0022-538X/83/090478-09$02.00/0
Copyright ©1983, AmericanSocietyforMicrobiology
Mutations in the Major
DNA-Binding Protein Gene
Simplex Virus Type 1 Result in Increased Levels
Gene Expression
of
Herpes
of Viral
PAULJ.GODOWSKIANDDAVID M. KNIPE*
Department of Microbiology and MolecularGenetics,HarvardMedical School,Boston,Massachusetts02115
Received1 March1983/ Accepted 7 June 1983
We have examined the effect oftemperature-sensitive mutations in the herpes simplex virus 1 DNA-binding protein gene on viral gene expression. We have
found thatatthenonpermissive temperature, thesynthesis of certain immediate early, early, and late viral polypeptides was greater in cells infected with the temperature-sensitive mutantsthan incellsinfected with thewild-type virus. This effect was independent of the requirement for this viral protein for viral DNA
replication. The alteredrate ofsynthesis of viral proteinswas duetoa thermola-bilegeneproduct. Cells infected with thesemutants atthepermissivetemperature and then shifted tothe nonpermissive temperatureexhibited enhanced levels of viral gene expression. The addition of actinomycin D at the time of the
temperature shift prevented the alteration in viral protein synthesis. Therefore, continuing transcription is required for this change ingeneexpression. Northern blot analysis of cytoplasmic RNA showed that the steady-state level of specific viral transcripts expressed from parental virus genomes was greater in cells infected by these mutants at the nonpermissive temperature. These results indicate that the major DNA-bindingprotein of herpes simplex virustype1actsas a negative regulator of viral gene expression by affecting the abundance of
cytoplasmic viral mRNAs.
Theregulation of herpes simplex virus (HSV)
geneexpression isacomplexprocessdependent onboth cellular and viral factors.Upon infection
of the host cell, a limited portion of the viral genome is transcribed by the host cell RNA polymerase IItoyieldimmediateearlymRNAs. Thesearetranslatedtoyield the immediate early proteins. These proteins alter transcription or
processing of viral mRNA such thata newclass
of mRNAs is produced. The proteins encoded by these mRNAs, the early proteins, are
re-quired for viral DNA replication. After viral DNAreplication, the synthesis of certain viral proteins is initiated, whereas the synthesis of otherproteins is enhanced. Theseare
collective-ly termed the late viral proteins (29).
The immediateearly proteinsaresynthesized atmaximal rateswithin afew hours after
infec-tion of the host cell. Thesynthesis of the imme-diate early proteins is reduced concomitantly with theincrease in synthesis of early proteins. Theinvestigation of the kinetics and the
require-ments for synthesis of viral polypeptides led Honess and Roizman (13, 14) to propose that earlygeneproducts negatively regulate
immedi-ate early gene expression. The specific early geneproducts involved in thisprocess werenot
identified. By analysis of immediate early
tem-perature-sensitive mutants, Watson and
Clem-ents(34) and Dixon and Schaffer (9) showed that inactivation of the infected cell polypeptide 4 (ICP4)genefunction led toincreased synthesis of immediate early RNA and proteins,
respec-tively. Thus, both immediate early and early gene productsmay negatively regulate
immedi-ateearlygeneexpression.
We have been studying the maturation and function ofICP8, the major HSV DNA-binding protein. This early protein accumulates in the cell nucleus where it bindstoviral DNA(19, 20). ICP8 is an essential function for viral DNA
replication (6). ICP8 bindsmoretightlyto single-stranded DNA thantodouble-stranded DNA in vitro (18, 25).
The complementation group 1-1 is likely to
contain mutants defective in ICP8. Mutants in thisgroup havelesions that map in or near the
ICP8 coding sequences (6, 37). These mutants
encodemajor DNA-binding proteins that exhibit alteredantigenic properties (37)orDNA-binding
478
on November 10, 2019 by guest
http://jvi.asm.org/
HSV DNA-BINDING PROTEIN AND GENE EXPRESSION
activities at the nonpermissive temperature (20).
In thisreport, we examinedthe effect of these
defectsinICP8 on viralgeneexpression.
MATERIALS AND METHODS
Cells, viruses, and inhibitors. The procedures used for propagating and titrating viral stocks have been described (19). Stocks of temperature-sensitive virus-es and wild-type viruses were titrated in parallel at 33°C. The origins of HSV type 1 (HSV-1) mP and mPtsHAl have been described (6, 12). HSV-1 strains KOS1.1 andKOS1.ltsl3(reference 15; L. Holland, R. Sandri-Goldin, J. C. Glorioso, and M. Levine, manu-script in preparation) were provided by R. Sandri-Goldin and M. Levine, University of Michigan, Ann Arbor. Strains KOS and KOStsC7 (27, 28) were pro-vided by P. Schaffer, Dana-Farber Cancer Institute, Boston, Mass. Sodium phosphonoacetate (PAA) was a gift of Abbott Labs, N. Chicago,Ill.PAA was used at 400,ug/ml,aconcentration which totally inhibits viral DNA replication in Vero cells (M. Quinlan and D. Knipe,unpublisheddata). PAAwasadded at thetime of infection and maintained throughout the incubation andlabeling period. Actinomycin D was obtained from Calbiochem, La Jolla,Calif.
