Copyright © 1992, American SocietyforMicrobiology
Identification and Characterization of the
Herpes
Simplex Virus
Type
1Virion Protein
Encoded by
the
UL35 Open Reading
Frame
DAVID S. McNABBANDRICHARDJ. COURTNEYt*
Department of Biochemistry andMolecularBiology, Louisiana State UniversityMedicalCenter, 1501 Kings Highway, P.O. Box 33932, Shreveport, Louisiana 71130-3932
Received 10 December1991/Accepted12February 1992
TheUL35openreadingframe (ORF)of herpessimplexvirustype 1(HSV-1) has beenpredictedfrom DNA
sequence analysistoencode asmallpolypeptidewith amolecularweight of12,095.Wehaveinvestigated the
protein productof the UL35 ORF by usingatrpE-UL35genefusiontoproduceacorrespondingfusionprotein
in Escherichia coli. The TrpE-UL35 chimeric protein was subsequently isolated and used as a source of
immunogen for theproduction of rabbitpolyclonal antiserum directed againstthe UL35 geneproduct.The
TrpE-UL35 antiserum was found to recognize a 12-kDa protein which was specifically present in HSV-1-infected cells. Byutilizingthe TrpE-UL35 antiserum, the kinetics ofsynthesis of the UL35 geneproductwas
examined, and these studies indicate that UL35 is expressed as a -Y2 (true late) gene. The 12-kDa protein recognized by theTrpE-UL35antiserumwasassociated with purified HSV-1virionsandtypeAand Bcapsids, suggestingthatthe UL35 ORFmayencode the12-kDacapsidproteinvariablydesignated p12, NC7,orVP26.
Toconfirmthisassignment,immunoprecipitation and immunoblotting studieswereperformedtodemonstrate that theTrpE-UL35antiserum reactswiththesamepolypeptide as anantiserumdirectedagainstthepurified
p12 capsid protein (anti-NC7) (G. H. Cohen, M. Ponce de Leon, H. Diggelmann, W. C. Lawrence, S. K. Vernon,and R.J.Eisenberg, J.Virol.34:521-531, 1980). Furthermore, the anti-NC7serumwasalso found
to reactwith theTrpE-UL35 chimeric protein isolated fromE. coli, providing additional evidence that the UL35geneencodesp12. Onthebasis of thesestudies,weconcludethatUL35representsatruelategenewhich
encodes the 12-kDacapsid proteinof HSV-1.
On the basis ofmorphological studies,theherpes simplex virus type 1 (HSV-1) virions have four distinct structural regions: an electron-opaque core which contains the viral
genome; an icosahedral capsid which encloses the
DNA-containing core; an electron-dense material, referred to as
the tegument, which surrounds the nucleocapsid; and an outerenvelope containingthe viralglycoproteins (37, 38, 45, 48). Amongthe 30 or more structural proteins which
com-prise the HSV-1virions (9, 21, 34, 44), onlyseven
polypep-tides are structural components of the nucleocapsids.
Gib-son and Roizman (16) identified six proteins (designated VP5, VP19c, VP21, VP22a, VP23, and VP24)ascomponents of capsids isolated from the nuclei of HSV-infected cells. Subsequently, Heilman et al. (20) and Cohen et al. (11) identifiedaseventhcapsid protein (designated p12 and NC7,
respectively) with amolecular weight of 12,000. The genes
encoding VP5 (13), VP19c (36, 49), VP22a (35), and VP23 (36) have been identified previously and are designated
UL19, UL38, UL26, and UL18, respectively. It has been suggested that VP21 may represent a processed form of
VP22a(8)and thusmayalso beencodedby the UL26gene.
If this is proven to be true, VP24 and p12 are the only remainingcapsid proteinswhich havenotbeenassignedtoa
specificgene. Theidentification of thegenesencoding each
of the capsid proteins is an essential step toward our
understanding ofherpesvirus assembly. Furthermore, such information isnecessary for designing experimental studies
* Correspondingauthor.
tPresent address:DepartmentofMicrobiologyandImmunology, PennsylvaniaStateUniversity CollegeofMedicine,500University Drive,P.O. Box850, Hershey, PA 17033.
of the function(s) of these structural polypeptides using molecular techniques.
HSV-1 has a linear double-stranded DNA genome of approximately 152 kb (28). The knowledge of the complete
DNAsequence of the HSV-1 genome(28)and the mapping
of thegenomiclocations of variousHSV-specific transcripts (see reference 47 for a summary) have greatly assisted
investigatorsinidentifying variousHSV-specificgene prod-ucts. Computer-assisted analyses of the DNA sequence of theHSVgenomehavesuggestedthepresence ofat least 72 separate open reading frames (ORFs) (28). Approximately one-third of the ORFs remain to be characterized with respect to the polypeptides they encode and to determine whethertheyarestructuralornonstructuralproteins. Inthe
studiesreported herein,we wereinterested inidentifyingthe
gene encodingthep12 capsid protein.
As notedabove, previous studieshavedemonstratedthat p12representsa12-kDaproteinwhich isassociated withthe
capsids isolated from the nuclei ofHSV-infected cells (11, 20). Recent studies by Newcombe and Brown (32) have demonstrated that p12 (termed VP26) and VP22a can be
quantitatively removed fromcapsids by extraction with 2.0 Mguanidine hydrochloride. Electronmicroscopic examina-tionof the extractedcapsidsrevealed the selective removal of the capsid pentonsand the internal coreproteins. Since
VP22a may reside within the capsid (31, 32), these results implythat p12maybe localized in oraround the pentonsof theHSVcapsids.Whetherp12 actuallyformsthe pentonsor
islocalized in closeproximitytothe pentons remains
unre-solved. Interestingly, previous studies by Knopf and Kaerner (25) suggest that a 12-kDa basic phosphoprotein
(termed BP2),which is present in HSV-1 virions and asso-2653
on November 9, 2019 by guest
http://jvi.asm.org/
2654 McNABB AND COURTNEY
ciatedwith chromatin isolated from HSV-1 infected cells is equivalent to p12 or NC7.
Since the gene encoding p12 had not been identified, we took advantage of these observations concerning the char-acteristics of BP2 and/orp12 to try to determine whether any of the uncharacterized ORFs within the HSV genome could encode a 12-kDa basic protein. Computer-assisted analyses of thepotential ORFs within the HSV-1 genome suggest that theUL35 ORF might encode such a protein. The UL35 ORF is predicted to encode a protein with a molecular weight of 12,095(28), which is consistent with the apparent molecular weight of p12 (11, 20). Additionally, computer analysis indicates that the predicted amino acid sequence of the UL35 gene product would produce a polypeptide with a calculated pl of 11.6 (29), which is consistent with the basic properties of BP2. The UL35 ORF is located at approxi-mately 0.463 to 0.465 map units on the HSV-1 genome. Analyses of HSV-1 transcripts located within this region of the viral genome suggest that the UL33, UL34, and UL35 ORFs are transcribed late in infection as a nested set of 3'-coterminal mRNAs (47); however, precise mRNA map-pingstudies need to be performed to confirm this observa-tion.Nevertheless, these results indicatethat the UL35 ORF represents a gene which is actively transcribed. Moreover, its appearance late in infection wopld be consistent with encoding a structural protein of the virion. On the basis of these observations, we reasoned that UL35 represented an attractive candidate for the gene encoding the p12 capsid protein of HSV-1.
