151M
and P157S (Fig. 1B). The Q151M replacement confers
low-level resistance to 3
⬘
-azido-3
⬘
-deoxythymidine (zidovudine
[AZT]), 2
⬘
,3
⬘
-dideoxyinosine (didanosine [ddI]), and 2
⬘
,3
⬘
-di-dehydro-3
⬘
-deoxythymidine (stavudine [d4T]) (25, 36). The
ad-dition of mutations at RT positions 62, 75, 77, and 116 in
combination with Q151M substantially increases the level of
resistance to these drugs both in vitro and in patients receiving
antiviral therapy (25, 36). The P157S mutation, originally
ob-served in a drug-resistant isolate of feline immunodeficiency
virus, confers resistance to (
⫺
)-

-2
⬘
,3
⬘
-dideoxy-3
⬘
-thiacytidine
(lamivudine [3TC]) in both feline immunodeficiency virus and
HIV-1 (61, 62). Mutation P157S or P157A is occasionally
ob-served in RT sequences from patients receiving nucleoside
analog therapy (15, 43, 46, 52).
Additional evidence for the role of motif B in substrate
selectivity comes from biochemical studies of HIV-1 RT
mu-tants. Specific substitutions at position Q151 affect nucleotide
insertion fidelity, discrimination against rNTPs, and the
incor-poration of nucleotide analogs (10, 23, 28, 33, 54, 74).
Replace-ments at motif B residues V148, W153, K154, P157, and F160
also affect fidelity and/or analog incorporation in cell-free
poly-merase assays (11, 18, 29, 33, 56, 57, 74). However, most of the
RT mutants examined in these experiments exhibit significant
reductions in catalytic activity and are therefore unlikely to
support HIV-1 replication (13, 48). Thus, the importance of
motif B for substrate analog susceptibility and accurate DNA
synthesis during viral replication remains largely unexplored.
To this end, we used “random scanning mutagenesis” to
construct pools of HIV-1 variants, and we subjected these
pools to a single passage in culture to identify motif B
muta-tions that preserve viral replication capacity. Individual
vari-ants were then screened for altered sensitivity to nucleoside
and pyrophosphate analogs. Our results show that many motif
B residues influence substrate analog susceptibility and that
* Corresponding author. Mailing address: University of Washington,
Department of Pathology, K-084 HSB, Box 357705, 1959 NE Pacific
St., Seattle, WA 98195. Phone: (206) 221-5650. Fax: (206) 543-3967.
E-mail: [email protected].
7169
on November 8, 2019 by guest
http://jvi.asm.org/
specific substitutions in this region confer hypersusceptibility
to structurally diverse nucleoside analog inhibitors. The
phe-notypes exhibited by these mutants suggest that motif B
influ-ences both the substrate selectivity and the primer unblocking
activity of HIV-1 RT.
MATERIALS AND METHODS
Inhibitors.RT inhibitors ddI, d4T, and phosphonoformic acid (foscarnet [PFA]) were purchased from Sigma-Aldrich Co., St. Louis, Mo. Inhibitors (R )-9-(2-phosphonylmethoxypropyl)adenine (tenofovir [PMPA]) and (1S,4R )-4-[2-ami-no-6-(cyclopropyl-amino)-9H-purin-9-yl]-2-cyclopentene-1-methanol (abacavir [ABC]) were obtained from Moravek Biochemicals Inc., Brea, Calif. AZT was purchased from Moravek and Sigma-Aldrich. 3TC was kindly provided by Ray-mond Schinazi of Emory University or purchased from Moravek.
Plasmids, cells, and virus.All mutant strains and random virus pools were derived from a modified version of the full-length pR9 HIV-1 clone (65) that lacks an ApaI site in the plasmid backbone (kindly provided by Uta von Schwedler, University of Utah). pBSpolwas created by subcloning an ApaI/ EcoRI fragment of pR9 (HIV-1NL4-3, nucleotides 2010 to 5743) into pBluescript
II KS(⫺) (Stratagene, La Jolla, Calif.). pBSpolam(containing an amber stop
codon substitution at RT position 154) and pBSpolClaI(containing the insertion
GATCGAT at RT codon 152 [ClaI site underlined]) were generated from pBSpolby site-directed mutagenesis (Muta-Gene phagemid mutagenesis kit; Bio-Rad Laboratories, Hercules, Calif.). pR9⌬polwas created from pR9 by replacing the ApaI/EcoRI fragment with an ApaI-ATCGATGCGGCCGC -EcoRI synthetic linker (unique ClaI and NotI sites are underlined and italicized, respectively).
293tsA1609neo(293T) (51) and HeLa–CD4–LTR–-galactosidase (HeLa-P4)
(5) cells were cultured (37°C, 5% CO2) in Dulbecco’s modified Eagle’s medium
(DMEM; Invitrogen Corp., Carlsbad, Calif.) supplemented with 4 mML -glu-tamine, 50 U/ml penicillin, 50g/ml streptomycin, and 10% fetal bovine serum (HyClone, Logan, Utah).
Random-scanning mutagenesis.Mutant plasmid libraries containing random substitutions at individual codons in RT motif B were constructed by oligonu-cleotide-mediated mutagenesis using pBSpolam(for G152, K154, P157, and Q161
mutant pools) or pBSpolClaI(for the remaining mutant pools) as template DNA.
The oligonucleotides used to generate single-codon random mutants (Operon, Alameda, Calif.) spanned HIV-1NL4-3nucleotides 2971 to 3030 for pools
ran-domized at sites V148 to Q151, nucleotides 2982 to 3029 for G152, nucleotides 2977 to 3034 for W153 and G155, nucleotides 2991 to 3030 for K154, nucleotides 2982 to 3038 for S156, and nucleotides 3002 to 3043 for P157 and Q161.
Nucle-otide sequences at each randomized site were as follows: V148, HNN/NVN; L149, NVN/RNN; P150, NDN/DNN; Q151, NBN/DNN; G152, HNN/HHN/ NHN; W153, NNH; K154, BNN/BNY/NNY; G155, HNN/NHN; S156, NWN/ SNN/VNR; P157, NDN/DNN; and Q161, NBN/DNN (where N is A/T/G/C, D is A/G/T, B is C/G/T, H is A/C/T, V is A/C/G, R is A/G, Y is C/T, and S is C/G). These resulted in the exclusion of wild-type amino acids at each target codon and also excluded variants L149F, Q151H, K154M, S156C, S156W, and Q161H from their respective random pools. Products from the mutagenesis reactions were electroporated into ElectroMAX DH10BEscherichia coli(Gibco BRL), plas-mids were isolated from pools of⬎104
independent transformants, and ApaI/ EcoRI fragments from the pools were cloned into pR9⌬pol. The resulting full-length pR9 mutant libraries were purified from pools of⬎104 independent
transformants using the Endo-Free maxiprep kit (QIAGEN Inc., Valencia, Calif.).
Transfections.To prepare wild-type virus stocks and random virus pools, CaPO4-pR9 DNA coprecipitates were prepared with 10g plasmid DNA as
described previously (6), except that the 2⫻BBS buffer was replaced with 2⫻ HEPES-buffered saline (270 mM NaCl, 10 mM KCl, 1.5 mM Na2HPO4· 2H2O,
11 mM dextrose, 40 mM HEPES, pH 7.05). 293T cultures were seeded into 10-cm plates and grown to approximately 25% confluence prior to the addition of CaPO4-DNA coprecipitates. Following a 3-hour incubation, culture
superna-tants were aspirated, 2 ml of 10% glycerol in phosphate-buffered saline (PBS) was added to each plate, and the cells were incubated at 37°C for 2 min. Cells were then rinsed twice with 6 ml of PBS, 10 ml of DMEM was added to each plate, and the cultures were returned to the incubator. Culture supernatants were harvested at approximately 42 h after transfection, filtered through 0.4-m sy-ringe filters, and stored in 1-ml aliquots in the vapor phase of liquid nitrogen for subsequent analysis. Stocks of individual HIV-1 mutants were produced by transfection of 293T cells as described above, except that the glycerol shock step was omitted. Cultures were instead treated with chloroquine at a final concen-tration of 25M in DMEM immediately before the DNA-CaPO4mixtures were
added. Supernatants were aspirated and replaced with fresh medium 1 day later and then harvested and frozen on the following day as described above. The titers of wild-type stocks produced by either protocol typically ranged from 2⫻ 105
to 5⫻105
focus forming units (FFU)/ml for frozen stocks and 1⫻106
to 4⫻ 106
FFU/ml for fresh preparations.