Labeling ofinfectedcells. Theinfection of Vero cell monolayers at amultiplicity of 20 PFU per cell and the labeling of these cultures with [35S]methionine (New England Nuclear Corp., Boston, Mass.) were per-formed in constant-emperature water baths as de-scribed previously (19). The 15-min labeling period was terminated by submerging the flasks in an ice bath. The cellswerewashed 3 times with phosphate-buffered saline at 0 to 4°C, scraped with a rubber policeman, and collected by centrifugation. The cell pelletwassuspended in electrophoresis sample buffer (19)containing 1 mM phenylmethylsulfonyl fluoride. Proteins from these sampleswereanalyzed by electro-phoresis in 9.25% diallyltartardiamide cross-linked polyacrylamide gelsasdescribedpreviously(19). Por-tions of eachsample containing equal quantities of cell lysatewereloadedontothegels. Theautoradiograms shown and those used for tracing were exposures within the linear response range for the film. The labeling of cultures for 5to30 min also showedlinear incorporation of label into viral polypeptide bands (datanotshown).Autoradiogramswerescannedwith
a Joyce-Loebl microdensitometer, and peak areas
weredetermined withaKeufel and Esser planimeter.
RNAisolationandcharacterization. Vero cell
mono-layerswereinfected with the indicated HSV-1strains andmaintained in culture mediumcontaining 400 ,ug of sodium PAA per ml. At 6 h postinfection, the cells wereharvested, and cytoplasmic RNA was isolated by
amodification of the method of Berk et al. (4). The cellswerelysedwith1%NonidetP-40(Sigma Chemi-calCo., St. Louis, Mo.) in0.15 MNaCl-0.02M Tris-hydrochloride (pH 7.9). Nuclei were separated from
thecytosol fraction bycentrifugationat2,000 xgfor
10minat4°C.Thecytosol fractionwaspoured intoan
equal volume ofasolutionof7Murea,0.35 MNaCl,
10 mMTris-hydrochloride (pH 7.4), 10 mMEDTA,
and4% SDS.This wasextracted twicewithanequal volume ofphenol (saturated with0.15 MNaCl, 10 mM Tris-hydrochloride (pH 7.6), and0.5 mMEDTA) and one-half volume ofchloroform-isoamyl alcohol (24:1).
Nucleic acids were precipitated by the addition of sodium acetate to 0.2 M and 2 volumes of ethanol followedby incubationat-20°Cfor 12 h. Samplesof each RNApreparation (10 or20Fg) were denatured andsubjectedtoelectrophoresis in 0.8% neutral
agar-ose gelscontaining 2.2 Mformaldehyde asdescribed previously (16), except thatethidium bromidewas not
included in the gel. The RNA was transferred to
nitrocellulose (Schleicher & Schuell Co., Keene, N.H.)in lOx SSC (1xSSC is 0.15 M NaClplus 15 mM sodiumcitrate [pH7.0]) (31). The filters were prein-cubated in 50% formamide (Fluka, Hauppauge, N.Y.)-5 x SSC-0.02% Denhardt solution (8)-S50 mM potassium phosphate buffer (pH 6.5)-250 ,ugof yeast RNA perml-0.25% sodiumdodecylsulfate for 17 hat 42°C. Hybridizationswereperformedfor 48 h with 32p-labeled DNAprobes (5 x 106 cpm pernitrocellulose strip) under these same conditions. The filters were
washedtwice in 2xSSC-0.1% sodium dodecyl sulfate
at roomtemperaturefor 30 min andoncein 0.1x SSC-0.1% sodium dodecyl sulfate at 50°C for 1 h in a circulating water bath. Autoradiography was per-formed withKodak XRP-5 filmat room temperature
or at -70°C with intensifying screens. Plasmid pBamHI-A' (7)was agiftof R.Costa and E. Wagner, UniversityofCalifornia,Irvine. Thisplasmidcontains the HSV-1 BamHI A' DNA fragment (map
coordi-nates 0.255 to 0.266) inserted in the BamHI site of pBR322. PlasmidpSG18SalA(20)containsanHSV-1 Sall DNAfragment(map coordinates 0.385to0.419, equivalenttotheSail 0fragmentof HSV-1 strain F DNA[21])inserted in the Sall site ofpBR325. Plasmid pSG-1 (10) was generously provided by M. Levine, University of Michigan, Ann Arbor. This plasmid contains the HSV-1 EcoRI JKDNAfragmentinserted in theEcoRI site of pBR325. The BamHI SPfragment is contained within the EcoRI JKfragmentandarises from thejunctionof the L and S components ofviral DNA(21).
Plasmid DNAs were isolated by CsCl equilibrium density gradient centrifugation oflysates from plas-mid-containing cells (5). The HSV-1 DNA inserts in theseplasmidswerereleased from thevectorDNAby digestion with the appropriate restriction enzyme. Restriction endonuclease digestions were performed under conditions specifiedby the supplier (New En-glandBiolabs, Beverly, Mass.). The DNAfragments containingtheHSV-1-specific sequenceswere isolat-ed by electroelution from neutral agarose gels (23). These were labeled by nick translation (26) with [32P]dCTP(800Ci/mmol;AmershamCorp., Arlington Heights,Ill.)to aspecific activityof 5 x
107
to7 x107
cpm/,lg.