In this article, wereport the expression of the UL35 ORF inEscherichiacoli as aTrpE-UL35chimericprotein and the use ofthis fusionprotein asanantigen for theproductionof a polyclonal antiserum which specifically recognizes the protein productof theUL35 ORF in HSV-infected cells. The UL35 gene product had an apparent molecular weight of 12,000, which is consistent with the calculated molecular weight of 12,095. The TrpE-UL35 antiserumwas also used to examine the kineticsof synthesis of the UL35 gene, and these studies revealed that the
UL35
gene product is syn-thesized as a true late (Y2) gene product. Immunoblottingexperiments also revealed that the 12-kDa protein recog-nized by the TrpE-UL35 antiserum was present in purified
HSV-1virions as well as type A andBcapsids. These results wereconsistent with thehypothesis that the UL35 gene may encode the 12-kDa capsid protein. This hypothesis was subsequently confirmedbydemonstrating the cross-reactiv-ity of anti-NC7 antiserum (prepared against the purified
12-kDacapsid protein) and the TrpE-UL35 antiserum.
MATERIALSANDMETHODS
Cell culture and virus. HEp-2 cells andhuman embryonic
lung fibroblasts (MRC-5) cellswere grown inEagle's mini-malessential medium containing0.075%sodium bicarbonate and supplemented with 10% newborn calf serum and 10% fetal calf serum, respectively. Stocks of the KOS strain of HSV-1 were prepared inMRC-5 cells, and all virus titrations wereconducted on African green monkey kidney (Vero) cell monolayers as previously described (6).
Enzymes and chemicals. Restriction endonucleases and DNA-modifying enzymes were obtained from Bethesda Re-search Laboratories, Inc. (Gaithersburg, Md.), New En-glandBiolabs (Beverly, Mass.), or Promega Corp. (Madison, Wis.) and were used according to the manufacturers' spec-ifications. Radiochemicals were purchased from Dupont, NEN Research Products (Boston, Mass.). Phosphonoacetic
acid(PAA),Coomassie brilliant blue R-250, and
3-3-indole-acrylic acid were obtained from Sigma Chemical Co. (St.
Louis, Mo.).
Antiserumreagents.Antisera used in thesestudies include hyperimmunemonospecific rabbit antisera toglycoproteins
BandC (14), ICP8(15), and VP5(29).Antiserumtothep12
capsid protein (anti-NC7)was kindly providedby Gary H. Cohen and Roselyn J. Eisenberg (Universityof
Pennsylva-nia, Philadelphia) and has been previouslydescribed (11).
Construction of a TrpE-UL35 expression plasmid. The pATH21 plasmid, encodinga truncated TrpE protein, was obtained from theAmerican Type Culture Collection
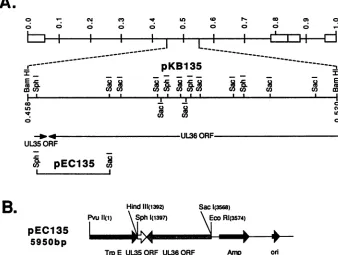
(Rock-ville, Md.) and has been previously described (26). This plasmid producesa37-kDafragment of E. coli anthranilate synthase encoded by the trpE gene. The plasmid pKB135
contains the 9.3-kb BamHI D genomic fragment ofHSV-1
(KOS) (Fig. 1A) cloned into the BamHI site ofpBR322 (5),
andwaspreviously constructedwithin ourlaboratory
(29).
The recombinant expression plasmid pEC135 was con-structedbyligationoftheUL35-containing2.2-kb
SphI-SacI
fragment of pKB135 into theSphI-SacI sites ofpATH21at the 3' end of the trpE gene (Fig. 1B). The plasmid was introduced into E. coli TB1 by the CaCl2 transformation procedure
(39).
Fusion protein isolation. The TrpE-UL35 fusion
protein
synthesized by E. coli TB1 formed insoluble aggregates whichwereisolated from celllysatesaspreviouslydescribed (26). Briefly, E. coli TB1-containing pEC135 was streaked ontoLuria-Bertani agarplatescontaining50 ,ug ofampicillin
perml and 20 ,g oftryptophanperml and grown
overnight
at37°C.Aloopof cells from theovernightculturewasused toinoculatemodified M9 mediumcontainingtryptophan
(26)
and grown withvigorousshakingat37°Cto
mid-logarithmic
phase. To induce the expression oftrpE-UL35, the entire culturewasdiluted 10-fold in modified M9 medium without
tryptophanand grownforanadditional 2 h. The culturewas
subsequentlytreated with 10 ,ug of
3-3-indoleacrylic
acid perml, andincubation wascontinued for 4 h.
The induced cells wereharvested bycentrifugationfor 5 minat3,300 xg,washedoncein 10 mMTris-HCI
(pH
7.5),
andpelletedagain.The cellpelletwasresuspendedin50 mM
Tris-HCl (pH 7.5)-5 mM EDTA-1mM
phenylmethylsulfo-nylfluoride-3 mg oflysozymeper mlandkeptonice for 2 h. Next, NaClwasadded to afinal concentrationof 0.3 M and mixedbygentlyinvertingthe tubes.Subsequently,
Nonidet P-40 was added to a final concentration of 0.7% and thelysate was gently mixed. To shear the DNA, the
samples
were sonicated with 30-s bursts until the
viscosity
was reduced. To separate the soluble andinsolublefractions,
thesamples were centrifuged for 10 min at 9,000 x g, and the insoluble pellet was
subsequently
washed once in 10 mMTris-HCl (pH 7.5)-l MNaCl and once in 10 mMTris-HCl
(pH 7.5). The insoluble pellet was
resuspended
in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) samplebuffer,boiled for 3min,and thencentrifuged
for 15 minat 16,000 xgto remove any insoluble material. The fusion proteinwas thenseparated bypreparative SDS-PAGE, and the gel was stained with 0.05%
(wt/vol)
Coomassie brilliant blue R-250 dissolved in dionized water (19). The gelwasbriefly destained with deionized water, the fusion protein was excised, and the gel fragments were stored at -80°C.
The TrpE and TrpE-UL35 proteins tobe used for subse-quent immunoblot analyses were eluted from the
gel
frag-ments essentially as described by Beemon and Hunter
(3).