HeLa-P4 passage protocol.HeLa-P4 cells were seeded at 105cells per 10-cm
plate and infected the next day with 104FFU in 2 ml DMEM containing 20g/ml
[image:2.585.114.478.72.233.2]DEAE-dextran (Sigma). After incubation at 37°C for 2 to 4 h, an additional 6 ml of DMEM was added to each plate, and incubation was continued overnight. The monolayers were then washed three times with 6 ml of PBS, and the cultures were replenished with 8 ml fresh medium, which was changed again after 2 days
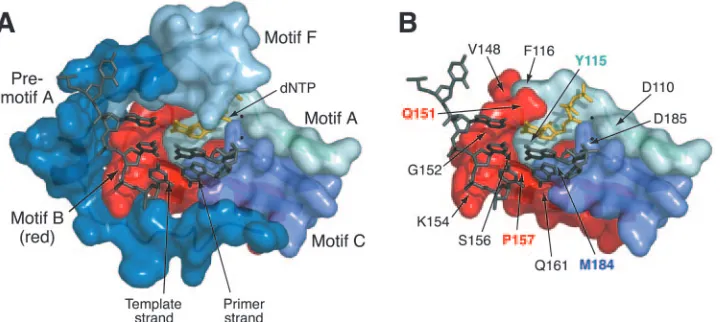
FIG. 1. Relationship of motif B to other structural components in the ternary complex of HIV-1 RT (Protein Data Bank entry 1RTD) (24).
(A) Surface representation of conserved structural motifs in the polymerase domain. Amino acids 107 to 118 (motif A), 147 to 169 (motif B), and
180 to 189 (motif C) are shown, as assigned by Poch et al. (53). Residues 102 to 106 (motif A), 143 to 146 (motif B), and 190 (motif C) are omitted
for clarity. Boundaries for motif F (residues 64 to 75) are based on recent alignments of viral RNA-dependent RNA polymerases (4, 76). The
region designated “premotif A” (residues 75 to 91) was originally identified in an alignment of HIV-1 RT with negative-stranded RNA virus
polymerases (45). (B) Location of several motif B amino acids and other residues discussed in the text. Motif F and premotif A have been removed
for clarity. Amino acid substitutions at residues Q151 and P157 (labeled in red) are known to confer resistance to nucleoside analogs (25, 36, 61).
Residues Y115 and M184 are key determinants of dNTP substrate selectivity in motifs A and C, respectively (40, 64). Mg
2⫹ions coordinated at
the active site are shown as small black spheres. These views were produced using MacPyMOL version 0.95 (http://pymol.sourceforge.org).
on November 8, 2019 by guest
http://jvi.asm.org/
of incubation. Culture supernatants were harvested on the fifth day after infec-tion, passed through 0.4-m filters, and treated with 420 U DNase I (Worthing-ton Biochemical) at 37°C for 20 min after we added MgCl2to a final
concen-tration of 10 mM. Viral particles were concentrated by layering 1 ml of culture supernatant onto a 250-l cushion of 20% sucrose in phosphate-buffered saline, followed by centrifugation at 15,500⫻gfor 90 min at 4°C. Virion pellets were resuspended in 100l of lysis buffer (7 M urea, 0.35 M NaCl, 4 mM EDTA, 10 mM Tris-HCl, pH 7.5, 1% sodium dodecyl sulfate) and stored at⫺20°C for subsequent RNA extraction and RT-PCR amplification. The remaining uncon-centrated culture supernatants were frozen in 1-ml aliquots in the vapor phase of liquid nitrogen.
RT-PCR and DNA sequencing.RT-PCR was performed in thin-walled tubes (Robbins), using the Access RT-PCR system (Promega), in 50-l reaction mix-tures containing 200M of each dNTP, 1 mM MgSO4, 5 U avian myeloblastosis
virus RT, 5 UTflDNA polymerase, 10l of 5⫻avian myeloblastosis virus-Tfl
buffer, 50 pmol each of primers H3 and BH1 (see below), and 1l of extracted viral RNA. Primers BH1 (5⬘-TATGGATCCCTTTTAGAATCTCCCTGTTTTC TGCC-3⬘; BamHI site underlined) and H3 (5⬘-AGTCAAGCTTGGATGGCCC AAAAGTTAAACAATGGCC-3⬘; HindIII site underlined) amplified a 0.8-kb fragment corresponding to nucleotides 2597 to 3483 of HIV-1NL4-3.
Thermocy-cling conditions in an MJ Research PTC-100 thermocycler were as follows: 48°C for 45 min, 94°C for 2 min, then 40 cycles of 94°C for 30 s, 60°C for 1 min, and 68°C for 2 min, and ending with 68°C for 7 min. Control reactions lacking RT were performed and analyzed in parallel to ensure that amplification was RNA dependent. When required, viral RNA samples were subjected to a second DNase I treatment to remove contaminating plasmid or proviral DNA (see above). RT-PCR products were digested with BamHI and HindIII, ligated into double-digested pBluescript II KS(⫺), transformed intoE. coli, and spread on agar plates containing ampicillin. Individual colonies were randomly picked from these plates, and plasmids were isolated and sequenced using primer pNL1 (5⬘-GACTTCAGGAAGTATACTGC-3⬘) and BigDye Terminator chemistry (Applied Biosystems, Foster City, Calif.).
Infectivity assay.Single-round infectivities were determined using HeLa-P4 indicator cells as previously described (5, 61). Briefly, HeLa-P4 cells were seeded into 96-well plates at 0.5⫻104cells/well and infected the following day with
serial dilutions of virus prepared in DMEM containing 20g/ml DEAE-dextran. After 40 h of growth, cultures were fixed and stained with 5-bromo-4-chloro-3-indolyl--D-galactopyranoside (X-Gal; Promega, Madison, Wis.). In this time frame, infected cells appeared as isolated groups of two to five contiguous Lac⫹ (blue) cells, indicating that the majority of foci were derived from a single cycle of virus replication. Titers of Lac⫹foci were normalized against HIV-1 capsid p24 concentration (HIV-1 enzyme-linked immunosorbent assay; PerkinElmer, Boston, MA) to determine the infectivities of mutants relative to that of the wild-type virus. The infectivity of the wild-type pR9-derived HIV-1NL4-3was
880⫾180 FFU/ng p24 (mean⫾standard error) for frozen stocks and 4,500⫾ 1,100 FFU/ng p24 for fresh supernatants. A variant containing the RT-inactivat-ing D185A mutation served as a negative control and yielded 0.014⫾0.004 FFU/ng p24 from frozen stocks.
Inhibitor sensitivity assay.For measurements of inhibitor sensitivity, HeLa-P4 cells were seeded into 96-well plates at a density of 0.5⫻104
cells/well and incubated overnight. Culture wells were dosed with various concentrations of inhibitor on the following morning, and the plates were then returned to the incubator for an additional 3 hours. Immediately prior to infection, virus stocks were diluted to 4,000 FFU/ml in DMEM containing 20g/ml of DEAE-dextran. Supernatants in the microtiter plates were aspirated, and 25l of the virus dilution was added directly to the monolayer in each well. Plates were then returned to the incubator for 3 hours. After this time, an additional 175l medium was added to each well, a second dose of inhibitor was added (at the same concentration as the first dose), and incubation was continued for two more days. Culture monolayers were fixed and stained as described above, and Lac⫹ foci were counted. Control cultures incubated in the absence of substrate analogs typically yielded 70 to 250 foci/well. Concentrations of analog required to inhibit focus formation by 50% of the untreated control value (50% effective concen-tration [EC50]) were calculated by linear regression of the resulting
dose-re-sponse data.
RESULTS
Random scanning mutagenesis of motif B.
To examine the
role of motif B in substrate analog sensitivity, we first
gener-ated pools of HIV-1 mutants containing random single amino
acid replacements at each of 11 motif B residues (60). This
strategy is an extension of alanine scanning mutagenesis (9)
and is referred to here as “random scanning mutagenesis.” The
benefit of random scanning mutagenesis is that all 19 amino
acid substitutions are introduced at each position, thus
permit-ting the detection of functional changes that are not produced
by simple alanine substitutions.