RESULTS
Synthesisofviralproteinsincellsinfected with aDNA-binding proteinmutant.Themajor
DNA-binding protein, ICP8,encoded by the group 1-1 mutant tsl3 is thermolabilefor binding toviral DNA (20). Others have shown that group 1-1 mutants aredefective forviral DNAreplication (6,37;L. Hollandetal.,manuscriptin
prepara-tion). Viral DNAreplication enhances late gene
expression, and late gene products inhibit early
VOL.47,1983 479
on November 10, 2019 by guest
http://jvi.asm.org/
480 GODOWSKI AND KNIPE
B
39.50C
M ts ts
*A:i4
330C
M
t sts-
--.e.-,w
et'-r.
,:,... 1
-49Pk,, ~~~~I.I -!- .::4
..
[image:3.492.53.456.76.370.2]..:
FIG. 1. Comparison of theratesofsynthesis of viral proteinsincellsinfected with HSV-1 tsl3orts+virus in
the presence ofPAA. Cells were infected with tsl3 (ts) or KOS1.1 (ts+)virus ormock-infected (M)in the presenceof PAA. At the times indicated, the cellswerelabeledfor 15minwith 6,uCiof[35S]methionineperml.
Extractswereprepared, and the viral proteinswereanalyzed by gel electrophoresisasdescribed in thetext.An
autoradiogram of the gel is shown. Someviralpolypeptides (ICP)areindicated. Thetwoforms ofICP8, differing
intheirdegree of intramolecular disulfide bonding (17),arealso indicated.
gene expression (29). Thepurposeof this study
was to determine whether ICP8 affects viral
gene expression independently of the require-mentof thisprotein for viral DNAreplication.
We compared the amounts of viral proteins synthesized during pulse-labeling periods in cells infected with tsl3 or its wild-type parent,
strain KOS1.1. To control for any effect that DNA replication might have on viral gene
expression, the infected cultureswereincubated
withPAA, an inhibitor of the viral DNA poly-merase. Replicate cultures were infected with
KOS1.1 ts+ or tsl3 viruses and maintained in
mediumcontaining PAAat330 or39.5°C. At 1-h
intervals between 3to8 hpostinfection, the cells
werepulse labeled with [35S]methionine, and the
labeled viral polypeptides were analyzed by
electrophoresis and autoradiography. At 39.5°C, cells infected with tsl3 synthesized increased
amountsof certain viralproteins relative tothe cells infected with thets+ virus (Fig. 1A). These included the early ICPs 6, 8, and 36 and the late ICPs 5, 11, 15, and 25. In addition, the
immedi-ate early ICP4 was synthesized in greater
amountsandatlater timesin cellsinfectedwith tsl3. At the permissivetemperature (33°C), the
amounts of viral proteins synthesized in cells infected with tsl3 and thets+ virusweresimilar (Fig. 1B). Thus, under conditions in whichviral DNA replication was uniformly inhibited in all cultures, the effect of thetemperature-sensitive mutationwastoincrease the level ofexpression of several viralgenes.
From this experiment, we quantitated the
magnitude of the difference in thepeakratesof synthesis of viral polypeptidesinthemutantand wild-type infected cells. By microdensitometry and planimetry of the autoradiograms (Fig. 1),
we determined theamount of labeled ICP8. At 39.5°C,theamountofsynthesisof ICP8 in cells infected with ts13 wasgreater at all times than that observed in cells infected with ts+ virus. Thepeak rate ofsynthesisof ICP8 (5 h postin-fection) was approximately threefold greater in
the mutant-infectedcells(Fig. 2A).At33°C,the
amount of ICP8 synthesized in cells infected with themutant and ts+ viruswas similar (Fig.
2B).
A
J. VIROL.
.-Q-MAP
-.,
.1 II
1, t, :, -,;"i, nI,.- '..!..
on November 10, 2019 by guest
http://jvi.asm.org/
HSV DNA-BINDING PROTEIN AND GENE EXPRESSION 481
A B
200
160
120
80
;40 =
0
IL_ ,,
0 c D
c
120-40
CL ~~~~~~a.a A C a a
HoursPostInfection
FIG. 2. Ratesofsynthesisof ICP8 in cells infected withDNA-negative mutants orts+ viruses. Infected cultureswerelabeledfor 15 minatthe timesindicated, and viral proteins were analyzed as in Fig. 1. The intensityof the ICP8 bandswasdeterminedby micro-densitometry andplanimetry. (AandB)Symbols: 0,
cells infected with tsl3; *, cells infected with ts+ virus. (C and D)Symbols:0,cells infected withtsC7; *, cellsinfected with KOS ts+. Cellswereincubated
at39.50C (A andC)or33°C (BandD).