Briefly, the gel fragments containing the proteins were J.VIROL.on November 9, 2019 by guest
http://jvi.asm.org/
A.
o - N C) t U) CO t- co 0) 0
I I I I
I
I I IJI
I---
pKB135
I_ _
E.c CU~~~~~~~~~~~~~~~~LLC
m I i c c ocIi CO
0
IV
I I 0
N
U)
-0 UL36ORF
UL35 ORF
X
pEC135
A
I i
Hind 111(1392) \ Sph1(1397) PM11(i)
pEC135
mm5950bp
Sac1(3m68)
\Eco RI(3s74)
_v-I:
Trp E UL35ORF UL36ORF Amp on
FIG. 1. Location in the HSV-1genomeof theUL35 geneand viral DNAfragments usedinthisstudy. (A) Beneath thediagramof the
HSV-1genome(top line), the BamHI D genomic fragment of HSV-1(KOS)presentinplasmid pKB135 (map coordinates 0.458 through 0.520) has beenexpandedtoshow the locations of the relevant restriction sitesused in this study. The location and direction of the UL35 and the
3' portionofthe UL36 ORFsareshownbeneathpKB135. Next is shown the location of the genomic fragment in plasmid pEC135 usedto
expressthe UL35 ORF in E. coli. (B) Linear diagram of the plasmidpEC135 usedtoexpressthe UL35 ORF in E. coli TB1asaTrpE-UL35
fusionprotein.The translational termination codon usedby TrpE-UL35 is that of the UL35gene.
resuspended in 1 ml of 0.05 M NH4HCO3 containing 0.1% SDS and homogenized by multiple passages through afine
wire mesh. The samples were diluted to a final volume of
approximately 1ml/100cm2ofgel with thesamebuffer. The
homogenate (containing afinal concentration of5%
2-mer-captoethanol)was boiled for 5 min and placed on a rotary shaker for 48 hat37°Cto allow theproteinsto diffuse from the gel fragments. The gel fragments were subsequently
pelleted by centrifugation at 12,000 xgfor 10min, and the supernatantwasremoved. Thegel fragmentswerewashed in 0.2 volume of thesamebuffer andpelleted, and the second
supernatant wasremoved and combined with the first. The
supernatants werelyophylized to dryness, and thesamples
wereresuspendedinSDS-PAGE sample buffer and storedat -200C.
Generation of TrpE-UL35 antiserum. In preparation for immunization, gel fragments containing theTrpE-UL35 chi-meric proteinwere combined with equal volumes of
phos-phate-bufferedsaline (PBS)andFreund's complete adjuvant (GIBCO Laboratories, Grand Island, N.Y.). This material
was injected intramuscularly at multiple sites into a female
New Zealand White rabbit. Boosterinjections ofantigen in Freund's incomplete adjuvantwere administered biweekly.
Approximately 75 ,g of antigen was used for the primary
injection; subsequent injectionscontained 30to50,ugof the chimericprotein. Bloodwasobtained from the marginalear
vein 10 to 14 days after the third and subsequent booster injections andwasallowedtoclotat37°C for15min, and the clot contracted overnight at 4°C. The resulting serum was
collectedby low-speed centrifugation (1,500 xgfor 10min at4°C)followedby high-speed centrifugation (12,000 xgfor 10 minat 40C)and storedat -20°C.
Metabolic labeling, immunoprecipitation, and SDS-PAGE. To label viralproteins, HEp-2 cells were mock infected or
infectedwith HSV-1(KOS) at amultiplicity of 10 PFU per
cell. After a 1-h adsorption at 37°C, the inoculum was
removed and the cellswereoverlaid with fresh maintenance
medium containing 2% calf serum. Prior to radiolabeling, cells were incubated for 1 h in minimal essential medium without methionine andcontaining 2% dialyzed calfserum.
Atvarious timespostinfection, cellswere radiolabeled with
[35S]methionine (50 to 100 ,uCi/ml; specific activity, 700.7 Ci/mmol; Dupont, NENResearch Products) and harvested by scraping; they were washed in cold PBS and
subse-quently used in immunoprecipitations. Whole cell pellets
were solubilized in cold radioimmunoprecipitation assay
buffer(50mMTris-HCI [pH 7.5],150 mMNaCl,1%Nonidet P-40, 1% deoxycholate, 0.1% SDS 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 100 ,ug oftolylsulfonyl phe-nylalanyl chloromethylketone[TPCK],2 mMbenzamidine), vortexed vigorously, incubated on ice for 5 min, and
dis-rupted by sonication. Extracts were then boiled for 4 min and clarified by centrifugation for 15 min at 16,000 x g.
Proteinassays weresubsequently performed for each
sam-ple by using the bicinchoninic acid protein assay reagent (Pierce, Rockford, Ill.) with bovine serum albumin as the
standard. Equal amounts of total cell protein were then incubated for 3 hat4°Cwith theappropriate antiserum,and subsequently, protein A-agarosebeads(50% [vol/vol]; Rep-ligen, Cambridge, Mass.) were added and the mixture was
allowedto incubate for 2 h at 4°C. The immunecomplexes
werewashed four times with coldradioimmunoprecipitation assaybuffer,andonefinal washwasperformedwithabuffer containing50 mM Tris-HCl(pH 7.5)and 500 mM NaCl. The
B.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.140.478.89.344.2]2656 McNABB AND COURTNEY
bound proteins were then eluted by boiling in SDS-PAGE sample buffer. Analysis of samples by SDS-PAGE was performed as describedpreviously (33). Samples were elec-trophoresed in 7, 10, 12, or15% polyacrylamide gels cross-linked with N,N-methylene-bisacrylamide (28.2:0.8 ratio of acrylamide to methylene-bisacrylamide). After electropho-resis, the slab gels either were directly visualized by staining with 0.25% (wt/vol) Coomassie blue R-250 or silver (30) or were fixed and dried onto Whatman 3MM filter paper (Whatman, Inc., Clifton, N.J.); the dried gels were then exposed to Kodak XARfilm (Eastman Kodak Co., Roches-ter, N.Y.) at -80°C. The [35S]methionine-labeled proteins were detected bythe use of a fluorographic enhancer (Flu-oro-Hance; Research Products International Corp., Mount Prospect, Ill.).
Immunoblotting. Immunoblotting was performed essen-tially as reported by Towbin et al. (46), with some modifi-cations (12). The immunoblots were incubated with the appropriate rabbit antiserum, and the reactiveproteinswere visualized byreaction with 125I-labeled protein A (Dupont, NEN Research Products) and then autoradiographed at -800C.
PAA inhibition of viral DNAsynthesis. HEp-2 cellswere incubated in minimal essential medium containing 2% calf serum and 300 ,ug of PAA for 2 h prior to HSV-1 (KOS)
infection, and this level was maintained throughout infec-tion.