We used two different strategies to optimize the yield of
random amino acid substitutions and minimize the proportion
of wild-type sequences at each target codon. In the first
ap-proach, we constructed a
pol
subclone that contained a lethal
amber stop codon at RT position K154 (pBSpol
am). Mutagenic
oligonucleotides were used to correct this stop codon and
simultaneously introduce random substitutions at RT codon
G152, K154, P157, or Q161. Following mutagenesis, the
pol
genes were ligated into a
pol-deleted HIV-1 construct
(pR9
⌬
pol) to produce full-length HIV-1 plasmid libraries,
each randomized at a specific codon in RT motif B. Sequence
analyses of 180 individual pR9 clones generated by this method
confirmed that each library contained a diverse array of amino
acid substitutions at the targeted motif B position. Pools
ran-domized at codon G152, K154, P157, or Q161 contained 12, 16,
17, or 14 of 20 possible amino acid residues, respectively, at the
target codon. However, each pool also contained a substantial
number of nonmutant clones that retained the K154 amber
stop codon (50% in the K154, P157, and Q161 pools and 85%
in the G152 pool). Although these K154 amber products did
not interfere with subsequent experimental steps, sequencing
of large numbers of clones was required in order to
character-ize the diversity of each library.
To increase mutagenesis efficiency, we devised a second
strategy for random mutagenesis of the remaining seven target
residues in motif B. This method started with a
pol
subclone
containing a unique ClaI restriction site at position 152 of RT
(pBSpol
ClaI). Oligonucleotide primers restored the wild-type
sequence at codon 152 while simultaneously introducing
ran-dom mutations at position 148, 149, 150, 151, 153, 155, or 156
of RT. After subcloning into pR9
⌬
pol
and amplification in
E.
coli, the resulting full-length HIV-1 plasmid libraries were
en-riched for mutant clones by being digested with ClaI, followed
by a third round of
E. coli
amplification. Sequencing of
indi-vidual clones generated by this second strategy showed that the
final proportion of mutant plasmids in the resulting libraries
was
⬎
90% and that the diversity of these libraries was similar
to the diversity achieved using the initial pBSpol
am-based
ap-proach (see above). Both mutagenesis strategies limited but
did not completely exclude wild-type clones, which were
present at frequencies of 5 to 12% in the plasmid libraries.
Selection of replication-competent mutants.
The full-length
mutant HIV-1 plasmid libraries were separately transfected
into 293T cells to produce mutant virus pools, each comprised
of variants with random replacements at a single codon in
motif B. Replication-competent viruses were selected from
these pools by subjecting the virus populations to a single 5-day
passage in HeLa-P4 cells (see Materials and Methods for
de-tails). Infectious mutants were then identified by sequencing
individual clones of RT-PCR products derived from the
result-ant HeLa-P4-passaged pools. Altogether, 52 different single
amino acid substitutions in motif B were detected following a
single passage in culture (Table 1). Eight of the 11 pools
(V148, L149, P150, Q151, K154, G155, P157, and Q161)
on November 8, 2019 by guest
http://jvi.asm.org/
yielded mutants with both conservative and nonconservative
replacements at the respective target codons. Two pools
(W153 and S156) yielded only a single variant, and only one
pool (G152) failed to produce infectious mutants.
To confirm that the variants identified in the
HeLa-P4-se-lected populations were viable, we performed two separate
analyses of viral replication. First, the K154, P157, and Q161
mutant virus pools were subjected to three additional passages
in culture, and the range of mutants present in these
subse-quent cultures was determined by sequencing 25 to 50 clones
from each passage interval. Of the 25 mutants identified in the
first passage of these mutant pools (Table 1), 23 were detected
in passage 2, 3, or 4 (60). Thus, the majority of mutants
ob-served after a single passage in culture were capable of
multi-ple cycles of replication. We also constructed 24 full-length
HIV-1 plasmid clones containing specific single amino acid
substitutions in motif B that were detected in the passage 1
supernatants. In most cases, the variants produced by these
clones retained
ⱖ
50% of wild-type infectivity (Fig. 2).
Alto-gether, 47 of the 52 mutants observed in the virus pools
fol-lowing a single passage in HeLa-P4 cells (Table 1) were
rep-lication competent, as evidenced by their infectivity as purified
clones (Fig. 2) and/or persistence in subsequent passages (60).
Residues throughout motif B affect nucleoside analog
sen-sitivity.
To assess the functional importance of motif B, we
initially measured the susceptibility of each
replication-compe-tent mutant shown in Fig. 2 to the cytidine nucleoside analog
3TC. Several mutations in motif B altered the sensitivity of the
virus to this analog (Fig. 3A and 4A). Overall, there was a
trend towards 3TC resistance among the variants analyzed
(Fig. 4A), although many substitutions were neutral and three
replacements (W153F, G155N, and G155Q) resulted in slight
hypersusceptibility to 3TC.
We also examined the susceptibility of each motif B mutant
to the thymidine analog AZT. As previously reported, the
Q151M mutation conferred moderate resistance to AZT, and
the Q151M/A62V/V75I/F77L/F116Y complex of mutations
conferred
⬎
50-fold resistance to the drug (25, 36). In contrast,
most of the other motif B mutants examined were
hypersus-ceptible to AZT (Fig. 3B and 4B). Specific substitutions at
positions V148, Q151, and G155 conferred 10- to 60-fold
in-creases in AZT sensitivity. Altogether, 20 of the 24 motif B
variants examined were hypersusceptible to AZT and/or
resis-tant to 3TC (Fig. 4). Specific mutations at codons V148, Q151,
and P157 conferred both AZT hypersusceptibility and 3TC
resistance.
We noted that a subset of AZT-hypersusceptible mutants
exhibited a substantial impairment in viral replication capacity
(Fig. 2). To further examine the relationship between
hyper-susceptibility and viral infectivity, we characterized the effects
of four specific mutations in the “primer grip” region of RT
(26). An F227A RT mutant was eightfold hypersensitive to
AZT and retained 27% of wild-type HIV-1 infectivity,
demon-strating that mutations outside of motif B can also confer AZT
hypersusceptibility. In contrast, variants W266F, W266Y, and
W266R exhibited 0.5, 27, and 92% of wild-type infectivity,
respectively, but
ⱕ
2-fold changes in AZT or 3TC sensitivity.
Similarly, the P150A replacement in motif B, which reduced
viral infectivity to 15% of the wild type, had no effect on AZT
sensitivity, while other variants that retained 80 to 100%
[image:4.585.41.284.85.217.2]rep-lication capacity were
ⱖ
10-fold hypersusceptible to AZT (e.g.,
[image:4.585.133.449.553.675.2]FIG. 2. Replication capacities of RT motif B mutants. 293T cells were transfected with the wild-type (WT) HIV-1 pR9 clone or with clones
containing specific substitutions in motif B. Titers in the resulting cultures were measured using HeLa-P4 cells and normalized to the concentration
of HIV-1 capsid p24 in the supernatant to determine the infectivity of each mutant relative to that of the wild-type virus. ND, not determined;
Q151M
⫹
4, multinucleoside-resistant mutant Q151M/A62V/V75I/F77L/F116Y (25).
*
, statistically different from the wild type by one-way analysis
of variance (
P
⬍
0.05).
TABLE 1. Amino acid substitutions observed in the mutant virus
pools following a single passage in HeLa-P4 cells
Positiona
Amino acid(s) (no. of clones)b
V148 ...
V
(
18
)
, I (1), S (2), C (3), T (1), R (1)
L149 ...
L
(
18
)
, I (6), T (1), M (2)
P150 ...
P
(
24
)
, A (2), K (1)
Q151... G (7), A (12), V (1), I (1), S (1), C (1), T (3), M (4) G152...G(19)
W153 ... W(25), F (1)
K154 ... G (10), A (2), V (7), L (5), S (6), C (4), T (9), R (1), N (1), W (1)
G155...G(11), A (4), V (1), L (1), S (1), C (1), M (2), N (3), Q (2) S156...S(20), A (25)
P157 ...P(18), G (13), A (2), L (1), S (2), C (4), T (3)
Q161...Q(8), G (8), A (4), V (3), L (2), S (3), T (1), M (3), E (4)
aPools of mutants were produced and manipulated as independent virus populations, each containing random mutations at the specified amino acid position in RT.
bRT-PCR products amplified from HeLa-P4 supernatants were cloned into a plasmid vector and sequenced. Numbers in parentheses indicate the number of clones containing wild-type (bold italics) or mutant (roman) amino acids at the targeted RT codon. No wild-type clones were observed in random mutant pool Q151 or K154. Data are retabulated from reference 60.
on November 8, 2019 by guest
http://jvi.asm.org/
Q151A and Q161G) (Fig. 2 and 4B). Taken together, these
data indicate that AZT hypersusceptibility does not generally
correlate with reduced viral replication capacity.