Comparison of late gene
expression
by ICP8mutants and replicating wild-type virus. The
expression
of HSV lategenes is- inhibited whenviral DNA replication is blocked (29). The
re-sultspresented abovedemonstratethatthe
inhi-bition of late gene
expression
fromparental
DNAmoleculeswasatleast
partially
overcomeby mutations in the
major
DNA-bindingprotein.
It wasof interest tocomparetheamountof late
gene
expression by
ICP8mutantswiththatofareplicating wild-type virus. Replicate cultures were infected at
39.5°C
with the ICP8 mutanttsHAl (6,
20)
or its parent strain mP in the presence orabsence ofPAA. At 6 hpostinfec-tion, viralpolypeptide synthesiswas
analyzed.
In cultures incubated with PAA, the amount
of
synthesis
ofearlypolypeptides
(e.g., ICPs6,
8, and 36), late
polypeptides
(e.g., ICPs5,
11, 15, and 25) and the immediateearly
ICP4 was greater in cellsinfectedwith tsHAl(Fig.
3,lanes 2and4). Thus, this ICP8 mutant also overpro-duced viralpolypeptides
at 39.50C. In culturesincubatedwithout PAA, synthesis of early pro-teins and of the immediate early ICP4 was greater in cells infectedwith tsHAl than in cells
infected with the ts+ virus. However, synthesis of the late ICPs 5, 15, and 25 was greatest in cells
infected with the ts+ virus (Fig. 3). Thus, al-though the leveloflate gene expression by ICP8 mutants was greaterthan that observed with a nonreplicating wild-type virus, it was not as great as that observed in cells in which viral DNA replication was occuring. These results extendearlier studies by Conley et al. (6)
show-ingthat ICP8 mutants are defective in the
accu-mulation oflategene products.
Characterization of other mutantsdefective for DNA replication. To determine whether an in-creased level of viral protein synthesis was a general characteristic of all mutants defective
forDNA replication, we examined the kinetics
.sL
+5
ti-f)
...---t,~
[image:4.492.45.239.61.316.2]: t.
FIG. 3. Synthesisof viralproteinsincellsinfected with HSV-1 tsHAl or is+ virus in the presence or absence of PAA. At 6 h postinfection at39.5'C, the cellswerelabeled for 15 min with
(35S]methionine,
and viralproteins
wereanalyzedasinFig.
1.Thesamplesshownwerefrom infection with tsHAl (ts)ormP(ts') with(+)orwithout(-)PAA.
VOL.47, 1983
m",-,:,-I--" "
Mww"Will... ..ogr
..Ipppw`
6.894-1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.492.301.400.250.609.2]of protein synthesis in cells infected with the
HSV-1 mutantKOStsC7. Thismutanthas been shownto encode athermolabileDNA
polymer-ase, and it fails to replicate its DNA at the
nonpermissivetemperature(2). Asamemberof complementationgroup1-3,itcomplements the
ICP8 mutant tsHAl (36). At 39.5 or 33°C, the amount of synthesis of ICP8 directed by the mutant orthe wild-type virus was similar(Fig. 2CandD). The ratesofsynthesis of otherviral
proteins in cells infected with tsC7 andts+virus
werealsosimilar(datanotshown). Theseresults
indicatethat amutational block in DNA
replica-tion alone is not sufficient to account for the increased rates of viral protein synthesis de-scribed above.
Changes inviralprotein synthesisafter a tem-perature shift. The major DNA-binding protein
encoded by tsl3 and tsHAl is thermolabile for
binding
toviral DNAin vivo. TheICP8 encoded by these mutants is released from viral DNAwithin 30minaftera shifttothenonpermissive
temperature (20). We determined whether the
genefunction involved inregulating the
expres-sion of viral genes could be inactivated by shifting cellstothe
nonpermissive
temperature.CellswereinfectedwithtsHAl orts+ virus and
incubatedat33°C.At 4h
postinfection,
after the initiation of early viralprotein synthesis,
the cellswere shiftedto39.5°C. Infected cells werelabeledatvarious times after the shift. Immedi-ately after the shift to
39.5°C,
viral protein synthesis was similar in cells infected with tsHAlorts+ virus(Fig.
4). At1hafter the shiftto 39.5°C, the rates of viral
protein
synthesiswere increased in cultures infected with either
virus(Fig.4).This increasewasalso observed in cultures left at 33°C (not
shown),
and thus this change wasnotrelatedtothetemperature shift.The levels of viral
protein synthesis
did notincrease further in cultures infected with ts+
virus (Fig. 4). However, the synthesis of viral proteins didincrease further in cultures infected with tsHAl. By 3 h afterthe temperature shift, thesynthesis of
certain
viralproteins
(e.g., ICPs4, 5, 6, 8, and 11)was enhanced in the
mutant-infected cells. The synthesis of ICP8 was
ap-proximately threefold greater than in cells in-fected with ts+ virus. Synthesis of ICP4 was detected only in cultures infected with tsHAl
and only after3 to 5 h at 39.5°C. These results indicate that tsHAl encodes a gene product
which either is thermolabile or has a very short half-life. Thedeficiencyof thisproductresults in
increased ratesof viral protein synthesis. Simi-lar results were obtained in temperature shift experiments involvingtsl3 (data notshown).