Purification ofextracellular virions. MonolayersofHEp-2
cells cultured in roller bottles (850 cm2) were infected at a multiplicity of 3 PFU per cell. After 1 h of adsorption at 37°C, maintenance mediumsupplementedwith2% newborn calfserumwas added. All incubations were carried out at
37°C. At 48 hpostinfection, virionswere
purified
byprevi-ouslydescribed methods (12). Briefly, extracellular virions wereharvested from the mediaat48 hpostinfectionand cell debris was removed by low-speed centrifugation. Virions werepelleted from the supernatant bycentrifugationfor1h at100,000 xg. The virion pelletwasgentlyresuspended in TNEbuffer(10mMTris-HCl [pH7.4], 100mMNaCl,1 mM
EDTA).Thevirussuspensionwasthenlayeredonto a20to
60%(wt/vol)continuoussucrosegradientandcentrifugedfor 20 hat50,000 xg.Aftercentrifugation, therefractive band
containingviruswasharvestedbypuncturingthe side of the tube withaneedleandsyringe.Therecovered virus suspen-sion was diluted, pelleted, and resuspended in water and stored at -80°C.
Isolation of intranuclear virus capsids. HEp-2 cells were infectedwith HSV-1 (KOS) at amultiplicity of 3 PFU per
cell, and typeA andBviral capsids were
purified
from the nuclear fraction essentially as described by Gibson and Roizman(16). Briefly, infectedcellswereharvested at22 h postinfectionandcollectedbycentrifugationat1,000 xg for 10 min.Thepelletwasresuspendedin 10 mMTris-HCl (pH7.2)-150 mM NaCl-2 mM MgCl2-1% Nonidet P-40. After incubation for30 min at 0°C, thenucleiwerepelleted from thesuspensionbycentrifugationat 1,000xg for 10min and lysed bytheaddition of0.5% deoxycholate in the presence of 50 ,ug of DNase I per ml for 15 min at 37°C. This was followedbyafurther 5-min incubationat0°Cin the presence of0.5%Brij58and 0.5 Murea. Theextract wasclarifiedby
centrifugationfor 10minat7,000 xg. The supernatantwas layeredontoa10 to40% (wt/wt) linearsucrosegradient and centrifugedat70,000xgfor 1 h at4°C.Aftercentrifugation, tworefractile bands located near the middle ofthe tube were collected and stored at -80°C.
RESULTS
ExpressionoftheUL35 geneinE.coli.On thebasis of the
predicted molecularweight and plof the UL35 gene
prod-uct, we reasoned that this ORF was a likely candidate to encode the 12-kDa basic
phosphoprotein
(BP2) previously
describedby Knopfand Kaerner
(25).
It also seemedplau-sible that BP2 was identical to the 12-kDa capsid
protein
previouslydescribedby Cohenetal.(11)
and Heilmanetal.(20). Thus, theobjectives ofthisstudywere to
identify
theprotein
product
of the UL35ORF,toexamine the kinetics ofsynthesis of the protein, and to determine whether the
polypeptide
encodedbythe UL35 ORFwasindeed thep12
capsid
protein.
Ourapproach
was toexpressthe UL35ORF as afusionprotein
in E. coli andtosubsequently
isolate the chimericprotein
touse as animmunogen
for theproduction
of rabbit
polyclonal
antiserum which wouldspecifically
recognize apolypeptide
present in HSV-infected cells.The expression of the UL35 ORF in E. coliwas
accom-plished by
constructing
atrpE-UL35
genefusionby
using
E.coli
expression
plasmid pATH21
(26).
TheTrpE-UL35
expression
plasmid (Fig. 1B),
designated pEC135,
was con-structedby
subcloning
the 2.2-kbSphI-SacI
fragment
ofpKB135
(Fig.
1A)
intopATH21,
as described in Materials and Methods. Theresulting
gene fusion contained 323 codons of thetrpE
genefused inframe with the 3' 91 codons of thepredicted
112-codonUL35 ORF.To
identify
thetrpE-UL35
geneproduct
in E.coli,
thefollowing
culturesweregrown and induced asdescribed in Materials and Methods:(i)
E. coliTB1(induced),
(ii)
E.coli
TB1
containing pATH21
(induced),
(iii)
E.coli
TB1 contain-ing pEC135(uninduced),
and(iv)
E. coli TB1containing
pEC135
(induced).
Afterinduction,
totallysates
werepre-pared
from each culture andsubsequently
separated
into soluble and insoluble fractions. Theproteins
present in each fraction wereseparated by
SDS-PAGE and visualizedby
Coomassie blue
staining
(Fig. 2).
E.coli
TB1containing
pATH21
produced
a 37-kDaprotein
underinducing
condi-tions(Fig. 2,
lanes2, 6,
and10)
whichwasabsent in E.coli
TB1 cells alone
(Fig. 2,
lanes1,
5,
and9).
The appearance of the 37-kDaprotein
underinducing
conditions is consistent with the apparent molecularweight
of the truncatedTrpE
protein
(26)
andsuggested
that thetrpE
genepromoterwasfunctioning
underinducing
conditions.Samples
from cellscontaining
pEC135
demonstrated the appearance ofa 40.5-kDaprotein
(Fig.
2, lanes 4,8,
and12)
which wasgreatly
diminished in uninduced cultures(Fig. 2,
lanes3,
7,
and11).
These results
implied
that the 40.5-kDapolypeptide
repre-sented theTrpE-UL35
chimericprotein.
Itshould be noted that theTrpE-UL35
polypeptide
migrated
faster than would bepredicted
from the calculated molecularweight;
however,
restriction
analysis
ofpEC135
indicated that the gene fusion was in the correctreading
frame(29).
The insolublepellet
containing
themajority
of theTrpE-UL35
protein
was solubilized in SDS-PAGEsample
buffer and resolvedby
preparative SDS-PAGE,
and the bandcorresponding
tothe fusionprotein
was excised from thegels
and used as theimmunogenfor
preparation
ofrabbitpolyclonal
antiserumas described in Materials and Methods.Identification of the UL35 gene product in HSV-infected cells. To examine the
reactivity
of theTrpE-UL35
antise-rum, HEp-2 cells were mock infected or infected with HSV-1 and radiolabeled with 100,uCi
of[35S]methionine
per ml from 8 to 12 hpostinfection.