Relationship between AZT hypersusceptibility and PFA
re-sistance.
Specific replacements at codons 88, 89, 90, 92, 156,
160, 161, and 164 of HIV-1 RT have previously been shown to
confer slight increases in AZT sensitivity (20, 39, 42, 67, 68).
These substitutions also confer resistance to PFA, an analog
that mimics the

-
␥
pyrophosphate group of the incoming
dNTP substrate. To further examine this phenotypic
relation-ship, we measured the sensitivities of 14 different
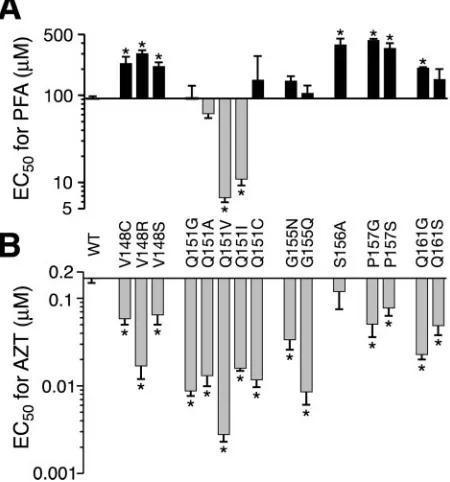
AZT-hyper-susceptible motif B mutants to PFA (Fig. 5). We also included
the S156A variant in these experiments as a positive control
for PFA resistance (67, 68). Six of the variants (V148C,
V148R, V148S, P157G, P157S, and Q161G) were resistant
to PFA (Fig. 5A) and therefore fit the aforementioned
pat-tern of AZT hypersusceptibility and PFA resistance. As
previously reported, the S156A mutation conferred fourfold
resistance to PFA without significantly affecting viral
sensi-tivity to AZT (67, 68).
We also observed several variants that did not follow the
AZT-hypersusceptible, PFA-resistant pattern (Fig. 5). For
ex-ample, mutants Q151G, Q151C, G155N, and G155Q displayed
6- to 20-fold hypersusceptibility to AZT but were not
signifi-cantly resistant to PFA. In addition, the Q151V and Q151I
variants showed increased sensitivity to both AZT and PFA
(Fig. 5 and Table 2). These results demonstrate that the
AZT-hypersusceptible phenotype is not necessarily coupled to PFA
resistance.
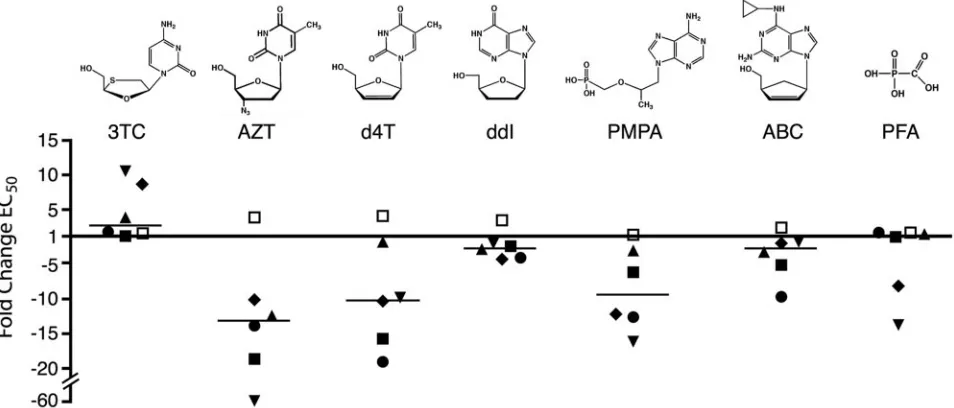
Variants hypersusceptible to other nucleoside analogs.
Sev-eral substitutions at position Q151 resulted in 10-fold or
greater hypersusceptibility to AZT (Fig. 4B), suggesting that
this residue is particularly important for substrate analog
sen-sitivity. We therefore determined the response of the Q151
mutants to a panel of structurally diverse nucleoside analog
inhibitors (Table 2 and Fig. 6). Consistent with previous
re-ports, the Q151M mutation resulted in low-level resistance to
d4T and ddI, and Q151M combined with A62V, V75I, F77L,
and F116Y conferred higher levels of resistance to these two
analogs (Table 2) (25, 36). In contrast, replacement of glycine,
alanine, valine, isoleucine, or cysteine at position 151 increased
HIV-1 sensitivity to several of the nucleoside analogs tested.
Mutants Q151G and Q151C were 5- to 10-fold
hypersuscep-tible to d4T, PMPA, and ABC (Table 2). Variants Q151V and
Q151I were also 10-fold hypersusceptible to d4T and PMPA
but showed only marginal hypersusceptibility to ABC. The
Q151A mutation resulted in two- to threefold increases in d4T,
PMPA, and ABC sensitivities. With the exception of
methio-nine, each of the Q151 substitutions also conferred modest
hypersusceptibility to ddI, with EC
50s two- to fivefold lower
than that of the wild type. Thus, several different substitutions
at Q151 conferred a multinucleoside-hypersusceptible
pheno-type (Fig. 6).
In addition to Q151 mutations, specific substitutions at other
motif B positions also conferred multinucleoside
hypersuscep-tibilty. Mutants V148R and G155Q, both of which were
hyper-susceptible to AZT (Fig. 4A), were also four- to sevenfold
hypersusceptible to PMPA (EC
50s of 1.4
⫾
0.2
M and 0.90
⫾
0.3
M for V148R and G155Q, respectively, versus 5.9
⫾
1.0
M for the wild-type virus). The G155Q variant also exhibited
three- to fivefold hypersusceptibility to d4T, ddI, and ABC but
no change in PFA sensitivity (data not shown).
In summary, substitutions at 9 of the 11 RT motif B positions
subjected to random mutagenesis altered viral susceptibility to
one or more nucleoside analogs, and several mutations
con-ferred altered sensitivity to the pyrophosphate analog PFA.
Thus, residues throughout RT motif B play important roles in
determining the substrate analog sensitivity of HIV-1.
FIG. 3. Representative dose-response data for the pyrimidine analogs 3TC (A) and AZT (B). Profiles for wild-type HIV-1 (dotted lines) and
several motif B variants (solid lines) are shown. Analog sensitivities were measured by quantitating the dose-dependent reduction of Lac
⫹foci in
HeLa-P4 cells. The percentages of solvent-only control foci are plotted as a function of nucleoside analog concentration. Curves were generated
using a sigmoidal regression equation (GraphPad Prism 4 software package [44]). The results for each strain are from a single assay, with two
determinations of focus formation per drug concentration. These data are representative of the responses observed for each mutant in multiple
independent experiments.
F
, P157S;
E
, W153F;
■
, wild type;
䊐
, Q151A;
ƒ
, Q151V;
Œ
, Q151M/A62V/V75I/F77L/F116Y;
‚
, Q161G;
䉬
, Q151M;
䉫
, G155Q.
on November 8, 2019 by guest
http://jvi.asm.org/
DISCUSSION
The polymerase domain of HIV-1 RT contains structural
motifs that are conserved in all reverse transcriptases and
RNA-dependent viral polymerases (Fig. 1) (4, 21, 76). Amino
acid residues within these motifs affect the fidelity of DNA
synthesis and influence the sensitivity of HIV-1 to substrate
analog inhibitors (40, 49, 64, 72). Motif B is particularly
inter-esting because of its central position in the core polymerase
structure (Fig. 1). Although a few mutations in HIV-1 RT
motif B have been shown to confer drug resistance (25, 36, 39,
61, 67), the role of this structure in viral sensitivity to
nucleo-side and pyrophosphate analogs is largely unknown. To
exam-ine the functional importance of motif B in the context of
replicating virus, we used random scanning mutagenesis to
introduce random single amino acid replacements at 11 motif
B codons. We then identified the range of replacements that
preserve HIV-1 replication in culture (Table 1 and Fig. 2) and
screened a subset of these infectious mutants for altered
sen-sitivity to substrate analog inhibitors. The results demonstrate
that residues throughout the targeted region of motif B affect
viral susceptibility to substrate analogs (Fig. 3 to 5) and that
specific substitutions, particularly at residue Q151, confer a
multidrug-hypersusceptible phenotype (Table 2 and Fig. 6).