To furtherexamine the mechanismby which
ICP8 affects viral gene expression, we deter-mined whether the alteration in viral protein
J. VIROL.
t
ts+
L-i.
--4
-3
-11
--- V
J :3 05O 3
[image:5.492.265.447.72.467.2]hrc) ovlowino4 shift 5 FIG. 4. Synthesis of viral proteins in infected cells after a temperature shift. Cells were infected with tsHAl(ts) ormP (ts+) at 33°C in the presence of PAA. At 4 h postinfection, the cultures were shifted to 39.50C.Atthetimes indicated after the shift, the cells were labeled with [35S]methionine, and the labeled proteins wereanalyzed as in Fig. 1.
synthesis after a temperature shift could be blocked by inhibiting transcription. In this
ex-periment, cells were infected with tsl3 or ts+
virusandincubatedat33°C for5h.Actinomycin
D was added to one halfof the cultures. All cultures were thenshiftedto39.50C. Theratesof
synthesisofICP8 were determined atthis time orafter a 4-hincubation periodatthis tempera-ture. Cells infected with ts+ virus synthesized
similar amountsof ICP8
immediately
after and 4 hafter the shift to39.5°C(Table 1). Cellsinfect-ed with tsl3 showed athreefoldincrease in the rate ofsynthesis of ICP8 after a4-h incubation
on November 10, 2019 by guest
http://jvi.asm.org/
HSV DNA-BINDING PROTEIN AND GENE EXPRESSION 483
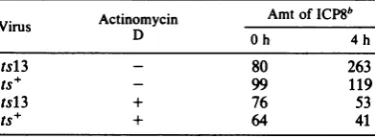
TABLE 1. Effect of actinomycin D on the overproduction of ICP8 after a temperature shifta
Virus Actinomycin Amt of
ICP8b
D Oh 4h
tsl3 - 80 263
ts+ - 99 119
tsl3 + 76 53
ts+ + 64 41
aCellswere infectedat 33°C for 5 h. At this time,
actinomycin D was added to one half of the cultures to aconcentration of 10 ,ug/ml. All cultures were then shifted to39°C for either 0 or 4 h and labeled for 15 min with[35S]methionine at 39°C.
b Theamountof ICP8 in arbitrary units determined by planimetry. These values represent the average of triplicate cultures.
period at39.5°C. However,theaddition of acti-nomycin D tocultures infected withts+ ortsl3 virus caused the synthesis of ICP8tobereduced to 64 and 70%o, respectively, of the initial rate by 4hafterthe temperatureshift.Thus,continuing transcription is needed fortheenhancedratesof ICP8 synthesis in cells infected with tsl3 at
39.5°C.These resultsalsoindicate that the
func-tional half-life of the ICP8 mRNA is similarin cellsinfected with tsl3 and ts+ viruses.
Accumulation of viral mRNA. Theobservation thattheinhibitionof transcription prevented the increased rates of protein synthesis in
mutant-infected cells suggested that the ICP8 function
may affect the level of viral mRNA accumula-tion. Weassayed the level of
viral-specific
cyto-plasmic RNA from cells infected with the mu-tant orts+ virusby Northern blot
hybridization.
Ashybridization probes,weused cloned HSV-1
DNA fragments derived from regions of the
genomethatencode ICPs
4,
5,
or8. The BamHI SPDNAfragmenthybridizes
witha4.7-kilobase (kb)transcript
that encodes ICP4(1, 22,
35).This probe alsocontainssequences
homolo-gous tothe3.0-kbtranscript encodingthe imme-diate
early
ICPO (1, 22). The BamHI A' DNAfragment hybridizes
to a major transcript that encodes ICP5 and to several minortranscripts(reference
7; E. Wagner,personal
communica-tion).
ThepSG18SalA DNA fragment hybridizesto a
major transcript
of4.5kb. Invitrotransla-tion of mRNA that hybridizes to the pSG18SalA DNAfragment yieldsa polypeptidewith a
mo-lecular weight of approximately 125,000. This
polypeptide is immunoprecipitatedwith a
mono-colonal antibody directed against ICP8 (L.
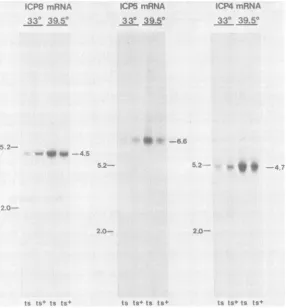
Ra-field and D. Knipe, manuscript inpreparation). With the ICP8 mRNA probe, weobserved hy-bridizationto amajor transcript of4.5 kb as well aswithseveralminor transcripts.The amountof all these transcriptswasgreaterincellsinfected
with
tsl3
at39.50C than incells
infected with thets+ virus. At 33°C, the amount of these tran-scripts was greaterin cells infected with the
ts'
virus (Fig. 5). Similarly, theamountsof the 6.6-kb transcript detected with the ICP5 mRNA probeandthe4.7-kbtranscript detected with the ICP4mRNAprobeweregreaterin cells infectedwith tsl3 at 39.50C (Fig. 5). With the ICP4 mRNA probe, we detected very low quantities ofthe3.0-kb mRNA that encodes ICPO in RNA isolated from cells infected with tsl3 or
ts+
virus.Therefore,wecouldnotdetermine
wheth-erthis mRNAwasalsooverproduced. Whenwe
compared the levels of
cytoplasmic
polyadeny-lated transcripts isolated from these cultures with these same probes, similar results were
obtained (P. J. Godowski and D.