Celllysates
wereprepared
andimmunoprecipitated
withpreimmune
serum or TrpE-UL35antiserum,
and theimmunoprecipitated
proteins
wereJ. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
1 2 3 4 5 6 7 8 9 10 11 12
92
5K-66 2K- VW =
45 OK- so _;* f
4TrpE-UL35
too -44TrpE
31
0K-
21.5K-M 3 4 5 6
43.0K-29.OK- _
18.4K-.143K
[image:5.612.337.534.77.323.2]14.3K- _;
FIG. 2. Expression of the TrpE-UL35 chimeric protein in E. coli. Extracts of E. coliTB1grown under inducing (lanes 1, 5, and 9) conditions, cells containing pATH21 grown under inducing (lanes 2, 6, and 10)conditions, andcells containing pEC135 grown under noninducing(lanes 3, 7, and 11) and inducing (lanes 4, 8, and 12) conditions were electrophoresed through an SDS-12% polyacryl-amide geland stainedwith Coomassie blue. Whole cells extracts are inlanes1 to4,andthesoluble proteins (lanes 5 to 8) and insoluble proteins (lanes 9 to 12) were isolated as described in Materials and Methods. Themigrationofthemolecular weight markers is shown
ontheleft, and the positions of the TrpE and TrpE-UL35 proteins areshownontheright.
resolvedbySDS-PAGE and visualized by fluorography (Fig. 3). TheTrpE-UL35 antiserum recognized a 12-kDa protein which was present in HSV-1-infected cell extracts (Fig. 3, lane 5) but not in mock-infected cells (Fig. 3, lane 6) or extracts which were immunoprecipitated with preimmune serum(Fig. 3, lanes 3 and4).These results suggest that the UL35 ORF encodes a protein with an apparent molecular weight of 12,000, which is consistent with the predicted molecularweight of 12,095. Furthermore, these data confirm that thepredicted UL35 ORF represents a viral gene which is actively expressed during HSV infection.
Time courseofexpression of the UL35 gene product. The expression of the UL35 gene during lytic HSV infection was analyzed by determining the kinetics of appearance of the UL35 gene product. Initially, pulse-labeling experiments wereperformedtoexamine therateofsynthesis of the UL35 geneproductduring the HSVreplicativecycle. HEp-2 cells weremock infectedorinfected with HSV-1, andatvarious timespostinfectionthecellswerepulse-labeled for1 hwith 50,uCi of
[35S]methionine
perml.Celllysateswerepreparedand immunoprecipitated with TrpE-UL35 antiserum, re-solvedby SDS-PAGE, and visualized byfluorography(Fig.
4). The UL35 gene product was initially detectable at 6 h
postinfection,and therateofsynthesis continuedtoincrease until 10hpostinfection, thereafterremaining relatively con-stantthrough 18 h postinfection. These results indicate that the UL35 gene product is synthesized late in infection, consistent with being a structural protein of the virion. Furthermore, the increasedrate ofsynthesis of the 12-kDa protein during the course ofHSV infection provides addi-tional evidencethat theTrpE-UL35 antiserumisrecognizing
avirally encoded protein.
Studieswerealsoperformedtoexamine theaccumulation oftheUL35 geneproductduringHSV infection.HEp-2cells
4.
412K
6.2K-3.0K-5
FIG. 3. Identification of the UL35 gene product in HSV-1 in-fectedcells.HEp-2cells were mockinfectedorinfected withHSV-1 and radiolabeled from 8 to 12 h postinfection with 100 ,Ci of
[35S]methionine per ml. At 12 h postinfection, cell lysates were
prepared andimmunoprecipitatedwithpreimmune serum or TrpE-UL35 antiserum. Theimmunoprecipitateswereresolvedby electro-phoresisthrough anSDS-15% polyacrylamidegel, and the proteins were visualized by fluorography. The odd-numbered lanes are HSV-infected extracts, and the even-numbered lanes are mock-infected extracts. Lanes 1 and 2 are HSV-infected and mock-infected cell extracts, respectively. Lanes3 and 4arecell extracts immunoprecipitatedwithpreimmune serum. Lanes 5 and 6 are cell extractsimmunoprecipitatedwithTrpE-UL35 antiserum. The mo-lecularweightmarkers(M)areshownontheleft.
were mockinfected orinfected with HSV-1, and atvarious times postinfection the cells were harvested and the cell lysates were subjected to immunoprecipitation with the TrpE-UL35 antiserum. Theproteinswereresolved by SDS-PAGE, and Western blot (immunoblot) analysis was per-formed with the TrpE-UL35 antiserum (Fig. 5B). For
com-parison and internal controls, a portion of the total cell lysates was separated by SDS-PAGE and Western blot analysiswas performed with antisera directed against ICP8
(, gene product), glycoprotein B
(yl
gene product), andglycoprotein C (Y2 gene product) (Fig. 5A). The kinetics of accumulation of these proteins during the HSV replicative
cycle has been previously reported (10, 41, 43). ICP8 was detectable earlyafter infection(6 h postinfection), reaching
maximumlevelsby11to 12 hpostinfection andmaintaining
these levelsthroughoutthe
replicative
cycle.Glycoprotein
B was also initially detectable at 6 h postinfection, obtainingmaximum levels by 12 h postinfection, and those levels remained constant through 18 h postinfection. Similarly, glycoproteinCwas weaklydetectable at 6 h
postinfection;
however, it continuedtoaccumulatethrough18h
postinfec-tion. The accumulation of the UL35 gene product was initially detected at 8 h postinfection, andthe
protein
con-tinued to accumulate through 18 hpostinfection.
Quantita-tive analysis of these data revealed that the UL35 gene product demonstrated an expressionpattern very similarto
416&*
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.65.291.79.254.2]2658 McNABB AND COURTNEY
3 6 8 10 12 15 18 M hpi -A- ICP8 --A-- gB --0 -- C UL35
29.0K- 18.4K-
14.3K
-
6.2K-
3.0K-FIG. 4. Kineticsof synthesis of the UL35 gene product. HEp-2 cells were mock infected (M) or infected with HSV-1, and at the indicated times postinfection, the cells were pulse-labeled with 50 ,uCi of [35S]methionine per ml for 1 h. After the 1-h pulse, cell extracts were harvested and immunoprecipitated with the TrpE-UL35 antiserum. Theimmunoprecipitateswereresolved by electro-phoresisthrough an SDS-15% polyacrylamide gel, and the proteins werevisualizedbyfluorography.Thenumbersabovethelanesrefer to the times (hours postinfection [hpi]) at which the cells were harvested after the 1-h pulse. Themock-infected cells were pulse-labeled at 18 h postinfection. The positions of the molecular weight markers are shown on the left.
that ofglycoprotein C, with detectable levels of synthesis observed at 8 hpostinfection and continued accumulation of the 12-kDa protein throughout the 18-h replicative, cycle examined(Fig. 6). These studies indicate that the UL35 gene product is expressed as a late gene. It should be noted that the 12-kDa UL35 gene product was observed at 3 and 6 h
postinfection; however, this was likely the result of input viralparticles since the UL35 gene product was found to be a structural protein of the virion (see Fig. 8). This is supported by two lines of evidence. First, the pulse-labeling studies whose results are shown in Fig. 4 indicate that detectable expression of the UL35 gene product was not observed until 6 h postinfection. Second, the presence of
A. 3 6 8 10 12 15 18 M hpi
ICP8* _ _
90 / x
O
80-z 3_,
70-60 /
w
H50-o
~40-30
-0 3 6 8 10 12 15 18
HOlRS POSTINFECTION
FIG. 6. Kinetics of accumulation of the UL35 gene product relative to otherHSV-encodedpolypeptides.Multiple exposuresof the autoradiographs shown in Fig. 5 were scanned with a laser densitometer. The values shownrepresenttherelativeabundanceof thedetected proteins atvarious times postinfection. Thesymbols representingeachpolypeptideareshownabovethegraph.
matureglycoprotein Cat3 and 6hpostinfectionimplies the presenceof detectableamountsof inputviralproteins.