[image:6.585.50.275.428.669.2]Previous efforts to identify determinants of dNTP selectivity
have primarily used conventional site-directed mutagenesis to
introduce single amino acid replacements in RT (40, 64).
FIG. 4. Susceptibilities of motif B mutants to 3TC (A) and AZT (B). Concentrations of nucleoside analog required to inhibit Lac
⫹focus
formation in HeLa-P4 cells by 50% (EC
50) were calculated by regression analyses of dose-response data (Fig. 3), as described in Materials and
Methods. The abscissa is set at the EC
50for wild-type (WT) virus; bars above and below the abscissa represent analog resistance and
hypersus-ceptibility, respectively. Each bar represents the mean
⫾
standard error from at least three independent experiments.
*
, significantly different from
the wild type by one-way analysis of variance (
P
⬍
0.05);
**
, significantly different from the wild type by a paired
t
test (
P
⬍
0.05). ND, not
determined. Variants Q151M
⫹
4 (Q151M/A62V/V75I/F77L/F116Y) and M184V, which exhibit high-level resistance to AZT and 3TC, respectively,
served as positive controls in these assays (25, 55, 69).
FIG. 5. Sensitivities of AZT-hypersusceptible mutants to PFA.
EC
50s for PFA (A) and AZT (B) were measured and graphed as
described in Materials and Methods and the legend to Fig. 3. Each bar
represents the mean
⫾
standard error from at least three independent
experiments.
*
, significantly different from the wild type (WT) by
one-way analysis of variance (
P
⬍
0.05).
on November 8, 2019 by guest
http://jvi.asm.org/
These variant RTs often exhibit severe impairments of
poly-merase function and are therefore unable to support viral
replication (13, 18, 23, 28, 48, 56, 57, 59, 74). Here, we used cell
culture to select replication-competent viruses from random
pools of HIV-1 RT mutants. A sampling of 24 culture-selected
mutants showed that most retained
ⱖ
50% of wild-type HIV-1
infectivity (Fig. 2). Thus, random scanning mutagenesis
cou-pled to selection in culture is a facile approach for identifying
catalytically active RT mutants. The strategies we used to
en-rich viable mutants and limit the proportion of wild-type
HIV-1 in the random virus pools can readily be applied to
other regions of the HIV-1 genome as well as other viruses for
which a full-length plasmid clone is available.
A notable finding from our study was the magnitude and
scope of substrate analog hypersusceptibility produced by
mu-tations in RT motif B. Substitutions at 8 of the 11 residues
analyzed increased the sensitivity of HIV-1 to one or more
inhibitors, and several variants displayed 10-fold or greater
analog hypersusceptibility (Fig. 4 and 5 and Table 2). Previous
studies of HIV-1 variants have reported low-level (i.e., two- to
fivefold) increases in viral sensitivity to nucleoside analogs (20,
39, 42, 61, 67, 68, 73), and replacements in conserved regions
of herpesvirus and bacteriophage
29 DNA polymerases
oc-casionally confer low-level hypersusceptibility to substrate
an-alogs (3, 7, 14, 19, 38, 70). Our data demonstrate that single
amino acid substitutions in HIV-1 RT motif B can produce
large increases in inhibitor sensitivity (Fig. 3 to 5) while
pre-serving viral replication capacity (Fig. 2). We anticipate that
other conserved RT motifs play a similar role in substrate
analog susceptibility.
Our analysis of motif B suggests that nucleoside analog
hypersusceptibility results from multiple biochemical
mecha-nisms that are not necessarily mutually exclusive. First, a subset
of motif B mutations may affect hypersensitivity to AZT by
impairing the “primer unblocking” activity of RT (41). This
inference is supported by the observation that specific
muta-FIG. 6. Hypersusceptibility of Q151 mutants to substrate analog inhibitors. Data from Fig. 4 and 5 and Table 2 are plotted to illustrate the
change (
n
-fold) in EC
50for each Q151 mutant relative to that of wild-type HIV-1. Values above and below the
x
axis are resistant and
hypersusceptible to drug, respectively. Horizontal bars indicate the median change (
n
-fold) in EC
50for each analog for the entire set of Q151
[image:7.585.53.530.479.683.2]mutants.
■
, Q151G;
䊐
, Q151M;
, Q151V;
Œ
, Q151A;
䉬
, Q151I;
F
, Q151C.
TABLE 2. Susceptibilities of Q151 RT mutants to substrate analogs
Virus
EC50(M)a
3TC AZT d4T ddI PMPA ABC PFA
Wild type
0.90
⫾
0.1 (1)
0.17
⫾
0.02 (1)
5.8
⫾
0.9 (1)
3.1
⫾
0.8 (1)
5.9
⫾
1 (1)
7.0
⫾
1 (1)
92
⫾
6 (1)
Q151G
0.79
⫾
0.3 (1)
0.0088
⫾
0.001 (0.1)
0.36
⫾
0.1 (0.1)
1.2
⫾
0.3 (0.4)
0.92
⫾
0.1 (0.2)
1.3
⫾
0.1 (0.2)
93
⫾
36 (1)
Q151A
3.4
⫾
0.5 (4)
0.013
⫾
0.003 (0.1)
2.8
⫾
2 (0.5)
1.1
⫾
0.1 (0.3)
1.8
⫾
0.2 (0.3)
2.0
⫾
0.5 (0.3)
62
⫾
8 (0.7)
Q151V
9.3
⫾
1 (10)
0.0028
⫾
0.001 (0.02)
0.58
⫾
0.2 (0.1)
1.4
⫾
0.1 (0.4)
0.36
⫾
0.1 (0.1)
3.4
⫾
0.5 (0.5)
6.7
⫾
0.9 (0.1)
Q151I
7.7
⫾
2 (9)
0.016
⫾
0.001 (0.1)
0.55
⫾
0.04 (0.1)
0.69
⫾
0.3 (0.2)
0.48
⫾
0.1 (0.1)
3.2
⫾
0.5 (0.5)
11
⫾
2.2 (0.1)
Q151C
1.5
⫾
0.6 (2)
0.012
⫾
0.002 (0.1)
0.30
⫾
0.05 (0.1)
0.73
⫾
0.2 (0.2)
0.46
⫾
0.1 (0.1)
0.71
⫾
0.2 (0.1)
150
⫾
130 (2)
Q151M
1.4
⫾
0.3 (2)
0.64
⫾
0.02 (4)
23
⫾
5 (4)
11
⫾
2 (3)
7.8
⫾
2 (1)
17
⫾
3 (2)
160
⫾
60 (2)
Q151M
⫹
4
b6.3
⫾
2 (7)
⬎
10 (
⬎
100)
⬎
100 (
⬎
20)
39
⫾
5 (13)
18
⫾
1 (3)
56
⫾
18 (8)
ND
ca
EC50s were obtained for HeLa-P4 cells as described in Materials and Methods. Numbers in parentheses indicate EC50s relative to that of the wild-type virus. Values
are the means⫾standard errors from three or more independent experiments. b
Multinucleoside-resistant mutant Q151M/A62V/V75I/F77L/F116Y (27, 37). c
ND, not determined.
on November 8, 2019 by guest
http://jvi.asm.org/
tions conferred both AZT hypersusceptibility and PFA
resis-tance (Fig. 5), a phenotypic pattern that has been correlated
with a loss of unblocking capacity (1, 42). Diminished primer
unblocking function may occur with or without compromised
RT polymerase activity, as suggested by the subset of
hyper-susceptible variants with reduced infectivity (Fig. 2 and 4).
Second, mutations in RT may confer nucleoside analog
hy-persusceptibilty by enhancing the efficiency of analog
incorpo-ration. Specific replacements at position Q151 increased the
sensitivity of HIV-1 to multiple substrate analogs (Table 2),
suggesting a direct effect on nucleotide selectivity (see below).