Knipe,
datanotshown).Thus,mutations in the
major
DNA-binding protein result in an increasedsteady
statelevel of the mRNA for ICPs
4,
5,
and8.DISCUSSION
Wehaveshown that cells infected with certain
HSV-1 group 1-1 mutantsexhibit increased lev-els of viral geneexpression.The members of this groupcontain mutations thatmapinor near the
coding sequences of the major DNA-binding protein. These mutations lead to alterations in
thecellular localization,DNA binding activities, orantigenic properties ofthe major
DNA-bind-ingprotein(20, 36). It therefore seems likely that
the alterations in viral gene expression are a
resultof the defect intheDNA-binding protein. As reported previously, ICP8exerts apositive
effect
on late gene expression, probably by promoting viral DNA replication (6). However,when viralDNAreplication was inhibited in all
infected cultures by incubation with PAA, the
DNA-bindingprotein inhibited the expression of
representative immediate early (ICP4), early
(ICPs6,8, and 36), and late (ICPs 5, 11, and 15)
proteins. Thus, under these conditions ICP8
acted as a negative regulator of viral gene
expression.
Wemeasured the rates of protein synthesis in
infected cells by determining the amount of incorporation of
[35S]methionine
into viralpoly-peptide bands
during
a short pulseperiod.
Thealtered levelsof viralgeneexpressionobserved
in cells infected with ICP8 mutants probably
reflect true differences in the amount of synthe-sis ofthese proteins because (i) viral proteins
accumulated to higher levels in these cells as
determined by Coomassie blue staining ofthe
gels (data not shown), and (ii) the level of cytoplasmicviral mRNA was increased in these cells. Thus, the differences in labeling of
poly-peptides are probably not explained by trivial
differences such as the uptake oflabelfromthe extracellular medium.
Role ofICP8 in the cascade regulation of HSV VOL.47,1983
on November 10, 2019 by guest
http://jvi.asm.org/
!CPS mr-4A
3
333°
39.5)
ICP4 nRINA
33'
'.39..
.5.
2.0-*S . ..' '
FIG. 5. Northern blot analysis of specifictranscripts from cells infected withwild-type or mutant virus. Samples of total cytoplasmic RNA from cells infected with tsl3 (ts) or KOS1.1 (ts+) at 33 or 39.5°C were
subjectedtoelectrophoresis informaldehyde-agarosegels and transferredtonitrocelluloseasdescribed in the
text.Todetect ICP8 mRNA, 20-,ugsamples ofRNA onthe blotwereprobed with32P-labeledpSG18SalADNA.
Autoradiographywasperformedat roomtemperature.TodetectICP5mRNA orICP4 mRNA, 10-,ug samples of RNA onblots were probed with labeledBamHIA' DNA orBamHlSP DNA,respectively. The size of themajor RNA species detected with eachprobe is indicated in kilobases ontherightof eachpanel. These sizeswere
determined relativetomigration of28S(5.2 kb)and18S (2.0kb)rRNAmarkersindicatedontheleft of each panel.
geneexpression. Intheir model forcascade
regu-lation of HSV gene expression, Honess and Roizman (13, 14) proposed that early
proteins
act as
negative
regulators of immediateearly
gene expression. We report evidence here that the ICP8 gene function affects the level of expression ofICP4, animmediateearlyprotein. Thus, this isthe firstearly genefunction tobe
identified that regulates the immediate early to
earlygenetransition.
ICP8is neededfor viral DNAreplicationand late gene expression. However, ICP8 inhibits
late gene expression from parental viral ge-nomes when viral DNA replication cannot oc-cur. Thelate genes encode the
majority
ofthevirion structural polypeptides (29). Thus, this
functionprevents the synthesis ofvirion struc-turalproteins untilprogeny DNAmolecules are
available forencapsidation.
Mechanism of action of ICP8 on viral gene
expression. We have shown that theamount of
mRNA encoding ICPs 4, 5, and 8 is higherin cellsinfectedwithICP8mutants.Thus,the ICP8
functionregulates thecytoplasmicaccumulation
ofviral mRNAs. We did not detectadifference inthefunctionalstability of viralmRNAsin cells
infected with these mutants in the presence of
actinomycinD.These data suggest that the ICP8
function regulates the transcription or
metabo-lism ofnewly synthesizedmRNA. Alternative-ly,this result couldreflectaneedfor continuous synthesis ofa product that acts together with ICP8 to control viral geneexpression.