Expression of the UL35 geneproduct requires viral DNA replication. The kinetics of synthesis of the UL35 gene product indicated that the protein is expressed late in infec-tion, suggesting that the UL35geneis regulated aseithera quasilate
(Yj)
or truelate (Y2)gene.The-Yl
genes aredefined as those genes whose products are expressed at reduced levels in the absence of viral DNA replication but are expressed atmaximum levelsafter the onsetof DNArepli-cation. Incontrast,the expression of_Y2 geneshasastringent requirement for viral DNA synthesis, and the genes arenot expressed in the absence of viral DNA replication. To precisely determine the temporal class of the UL35 gene, experiments were performed to examine the expression of
[image:6.612.322.558.73.238.2]B. 3 6 8 10 12 15 18 M hpi 18.4K-
f4.3K-.. ^ _* 12K
6.2K-
3.oK-.~~~~~I
pgc
~
or
rri
g
FIG. 5. Time course of the UL35 gene product accumulation in HSV-infected cells. HEp-2 cells were mock infected (M) or infected with HSV-1, and at various times after infection, the cell extracts were harvested and the samples were electrophoresed on SDS-7% polyacrylamidegels(A) or the extracts were immunoprecipitated with TrpE-UL35 antiserum and the immunoprecipitates were resolved on
anSDS-15%polyacrylamide gel (B). After electrophoresis, the proteins were transferred to nitrocellulose and immunoblotted with antiserum against ICP8, glycoprotein B (gB), or glycoprotein C (gC) (A) or TrpE-UL35 antiserum (B). The reactive proteins were visualized by incubation with
125I-labeled
protein A and subsequent autoradiography. The precursors containing high levels of mannose (pgB and pgC) and thefullyprocessed forms of gB and gC are indicated. The numbers above the lanes refer to the hours postinfection (hpi) that the cells were harvested.Themock-infected cells (M) were harvested at 18 h postinfection. The positions of the molecular weight markers are shown on the left.J. VIROL.
Alam,
''badm.JimiL
9B0- "VW* F;
gBol n
p
wIwVW,
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.132.500.498.643.2]A.
-PAA + PAA
M I M
ICP80 _
B.
gBs
pgBI
a'-C.
gc.
pgC
.D.
[image:7.612.119.248.76.370.2]12K
FIG. 7. Synthesisof theUL35geneproductin thepresenceand
absence of viral DNAreplication. HEp-2 cellsweremock infected (M)orinfected with HSV-1(I)in thepresence(+PAA)orabsence
(-PAA) of300 ,ugof PAA perml as described in Materials and
Methods. The cellswere radiolabeled with100 ,uCiof
[35S]methio-ninepermlbetween 3 and 18 hpostinfection.The cellswerelysed
inradioimmunoprecipitationassaybuffer,andaliquotswerereacted with antiserumagainstICP8(A), glycoproteinB(gB) (B),
glycopro-tein C (gC) (C), or the UL35 gene product (D). The precursors
containing highlevels ofmannose(pgBandpgC)arealso indicated. Theimmunoprecipitated proteinswereresolvedby electrophoresis
throughSDS-7%polyacrylamide (A, B, andC)orSDS-15% poly-acrylamide (D) gelsand visualizedby fluorography.
the UL35 gene in the absence ofviral DNA replication. In experiments whose dataare not shown, it was determined
that 300 ,ug of PAA added 2 h prior to infection and maintainedthroughoutinfectionwassufficienttoblock viral DNAsynthesis.Tostudythe effect of viral DNAreplication
onthesynthesisof theUL35geneproduct, HEp-2cellswere
mock infected or infected with HSV-1 in the presence or
absence of PAA, incubated in the presence of 100 ,uCi of [35S]methioninepermlbetween 3 and 18hpostinfection,and harvestedat 18hpostinfection.Theproteinswere
immuno-precipitatedfrom theextracts,resolvedby SDS-PAGE,and visualizedby fluorography. ThesynthesisofICP8, aknown early (1)geneproduct (17, 18),wasonly slightlydiminished inthe absence of viralDNAreplication (Fig. 7A), indicating that the DNAsynthesis inhibitor was notsuppressingviral gene expression in a nonspecific manner. Synthesis of the
control protein, glycoprotein B, was diminished approxi-matelythreefoldinthe absenceof viral DNAsynthesis (Fig. 7B), which is consistent with its assignment as a Yl gene
product (22, 37). The expression of the second control protein, glycoprotein C, was completely abolished in the
absence of viral DNAreplication (Fig. 7C),which is
consis-tentwith its classification as a
Y2
gene product (22, 23). The synthesis of the UL35 gene product was also completely abolished in the absence of viral DNA replication (Fig. 7D). These results demonstrate that the expression of the UL35 gene is absolutely dependent on viral DNA replication, indicating that UL35 represents aY2
(true late) gene.Association of the UL35 gene product with purified HSV virions and capsids. Our original hypothesis was that the UL35 ORF may encode the p12 capsid protein. The classi-ficationof UL35 as a
Y2
genefurther suggests that the UL35 gene product may be a structural protein. Since previous studies have shown that p12 is associated with both type A and B intranuclear capsids (7, 11, 20), and by inference should also beassociated with whole virions, we wondered whether the 12-kDa product of the UL35 ORF was also associated with all three types of particles. If the product of theUL35 ORF isassociated with the three types of particles, theUL35 ORF may encode p12.To address thispossibility, HSV-1 virions and type A and B capsids were isolated as described in Materials and Methods. Inexperiments whose results are not shown, the
purified virions or A and B capsids were resolved by SDS-PAGE and visualized by Coomassie blue or silver
stainingto assessthepurity of each preparation. The struc-tural protein profiles were similar to those described by Spear and Roizman(44) and Gibson and Roizman(16), with minimal contamination by cellular proteins. To determine whether the UL35 gene product was associated with the
purified virus particles, the proteins were separated by SDS-PAGE and electrophoretically transferredto nitrocel-lulose, and immunoblot analysis was performed with the
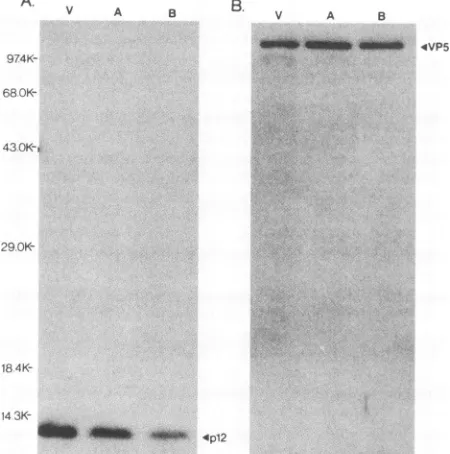
TrpE-UL35antiserum(Fig. 8A).These results demonstrated that the 12-kDaproteinencodedby the UL35 gene is present inpurifiedvirionsaswellastypeAandBcapsids, implying
that the UL35 gene may encode the p12 capsid protein. It should be noted that the SDS-PAGE gels were loaded quantitatively intermsof virusparticles by using theamount of VP5 (the major capsid protein) present in each particle
preparationasthecommon reference. Toconfirm that each sample contained equivalent amounts of viral particlesthe same blot was reprobed with anti-VP5 antiserum, and the results are shown in Fig. 8B. Unexpectedly, these studies demonstrated that less of the 12-kDaproteinwaspresentin isolated type A and B capsids than in purified virions. By
using densitometric scanning of the autoradiograms, these datawere examined quantitatively (Table 1). These results indicate that isolated virions contained 2.5-fold more p12
than did purified type A and B capsids. At present, we cannot provide an explanation for this observation; how-ever, experiments to define the precise location of the 12-kDa protein within the viral capsid are currentlyunder way, and these studies mayprovide someinsights into this
finding.