Other mutations in motif B are likely to impart nucleoside
analog hypersusceptibility indirectly by repositioning residues
Y115 and M184 (Fig. 1B), which are key determinants of
substrate specificity (40, 64). Taken together, our data suggest
that motif B influences both the efficiency of the primer
un-blocking reaction and the selectivity of nucleotide
incorpora-tion by HIV-1 RT. Thus, motif B likely contributes to
impor-tant enzyme-substrate interactions at multiple steps in the
catalytic cycle of polymerization.
Our results demonstrate that residue Q151 is particularly
important for substrate analog sensitivity (Table 2 and Fig. 6).
Substitutions at this position presumably affect the interaction
between RT and the incoming dNTP (Fig. 1) (24), thereby
directly influencing analog binding and/or polymerization (10).
It is well established that the Q151M mutation contributes to
nucleoside analog resistance in HIV-1 and simian
immunode-ficiency virus (25, 36, 71). Here, we show that other Q151
substitutions confer broad-spectrum hypersusceptibility to
structurally diverse nucleoside analogs, with up to a 60-fold
increase in analog sensitivity. Moreover, we show that a
sub-set of Q151 mutations also imparts hypersusceptibility to the
pyrophosphate analog PFA. These data suggest that specific
Q151 replacements in HIV-1 RT confer a general relaxation
of polymerase active-site stringency. Additional experiments
are required to examine the relationship between multidrug
hypersusceptibility and other aspects of RT substrate
selec-tivity, such as mispair formation and rNTP versus dNTP
discrimination.
Residues that are structurally analogous to Q151 of HIV-1
RT also influence nucleoside inhibitor sensitivity in other
DNA polymerases. In family A (E. coli
DNA polymerase
I-re-lated) enzymes, a conserved aromatic residue (phenylalanine
or tyrosine) that is positioned similarly to Q151 in the polymerase
active site strongly influences dNTP versus dideoxynucleoside
triphosphate selectivity (2, 35, 66). Substitutions at a structurally
equivalent asparagine residue in family B (mammalian DNA
polymerase
␣
-related) polymerases also affect fidelity and/or
nu-cleoside analog sensitivity (27, 31, 47). Taken together, these data
indicate that residues analogous to Q151 influence the inhibitor
sensitivities of polymerases from diverse organisms.
In patients receiving antiviral therapy, drug treatment
occa-sionally selects for variants that are resistant to one or more
components of the administered regimen but are
hypersuscep-tible to other inhibitors. For example, mutations that emerge
in vivo in response to 3TC, ddI, PFA, or certain nonnucleoside
reverse transcriptase inhibitors can confer hypersusceptibility
to AZT and “resensitize” AZT-resistant viruses (20, 30, 32, 42,
63, 68, 75). Increased viral sensitivity to protease inhibitors and
nonnucleoside reverse transcriptase inhibitors in clinical
iso-lates of HIV-1 has also been reported (16, 34, 37, 58). Our
analysis suggests that there are a number of RT mutations that
confer hypersusceptibility to AZT and other nucleoside
ana-logs. These mutations may contribute to observed differences
in drug sensitivity among “wild-type” HIV-1 isolates (22, 50)
and could potentially influence the efficacy of
nucleoside-con-taining antiretroviral regimens.
ACKNOWLEDGMENTS
We thank Tom North, Masanori Ogawa, and Tina Albertson for
critical reading of the manuscript and Crystal Pyrak for excellent
tech-nical assistance.
This work was supported by Public Health Service grants R01
AI34834 to B.D.P. and F32 AI10139 to R.A.S.
REFERENCES
1.Arion, D., N. Sluis-Cremer, and M. A. Parniak.2000. Mechanism by which phosphonoformic acid resistance mutations restore 3⬘-azido-3⬘ -deoxythymi-dine (AZT) sensitivity to AZT-resistant HIV-1 reverse transcriptase. J. Biol. Chem.275:9251–9255.
2.Astatke, M., N. D. Grindley, and C. M. Joyce.1998. How E. coli DNA polymerase I (Klenow fragment) distinguishes between deoxy- and dideoxynucleotides. J. Mol. Biol.278:147–165.
3.Bestman-Smith, J., and G. Boivin.2003. Drug resistance patterns of recom-binant herpes simplex virus DNA polymerase mutants generated with a set of overlapping cosmids and plasmids. J. Virol.77:7820–7829.
4.Bruenn, J. A.2003. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res.31:1821–1829. 5.Charneau, P., G. Mirambeau, P. Roux, S. Paulous, H. Buc, and F. Clavel. 1994. HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol.241:651–662.
6.Chen, C., and H. Okayama.1987. High-efficiency transformation of mam-malian cells by plasmid DNA. Mol. Cell. Biol.7:2745–2752.
7.Cihlar, T., M. D. Fuller, and J. M. Cherrington.1998. Characterization of drug resistance-associated mutations in the human cytomegalovirus DNA polymerase gene by using recombinant mutant viruses generated from over-lapping DNA fragments. J. Virol.72:5927–5936.
8.Coffin, J. M., S. H. Hughes, and H. E. Varmus (ed.).1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
9.Cunningham, B. C., and J. A. Wells.1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science244: 1081–1085.
10.Deval, J., B. Selmi, J. Boretto, M. P. Egloff, C. Guerreiro, S. Sarfati, and B. Canard.2002. The molecular mechanism of multidrug resistance by the Q151M human immunodeficiency virus type 1 reverse transcriptase and its suppression using alpha-boranophosphate nucleotide analogues. J. Biol. Chem.277:42097–42104.
11.Diamond, T. L., G. Souroullas, K. K. Weiss, K. Y. Lee, R. A. Bambara, S. Dewhurst, and B. Kim.2003. Mechanistic understanding of an altered fidelity simian immunodeficiency virus reverse transcriptase mutation, V148I, identified in a pig-tailed macaque. J. Biol. Chem.278:29913–29924.
12.Ding, J., K. Das, Y. Hsiou, S. G. Sarafianos, A. D. Clark, A. Jacobo-Molina, C. Tantillo, S. H. Hughes, and E. Arnold.1998. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol.284:1095–1111.
13.Gao, G., and S. P. Goff.1998. Replication defect of Moloney murine leuke-mia virus with a mutant reverse transcriptase that can incorporate ribonucle-otides and deoxyribonucleribonucle-otides. J. Virol.72:5905–5911.
14.Gibbs, J. S., H. C. Chiou, K. F. Bastow, Y. C. Cheng, and D. M. Coen.1988. Identification of amino acids in herpes simplex virus DNA polymerase in-volved in substrate and drug recognition. Proc. Natl. Acad. Sci. USA85: 6672–6676.
15.Gonzales, M. J., T. D. Wu, J. Taylor, I. Belitskaya, R. Kantor, D. Israelski, S. Chou, A. R. Zolopa, W. J. Fessel, and R. W. Shafer.2003. Extended spectrum of HIV-1 reverse transcriptase mutations in patients receiving multiple nucleoside analog inhibitors. AIDS17:791–799.
16.Gonzalez, L. M., R. M. Brindeiro, M. Tarin, A. Calazans, M. A. Soares, S. Cassol, and A. Tanuri.2003. In vitro hypersusceptibility of human immuno-deficiency virus type 1 subtype C protease to lopinavir. Antimicrob. Agents Chemother.47:2817–2822.
17.Gotte, M.2004. Inhibition of HIV-1 reverse transcription: basic principles of drug action and resistance. Expert Rev. Anti-Infect. Ther.2:707–716. 18.Gutierrez-Rivas, M., A. Ibanez, M. A. Martinez, E. Domingo, and L.
Menendez-Arias.1999. Mutational analysis of Phe160 within the “palm” subdomain of human immunodeficiency virus type 1 reverse transcriptase. J. Mol. Biol.290:615–625.
on November 8, 2019 by guest
http://jvi.asm.org/
19.Hall, J. D., and S. Woodward.1989. Aphidicolin resistance in herpes simplex virus type 1 appears to alter substrate specificity in the DNA polymerase. J. Virol.63:2874–2876.