There are two general models to explainthe
effect ofthe ICP8functiononviral gene
expres-sion.Inthefirst, ICP8 inhibits theexpression of
the immediate earlygenes. Lower levelsofthe
immediate early proteins wouldthen cause
low-17 ,PR %.^-,p
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.492.114.401.78.387.2]HSV DNA-BINDING PROTEIN AND GENE EXPRESSION
erlevels of expressionofallthe laterviral gene products. In this model, ICP8 would affect its
own expression only indirectly. In the second model, ICP8inhibitstheexpression of the
differ-ent viralgenes by a common mechanism. This
could be mediated by the bindingof ICP8 to viral
DNA or to mRNA transcripts, for example. In thismodel, ICP8affects its own expression more directly. This second model is supported by the observation that after a temperature shift, the expressionof several viral genes, including that encoding protein 4, increased coordinately. Ei-therofthe two models proposed could
incorpo-ratetheobservationthatafter a shift to39.5°C of cells infected with tsHAl or tsl3, ICP8 is re-leased from viral DNA (20).
Similar alterations in the level of viral gene expressionareobserved instrains of adenovirus that encode a defective 72K DNA-binding pro-tein. This protein, like the HSV-1 ICP8, is essential forviralDNAreplication (33) and has a high affinity for single-stranded DNA (30). The 72Kprotein plays amultifunctional role in
con-trolling adenovirusgeneexpression. Itrepresses the transcription ofearly region 4 (24) and has been implicated in controlling the stability of
early viral mRNAs (3). Mutations that alleviate
the block ofadenovirus lategene expression in monkey kidney cells map in the viral gene for
the 72K protein (17). The simian virus 40 T antigen, an essential protein for simian virus 40 DNA replication, alsonegatively regulatesearly
viral transcription. Both oftheseevents involve the binding of T antigen to specific sequences
near the origin ofreplication (11, 32). Further
studies are in progressto determine the precise
mechanism by which ICP8 affects HSV gene
expression.
ACKNOWLEDGMENTS
We thank LoriRafield for technical advice concerning the
Northernblothybridizations. We also thank Anne Spang and Margaret Levin for their assistance and Rosemary Bacco for thepreparationof the manuscript.
This work was supported by Public Health Service grant CA26345 from the National Cancer Institute. P.J.G. was a
predoctoral trainee supported by the National Institutes of Health training grant 5T32GM071%. D.M.K. is a Cancer Research Scholarof the Massachusetts Division of the Ameri-canCancer Society.
LITERATURE CITED
1. Anderson, K. P., R. H. Costa, L. E.Holland, and E. K. Wagner. 1980. Characterization of herpes simplex virus type1 RNApresent in the absence of de novo protein synthesis. J. Virol. 34:9-27.
2. Aron, G. M., D. J. M.Purifoy, and P. A. Schaffer. 1975. DNA synthesis and DNA polymerase activity of herpes simplex virus type 1 temperature-sensitive mutants. J. Virol.16:498-507.
3. Babich, A., and J. R.Nevins. 1981. The stability of early adenovirus mRNA iscontrolled by the viral 72 kd DNA-binding protein. Cell 26:371-379.
4. Berk, A. J., F. Lee, T. Harrison,J.Wiiams, and P.A. Sharp. 1979. Pre-early adenovirus 5gene product regu-lates synthesis of early viral messenger RNAs. Cell 17:935-944.
5. Clewell,D. B., and D. R. Helinski.1972.Effectofgrowth conditions on the formation of therelaxation complexof supercoiled ColEl deoxyribonucleic acid and protein in Escherichia coli. J. Bacteriol. 110:1135-1146.
6. Conley, A. J., D. M. Knipe, P. C. Jones, and B.Rolzman. 1981. Molecular genetics of herpes simplex virus. VII. Characterization ofa temperature-sensitive mutant pro-duced by in vitro mutagenesis and defective in DNA synthesis and accumulation of gamma polypeptides. J. Virol. 37:191-206.
7. Costa, R. J., B. Gadyathri, K. P. Anderson, B. H. Gay-lord, and E. K. Wagner. 1981. Characterization of a major late herpes simplex virus type 1 mRNA. J. Virol. 38:483-4%.
8. Denhardt, D. T. 1966. A membranefiltertechnique for the detection of complementary DNA. Biochem. Biophys. Res. Commun. 23:641-646.
9. Dixon, R. A. F., and P. A. Schafer. 1980. Fine-structure mapping and functional analysis of temperature-sensitive mutants in the gene encoding the herpes simplex virus type 1 immediate early protein VP 175. J. Virol. 36:189-203.
10. Goldin, A. L., R. M.Sandri-Goldin,M.Levine,and J. C. Glorloso. 1981. Cloning of herpes simplex virus type 1 sequences representing the whole genome. J. Virol. 38:50-58.
11. Hansen, U., D. G. Tenen, D. M. Livingston, and P. A. Sharp. 1981. T antigen repression of SV40 early transcrip-tion from two promoters. Cell 27:603-612.
12. Hoggan, M. D., and B.Roizman. 1959. The isolation and properties of a variant of herpes simplex producing multi-nucleatedgiant cells in monolayer cultures in the presence of antibody. Am. J. Hyg. 70:208-219.
13. Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirusmacromolecular synthesis. I. Cascade regula-tion of thesynthesis of three groups of proteins. J. Virol. 14:8-19.
14. Honess, R. W., and B. R. Roizman. 1975. Regulation of herpesvirus macromolecular synthesis: sequential transi-tion of polypeptide synthesis requires functransi-tional viral polypeptides. Proc. Natl. Acad. Sci. U.S.A. 72:1276-1280.