Anti-NC7 andanti-TrpE-UL35antiserarecognizethe same polypeptide. The association of the12-kDaUL35 gene
prod-uct with purified virions as well as type A and B capsids stronglysuggests that the UL35 gene encodesthep12 capsid protein.Toconfirm thisassignment, weobtained antiserum
preparedagainstthepurified
p12
capsidprotein (designated
anti-NC7, kindly provided by G. Cohen and R.Eisenberg)
andperformedstudiestodetermine whether anti-NC7 would cross-react with the same polypeptide
recognized by
theTrpE-UL35 antiserum. If the
protein product
of the UL35 ORF is p12, the antiserumprepared
to thepurified p12
capsid protein should react with the same 12-kDaprotein
recognized bythe TrpE-UL35antiserum.on November 9, 2019 by guest
http://jvi.asm.org/
2660 McNABB AND COURTNEY A.
974K-68
0K-V A B. v A B
__WANNO'ONEU 4VP5
430OK-,
29.0K-18
4K-14
3K-OW' 4P12
FIG. 8. Association of the UL35 geneproductwith HSVvirions and intranuclear capsids. HSV-1 virions and intranuclear capsids (types A and B) were purified as described in Materials and Methods. The viralpolypeptides present ineachpreparationwere resolved by electrophoresis through an SDS-12% polyacrylamide gel, and the proteinsweretransferred to nitrocellulose and immu-noblotted with either the TrpE-UL35 antiserum (A) or anti-VP5 serum(B).The reactiveproteinswerevisualizedbyincubation with
125I-labeled protein A and subsequent autoradiography. Panels A and B showautoradiographsofthe samenitrocellulose sheet sepa-rately probed with each antiserum. The positions of the molecular weight markers are shown on the left, and thepositionsofp12and VP5 areshownonthe right. Abbreviations: V,purified virions;A, type Acapsids; B, type Bcapsids.
Totestthecross-reactivityof theseantisera,HEp-2 cells weremockinfectedor infected with HSV-1 and celllysates were prepared at 18 h postinfection. The lysates were
immunoprecipitated with either anti-NC7 or
anti-TrpE-UL35 serum, and theimmunoprecipitateswereresolvedby SDS-PAGE and electrophoretically transferred to nitrocel-lulose. The samples were then subjected to immunoblot
analysiswith the opposite antiserum. The results for these analysesareshown inFig. 9 and clearly demonstrate that the anti-NC-7 antiserum and the TrpE-UL35 antiserum recog-nize thesame12-kDa protein. As acontrol, purified HSV-1
virionswerealsoanalyzed in parallel toconfirm the
reactiv-ityof each antiserum.
TABLE 1. Relativeamounts ofp12associated with HSV-1 virions and type A and Bcapsidsa
Particle type Amt of:
p12/VP5
VP5 p12 ratio
HSV-1virions 1.3 3.2 2.5
Type Acapsids 1.7 2.0 1.2
TypeBcapsids 1.4 1.2 0.9
aThe procedures for gel electrophoresis and immunoblotting are described
in Materials and Methods. The autoradiographs shown in Fig. 8 were
quantitated by usinganLKBUltroscanXLlaserdensitometer.Therelative
values and the ratio ofp12/VP5areshown.
To further demonstrate that the anti-NC7antiserum reacts with the protein productof the UL35 gene, the TrpE-UL35 chimeric protein was isolated from E. coli extracts by preparative SDS-PAGEand theproteinwaseluted fromthe gel fragments as described in Materials and Methods. The isolated TrpE-UL35 polypeptide was resolved by SDS-PAGE, and immunoblot analysiswas performed with pre-immune,anti-TrpE-UL35, and anti-NC7sera.Asacontrol, theTrpEprotein alone was isolated in an identical manner and analyzed in parallel. A Coomassie blue-stained SDS-PAGE gel of the isolated proteins is shown in Fig. 10A. Immunoblot analysis with preimmune serumfailed to dem-onstrate reactivity to either protein (Fig. lOB). As antici-pated, theTrpE-UL35antiserum reacted with both theTrpE andTrpE-UL35 proteins (Fig. 10C); however,the anti-NC7 antiserumonly recognizedtheTrpE-UL35fusionprotein but not the TrpE protein alone (Fig. 10D). Since the only differencebetween TrpEandTrpE-UL35 is the addition of the 91 amino acid residues contributed by the UL35 gene product, these results indicate that the anti-NC7 antiserum recognizes the viralpolypeptideencodedby the UL35 gene. On the basis of thesedata,weconclude that the UL35 gene encodes thep12 capsid proteinofHSV-1.
DISCUSSION
In the studies described in this report,wehaveexpressed theUL35 ORF of HSV-1 in E.colias aTrpE-UL35chimeric protein.The isolatedTrpE-UL35polypeptidewasused as an
immunogen toproducearabbitpolyclonal antiserum which wouldspecificallyreactwith theputativeprotein productof the UL35 ORF in HSV-infected cells. We found that this antiserum reactedspecificallywitha12-kDaproteinpresent only in HSV-infected cells. The apparent size of theprotein
was in excellent agreement with the molecular weight of
12,095 predicted for the UL35 gene product (28). The identification of thepredicted product of the HSV-1 UL35 ORF confirms the reading frame of this ORF and demon-strates that UL35 represents an actively expressed gene
during HSVinfection. Analysisof the kinetics ofsynthesis
of the UL35 gene product indicated that the protein is
synthesized late in infection, with an accumulation pattern similartothat ofglycoproteinC. Additional studies revealed that thesynthesisof the UL35 gene productwasabsolutely dependent on prior viral DNA replication, implying that UL35 isa truelate(_Y2)gene.