20.Hammond, J. L., D. L. Koontz, H. Z. Bazmi, J. R. Beadle, S. E. Hostetler, G. D. Kini, K. A. Aldern, D. D. Richman, K. Y. Hostetler, and J. W. Mellors. 2001. Alkylglycerol prodrugs of phosphonoformate are potent in vitro inhib-itors of nucleoside-resistant human immunodeficiency virus type 1 and select for resistance mutations that suppress zidovudine resistance. Antimicrob. Agents Chemother.45:1621–1628.
21.Hansen, J. L., A. M. Long, and S. C. Schultz.1997. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure5:1109–1122. 22.Harrigan, P. R., J. S. Montaner, S. A. Wegner, W. Verbiest, V. Miller, R.
Wood, and B. A. Larder.2001. World-wide variation in HIV-1 phenotypic susceptibility in untreated individuals: biologically relevant values for resis-tance testing. AIDS15:1671–1677.
23.Harris, D., N. Kaushik, P. K. Pandey, P. N. Yadav, and V. N. Pandey.1998. Functional analysis of amino acid residues constituting the dNTP binding pocket of HIV-1 reverse transcriptase. J. Biol. Chem.273:33624–33634. 24.Huang, H., R. Chopra, G. L. Verdine, and S. C. Harrison.1998. Structure of
a covalently trapped catalytic complex of HIV-1 reverse transcriptase: im-plications for drug resistance. Science282:1669–1675.
25.Iversen, A. K., R. W. Shafer, K. Wehrly, M. A. Winters, J. I. Mullins, B. Chesebro, and T. C. Merigan.1996. Multidrug-resistant human immunode-ficiency virus type 1 strains resulting from combination antiretroviral ther-apy. J. Virol.70:1086–1090.
26.Jacobo-Molina, A., J. Ding, R. G. Nanni, A. D. Clark, Jr., X. Lu, C. Tantillo, R. L. Williams, G. Kamer, A. L. Ferris, P. Clark, A. Hizi, S. H. Hughes, and E. Arnold.1993. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å reso-lution shows bent DNA. Proc. Natl. Acad. Sci. USA90:6320–6324. 27.Kamiyama, T., M. Kurokawa, and K. Shiraki.2001. Characterization of the
DNA polymerase gene of varicella-zoster viruses resistant to acyclovir. J. Gen. Virol.82:2761–2765.
28.Kaushik, N., T. T. Talele, P. K. Pandey, D. Harris, P. N. Yadav, and V. N. Pandey.2000. Role of glutamine 151 of human immunodeficiency virus type-1 reverse transcriptase in substrate selection as assessed by site-directed mutagenesis. Biochemistry39:2912–2920.
29.Klarmann, G. J., R. A. Smith, R. F. Schinazi, T. W. North, and B. D. Preston. 2000. Site-specific incorporation of nucleoside analogs by HIV-1 reverse transcriptase and the template grip mutant P157S. Template interactions influence substrate recognition at the polymerase active site. J. Biol. Chem. 275:359–366.
30.Larder, B. A.1992. 3⬘-Azido-3⬘-deoxythymidine resistance suppressed by a mutation conferring human immunodeficiency virus type 1 resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Che-mother.36:2664–2669.
31.Larder, B. A., S. D. Kemp, and G. Darby.1987. Related functional domains in virus DNA polymerases. EMBO J.6:169–175.
32.Larder, B. A., S. D. Kemp, and P. R. Harrigan.1995. Potential mechanism for sustained antiretroviral efficacy of AZT-3TC combination therapy. Sci-ence269:696–699.
33.Larder, B. A., S. D. Kemp, and D. J. Purifoy.1989. Infectious potential of human immunodeficiency virus type 1 reverse transcriptase mutants with altered inhibitor sensitivity. Proc. Natl. Acad. Sci. USA86:4803–4807. 34.Leigh Brown, A. J., S. D. Frost, B. Good, E. S. Daar, V. Simon, M. Markowitz,
A. C. Collier, E. Connick, B. Conway, J. B. Margolick, J. P. Routy, J. Corbeil, N. S. Hellmann, D. D. Richman, and S. J. Little.2004. Genetic basis of hyper-susceptibility to protease inhibitors and low replicative capacity of human im-munodeficiency virus type 1 strains in primary infection. J. Virol.78:2242–2246. 35.Lim, S. E., M. V. Ponamarev, M. J. Longley, and W. C. Copeland.2003. Structural determinants in human DNA polymerase gamma account for mitochondrial toxicity from nucleoside analogs. J. Mol. Biol.329:45–57. 36.Maeda, Y., D. J. Venzon, and H. Mitsuya.1998. Altered drug sensitivity,
fitness, and evolution of human immunodeficiency virus type 1 with pol gene mutations conferring multi-dideoxynucleoside resistance. J. Infect. Dis.177: 1207–1213.
37.Martinez-Picado, J., T. Wrin, S. D. Frost, B. Clotet, L. Ruiz, A. J. Brown, C. J. Petropoulos, and N. T. Parkin.2005. Phenotypic hypersusceptibility to multiple protease inhibitors and low replicative capacity in patients who are chronically infected with human immunodeficiency virus type 1. J. Virol. 79:5907–5913.
38.Matsumoto, K., C. I. Kim, H. Kobayashi, H. Kanehiro, and H. Hirokawa. 1990. Aphidicolin-resistant DNA polymerase of bacteriophage phi 29 APHr71 mutant is hypersensitive to phosphonoacetic acid and butylphe-nyldeoxyguanosine 5⬘-triphosphate. Virology178:337–339.
39.Mellors, J. W., H. Z. Bazmi, R. F. Schinazi, B. M. Roy, Y. Hsiou, E. Arnold, J. Weir, and D. L. Mayers.1995. Novel mutations in reverse transcriptase of human immunodeficiency virus type 1 reduce susceptibility to foscarnet in laboratory and clinical isolates. Antimicrob. Agents Chemother.39:1087– 1092.
40.Menendez-Arias, L.2002. Molecular basis of fidelity of DNA synthesis and
nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol.71:91–147.
41.Meyer, P. R., S. E. Matsuura, A. M. Mian, A. G. So, and W. A. Scott.1999. A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell4:35–43. 42.Meyer, P. R., S. E. Matsuura, D. Zonarich, R. R. Chopra, E. Pendarvis, H. Z. Bazmi, J. W. Mellors, and W. A. Scott.2003. Relationship between 3⬘ -azido-3⬘-deoxythymidine resistance and primer unblocking activity in foscarnet-resistant mutants of human immunodeficiency virus type 1 reverse transcrip-tase. J. Virol.77:6127–6137.
43.Milazzo, L., S. Rusconi, L. Testa, S. La Seta-Catamancio, M. Galazzi, S. Kurtagic, P. Citterio, M. Gianotto, A. Grassini, F. Adorni, A. d’Arminio-Monforte, M. Galli, and M. Moroni.1999. Evidence of stavudine-related phenotypic resistance among zidovudine-pretreated HIV-1-infected subjects receiving a therapeutic regimen of stavudine plus lamivudine. J. Acquir. Immune Defic. Syndr.22:101–103.
44.Motulsky, H. J., and A. Christopoulos.2003. Fitting models to biological data using linear and nonlinear regression. A practical guide to curve fitting. GraphPad, Inc., San Diego, Calif.
45.Muller, R., O. Poch, M. Delarue, D. H. Bishop, and M. Bouloy.1994. Rift Valley fever virus L segment: correction of the sequence and possible func-tional role of newly identified regions conserved in RNA-dependent poly-merases. J. Gen. Virol.75:1345–1352.
46.Nijhuis, M., R. Schuurman, D. de Jong, R. van Leeuwen, J. Lange, S. Danner, W. Keulen, T. de Groot, and C. A. Boucher.1997. Lamivudine-resistant human immunodeficiency virus type 1 variants (184V) require mul-tiple amino acid changes to become co-resistant to zidovudine in vivo. J. In-fect. Dis.176:398–405.
47.Ogawa, M., S. Limsirichaikul, A. Niimi, S. Iwai, S. Yoshida, and M. Suzuki. 2003. Distinct function of conserved amino acids in the fingers of Saccharo-myces cerevisiae DNA polymerase alpha. J. Biol. Chem.278:19071–19078. 48.Olivares, I., V. Sanchez-Merino, M. A. Martinez, E. Domingo, C.
Lopez-Galindez, and L. Menendez-Arias.1999. Second-site reversion of a human immunodeficiency virus type 1 reverse transcriptase mutant that restores enzyme function and replication capacity. J. Virol.73:6293–6298. 49.Parikh, U., C. Calef, B. Larder, R. Schinazi, and J. W. Mellors.2001.