15. Hughes, R. G., and W. H. Munyon. 1975. Temperature-sensitivemutants of HSV 1 defective in lysisbut not in transformation. J. Virol. 16:275-283.
16. Hummel,M., and E.Kieff.1982. Epstein-Barr virus RNA VIII: viral RNA in permissively infected B95-8 cells. J. Virol. 43:262-272.
17. Klessig, D. F., and T. Grodzicker. 1979. Mutations that allow human Ad2 and AdS to express late genes in monkey cells map in the viral gene encoding the 72K DNAbinding protein. Cell 17:957-966.
18. Knipe, D. M., M. P. Quinlan, and A. E. Spang. 1982. Characterization of twoconformational forms of the ma-jor DNA-binding proteinencoded by herpes simplex virus
1. J.Virol.44:736-741.
19. Knipe,D. M., and A. E. Spang. Definition of a series of stagesin the association of two herpesviral proteins with the cell nucleus. J. Virol. 43:314-324.
20. Lee, C. K.,and D. M.Knipe.1983. Thermolabile in vivo DNA-binding activity associatedwith a protein encoded by mutants of herpes simplex virus type 1. J. Virol. 46:909-919.
21. Locker, H., and N.Frenkel. 1979. BamI,KpnI,andSall restriction enzyme mapsof the DNAs of herpes simplex virus strains Justin andF: occurrence of heterogeneities in definedregions ofthe viral DNA. J. Virol. 32:429-441. 22. Mackem,S., and B.Roizman.1980. Regulation of
herpes-virus macromolecular synthesis: transcription-initiation sites and domains of alphagenes. Proc. Natl. Acad. Sci. U.S.A. 77:7122-7126.
485 VOL.47,1983
on November 10, 2019 by guest
http://jvi.asm.org/
23. McDonell, M.W., M. N. Simon, and F. W.Studier. 1977. Analysis ofrestriction fragments of T7 DNA and determi-nationof molecular weights by electrophoresis in neutral andalkaline gels. J. Mol. Biol. 110:119-146.
24. Nevins, J. R.,and J. J.Winkler.1980. Regulation of early adenovirus transcription:aprotein product of early region 2specificallyrepressesregion 4 transcription. Proc. Natl. Acad.Sci. U.S.A. 77:1893-1897.
25. Powell, K. L., and D. J. M.Purifoy. 1976.DNA-binding proteins of cells infected by herpes simplex virustype1 andtype2.Intervirology 7:225-239.
26.Rigby, P. W. J., M. Dleckmann, C. Rhodes,andP. Berg. 1977. Labeling deoxyribonucleic acid to high specific activityin vitroby nick translation with DNA polymerase 1.J.Mol. Biol. 113:237-251.
27.Schaffer, P. A., G. M. Aron,N.Blawal,andM. Benyesh-Melnick. 1973. Temperature-sensitive mutants ofherpes simplex virus type 1: isolation, complementation and partialcharacterization. Virology 52:57-71.
28. Smith, K.0.1964.Relationship between the envelope and
theinfectivity of herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814-816.
29. Spear, P.,andB.Roizman. 1980, Herpessimplex virus,p.
615-745. In J. Tooze (ed.), DNA tumor viruses. Cold Spring HarborLaboratory, Cold Spring Harbor, N.Y. 30. Supwara,K., Z.Gilead,and M.Greene. 1977.
Purifica-J. VIROL.
tion and molecularcharacterizationof adenovirus type 2
DNA-binding protein.J.Virol. 21:338-346.
31. Thomas, P. 1980. Hybridization ofdenatured RNA and smallDNAfragmentstransferred tonitrocellulose. Proc. Natl. Acad. Sci. U.S.A. 77:5202-5205.
32. T3hn,R.1978. ThebindingsiteonSV40 DNA fora
T-antigen-related protein.Celi 13:165-179.
33. van der Vilet, P. C., and J.S. Sussenbach. 1975. An adenovirustype 5 genefunctionrequiredfor initiation of viral DNAreplication. Virology67:415-426.
34. Watson, R. J., and J. B. Clements.1980. A herpessimplex virustype 1functioncontinuouslyrequiredforearlyand late virus RNAsynthesis.Nature.(London)285:329-330. 35. Watson, R. J., C. M. Preston, and J. B. Clements. 1979.
Separationandcharacterizationofherpes simplexvirus type 1immediateearlymRNAs. J. Virol. 31:42-52. 36. Weller, S. K., K. J. Lee, D. J. Sabourin, and P. A.
Schaffer. 1983. Geneticanalysisoftemperature-sensitive mutants which define the gene for the major herpes
simplex virus type 1 DNA-binding protein. J. Virol. 45:354-366.
37. Wolf, H. and B.Roizman. 1977. Theregulation of structur-alpolypeptide synthesisinherpes simplexvirus type land 2 infectedcells,p. 327. In G. DeThe,W.Henle,and F.
Rapp (ed.), Oncogenesis and herpesviruses III. Interna-tional Agency forResearch on Cancer,Lyon.