Mutations in retroviral genes associated with drug resistance, p. 191–277.In
C. Kuiken, B. Foley, B. Hahn, P. Marx, F. McCutchan, J. Mellors, S. Wo-linski, and B. Korber (ed.), HIV-1 sequence compendium. Theoretical Biol-ogy and Biophysics Group, Los Alamos National Laboratory, Los Alamos, N.Mex.
50.Parkin, N. T., N. S. Hellmann, J. M. Whitcomb, L. Kiss, C. Chappey, and C. J. Petropoulos.2004. Natural variation of drug susceptibility in wild-type human immunodeficiency virus type 1. Antimicrob. Agents Chemother.48: 437–443.
51.Pear, W. S., G. P. Nolan, M. L. Scott, and D. Baltimore.1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA90:8392–8396.
52.Picard, V., E. Angelini, A. Maillard, E. Race, F. Clavel, G. Chene, F. Ferchal, and J. M. Molina.2001. Comparison of genotypic and phenotypic resistance patterns of human immunodeficiency virus type 1 isolates from patients treated with stavudine and didanosine or zidovudine and lamivudine. J. In-fect. Dis.184:781–784.
53.Poch, O., I. Sauvaget, M. Delarue, and N. Tordo.1989. Identification of four conserved motifs among the RNA-dependent polymerase encoding ele-ments. EMBO J.8:3867–3874.
54.Rezende, L. F., K. Curr, T. Ueno, H. Mitsuya, and V. R. Prasad.1998. The impact of multidideoxynucleoside resistance-conferring mutations in human immunodeficiency virus type 1 reverse transcriptase on polymerase fidelity and error specificity. J. Virol.72:2890–2895.
55.Schinazi, R. F., R. M. Lloyd, Jr., M. H. Nguyen, D. L. Cannon, A. McMillan, N. Ilksoy, C. K. Chu, D. C. Liotta, H. Z. Bazmi, and J. W. Mellors.1993. Characterization of human immunodeficiency viruses resistant to oxathio-lane-cytosine nucleosides. Antimicrob. Agents Chemother.37:875–881. 56.Sharma, B., N. Kaushik, K. Singh, S. Kumar, and V. N. Pandey.2002.
Substitution of conserved hydrophobic residues in motifs B and C of HIV-1 RT alters the geometry of its catalytic pocket. Biochemistry41:15685–15697. 57.Sharma, B., N. Kaushik, A. Upadhyay, S. Tripathi, K. Singh, and V. N. Pandey.2003. A positively charged side chain at position 154 on the beta8-alphaE loop of HIV-1 RT is required for stable ternary complex formation. Nucleic Acids Res.31:5167–5174.
58.Shulman, N., A. R. Zolopa, D. Passaro, R. W. Shafer, W. Huang, D. Katzenstein, D. M. Israelski, N. Hellmann, C. Petropoulos, and J. Whitcomb. 2001. Phenotypic hypersusceptibility to non-nucleoside reverse transcriptase inhibitors in treatment-experienced HIV-infected patients: impact on virological response to efavirenz-based therapy. AIDS15:1125–1132.
59.Singh, K., N. Kaushik, J. Jin, M. Madhusudanan, and M. J. Modak.2000. Role of Q190 of MuLV RT in ddNTP resistance and fidelity of DNA synthesis: a molecular model of interactions with substrates. Protein Eng. 13:635–643.
60.Smith, R. A., D. J. Anderson, and B. D. Preston.2004. Purifying selection
on November 8, 2019 by guest
http://jvi.asm.org/
masks the mutational flexibility of HIV-1 reverse transcriptase. J. Biol. Chem.279:26726–26734.
61.Smith, R. A., G. J. Klarmann, K. M. Stray, U. K. von Schwedler, R. F. Schinazi, B. D. Preston, and T. W. North. 1999. A new point mutation (P157S) in the reverse transcriptase of human immunodeficiency virus type 1 confers low-level resistance to (⫺)--2⬘,3⬘-dideoxy-3⬘-thiacytidine. Antimi-crob. Agents Chemother.43:2077–2080.
62.Smith, R. A., K. M. Remington, B. D. Preston, R. F. Schinazi, and T. W. North.1998. A novel point mutation at position 156 of reverse transcriptase from feline immunodeficiency virus confers resistance to the combination of (⫺)--2⬘,3⬘-dideoxy-3⬘-thiacytidine and 3⬘-azido-3⬘-deoxythymidine. J. Virol. 72:2335–2340.
63.St. Clair, M. H., J. L. Martin, G. Tudor-Williams, M. C. Bach, C. L. Vavro, D. M. King, P. Kellam, S. D. Kemp, and B. A. Larder.1991. Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse tran-scriptase. Science253:1557–1559.
64.Svarovskaia, E. S., S. R. Cheslock, W. H. Zhang, W. S. Hu, and V. K. Pathak. 2003. Retroviral mutation rates and reverse transcriptase fidelity. Front. Biosci.8:d117–d134.
65.Swingler, S., P. Gallay, D. Camaur, J. Song, A. Abo, and D. Trono.1997. The Nef protein of human immunodeficiency virus type 1 enhances serine phos-phorylation of the viral matrix. J. Virol.71:4372–4377.
66.Tabor, S., and C. C. Richardson.1995. A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc. Natl. Acad. Sci. USA 92:6339–6343.
67.Tachedjian, G., D. J. Hooker, A. D. Gurusinghe, H. Bazmi, N. J. Deacon, J. Mellors, C. Birch, and J. Mills.1995. Characterisation of foscarnet-resistant strains of human immunodeficiency virus type 1. Virology212:58–68. 68.Tachedjian, G., J. Mellors, H. Bazmi, C. Birch, and J. Mills.1996.
Zidovu-dine resistance is suppressed by mutations conferring resistance of human immunodeficiency virus type 1 to foscarnet. J. Virol.70:7171–7181.
69.Tisdale, M., S. D. Kemp, N. R. Parry, and B. A. Larder.1993. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3⬘-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc. Natl. Acad. Sci. USA90:5653–5656.
70.Tsurumi, T., K. Maeno, and Y. Nishiyama.1987. A single-base change within the DNA polymerase locus of herpes simplex virus type 2 can confer resis-tance to aphidicolin. J. Virol.61:388–394.
71.Van Rompay, K. K., J. L. Greenier, M. L. Marthas, M. G. Otsyula, R. P. Tarara, C. J. Miller, and N. C. Pedersen.1997. A zidovudine-resistant simian immunodeficiency virus mutant with a Q151M mutation in reverse tran-scriptase causes AIDS in newborn macaques. Antimicrob. Agents Chemother. 41:278–283.
72.Vivet-Boudou, V., J. Didierjean, C. Isel, and R. Marquet.2006. Nucleoside and nucleotide inhibitors of HIV-1 replication. Cell. Mol. Life Sci.63:163– 186.
73.Wainberg, M. A., M. D. Miller, Y. Quan, H. Salomon, A. S. Mulato, P. D. Lamy, N. A. Margot, K. E. Anton, and J. M. Cherrington.1999. In vitro selection and characterization of HIV-1 with reduced susceptibility to PMPA. Antivir. Ther.4:87–94.
74.Weiss, K. K., S. J. Isaacs, N. H. Tran, E. T. Adman, and B. Kim.2000. Molecular architecture of the mutagenic active site of human immunodefi-ciency virus type 1 reverse transcriptase: roles of the beta 8-alpha E loop in fidelity, processivity, and substrate interactions. Biochemistry39:10684–10694. 75.White, K. L., N. A. Margot, J. K. Ly, J. M. Chen, A. S. Ray, M. Pavelko, R.
Wang, M. McDermott, S. Swaminathan, and M. D. Miller.2005. A combi-nation of decreased NRTI incorporation and decreased excision determines the resistance profile of HIV-1 K65R RT. AIDS19:1751–1760.
76.Xu, X., Y. Liu, S. Weiss, E. Arnold, S. G. Sarafianos, and J. Ding.2003. Molecular model of SARS coronavirus polymerase: implications for bio-chemical functions and drug design. Nucleic Acids Res.31:7117–7130.
on November 8, 2019 by guest
http://jvi.asm.org/