The association of the UL35 gene product with purified
virions aswellas typeA andBcapsids suggestedthat this
polypeptidemightrepresentthepreviouslyidentified 12-kDa
capsid proteinvariablydesignated p12, NC7,and VP26(11,
20, 32). This assignment was confirmed by using two ap-proaches. First, immunoprecipitation and immunoblotting
studieswereperformed to demonstratethat antiserum pre-paredagainstthepurified 12-kDacapsid protein (anti-NC7)
cross-reacted with the same 12-kDa protein recognized by
theTrpE-UL35 antiserum. Second, the anti-NC7 antiserum was also shown to react with the isolated TrpE-UL35
polypeptide synthesizedinE. coli.With the identification of UL35 asthe gene encoding p12, theonly remaining capsid
proteins which havenotbeenassignedto aspecificgeneare VP21 andVP24. Asstatedpreviously, VP21 may represent aprocessed form of VP22a(8), thereby also being encoded
bythe UL26 gene(35).ThusVP24,a25-kDacapsid protein,
may represent theonlyremaining capsid protein which has notbeenassignedto a specific gene.
With thegeneral structure oftheherpesvirus capsid now J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.69.294.83.310.2] [image:8.612.61.299.630.693.2]anti-TrpE UL35 onti-NC7
M M V B.
anti-TrpE--UL35 onti-NC 7
M M
29.0K-
18.4K-
14.3K-p12k 4._
4..
t4.3K-
6.2K-
3.0K-
6-FIG. 9. Cross-reactivity of anti-TrpE-UL35and anti-NC7. HEp-2 cellsweremockinfected (M)orinfectedwith HSV-1 (I), and celllysates werepreparedat18hpostinfection. Aliquots ofthe lysateswerereacted with either anti-TrpE-UL35oranti-NC7, and theimmunoprecipitates were resolved by electrophoresis on SDS-15% polyacrylamide gels. The proteins were transferred to nitrocellulose membranes and immunoblotted with the TrpE-UL35 antiserum(A) oranti-NC7 (B). The reactive proteinswerevisualizedbyincubationwith 1'251-labeled
protein A andautoradiography. The antisera usedfor theimmunoprecipitationsareindicated above each pair oflanes.PurifiedHSV-1virions
(V)wereanalyzed in parallelasacontrol. The positionsofthe molecular weight markersareshownonthe left of each panel.
reasonably well defined (1, 7, 40), thecurrentchallenge isto understand the precise location and function(s) of these
seven proteins which comprise the HSV capsids and to understandtheir role in the assembly and maturation ofthe herpesvirion. In our studies, we have observed a 2.5-fold
increase in the amountofp12 associated with HSV virions
versusintranuclear type Aand B capsids (Fig. 8and Table 1). Studies to explain this observation are currently in
progress, but the presence of more p12 associated with
purified virions induced usto raise somehypotheses about the possiblelocation and function of p12 within the virion.
It has been previously demonstrated that preparations of typeA capsids appear tocontain little or noviralDNA (7,
16), and preparations of type B capsids appear to be a
A.
12
B.
1 2mixture of DNA-containing and empty capsids (27, 42). These findings differ from those for isolated virions, which represent apopulation highly enriched for DNA-containing
particles. If p12 is somehow associated with the viral DNA, thenthepresenceof lessp12inisolatedtypeAand B capsids could be explained by the variableamountsof viral DNA in these particle preparations. The possibility of p12 being associated with the viral DNA is supported by several independent studies. Knopfand Kaerner (25) have demon-strated that an acid-soluble 12-kDa basic phosphoprotein, designated BP2, was foundto be associated with the chro-matin isolated from HSV-infected cells extracted with 0.25 M HCl and with similarly acid-extracted HSV virions. On the basis ofcurrentinformation,wecannotdirectly conclude
C. 1 2
43.0K-'U
i __
D.
l
2
31.0K-FIG. 10. Reaction of anti-NC7 with the TrpE-UL35 chimeric protein synthesized in E. coli. The TrpE and TrpE-UL35 proteins synthesizedin E. coliwereisolatedfrompreparativeSDS-polyacrylamide gelsasdescribed in Materials and Methods. Theisolatedproteins
wereelectrophoresed onSDS-10%polyacrylamide gels and stained with Coomassieblue (A)ortransferred tonitrocellulose. Immunoblot analysesweresubsequently performedwith rabbitpreimmuneserum(B), anti-TrpE-UL35serum(C),oranti-NC7serum(D),and thereactive
proteinswerevisualized by incubationwith 1251-labeled protein A and autoradiography. Lanes 1 and 2 of each panel containTrpE and TrpE-UL35, respectively. Thepositionsofmolecularweightmarkersareshownonthe left ofpanelA.
A.
29.0K-
18.4K-V
6.2K-
3.0K-4.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.125.491.77.244.2] [image:9.612.144.467.487.670.2]2662 McNABB AND COURTNEY
that the UL35 gene
product
is BP2; however, computeranalysis
ofthepredicted
amino acid sequence of the UL35 geneproduct
does indicate that the geneproduct
isex-tremely
basic,
withapl
of 11.6(29).
Wehavealsofound that the UL35 geneproduct
is a phosphoprotein which can be resolved intomultiple phosphorylated
species by acid-ureagel
electrophoresis (29).
Thesefindings
are consistent with the observations ofKnopf
and Kaerner(25),
which demon-strated that BP2 could beseparated
into threephosphopro-tein
species by
acid-ureagel electrophoresis;
therefore, itis conceivable thatp12
and BP2 may represent the samepolypeptide. Additionally,
studiesby Bayliss
etal.(2)
have identified16DNA-binding proteins,
presentin HSV-infectedcells,
which bind to DNA-cellulose. One of thesepolypep-tides, designated BP15,
hadamolecularweight
of12,000andappeared
to bind double-stranded DNA. It is conceivable that this 12-kDaprotein
may represent thep12 capsid
protein; however,
similarDNA-binding
studies and assays forp12
will have to beperformed
to demonstrate thisdefinitively.
Finally,
studiesof theDNA-binding proteins
ofherpesvirus
saimiriby
Blair and Honess(4)
have demon-strated thepresence ofa 12-kDa basicnucleocapsid protein
which appears to bind to herpesvirus saimiri DNA in vivo andinvitro. We donot know whether the12-kDa
protein
ofherpesvirus
saimiri represents a homolog top12
of HSV;however,
theproteins
have several properties which arestrikingly
similar.First,
bothproteins
are extremely basic and have been described ashistonelike(4,
11,25).
Second,they
are both components of the viralnucleocapsids (4, 11,20). Finally,
bothproteins
appear tobe true late (Y2) geneproducts, requiring
viralDNAreplication
for theirsynthesis(4
andFig.
7of thisreport).
Atpresent,wehavenodirect
proof
thatp12
interactswith the viral DNA within thenucleocapsid;
however, these observations indicate that thispossibility
warrants furtherinvestigation.
Current studies in ourlaboratory
arefocused ondetermining
whetherp12
canbind toHSVDNAinvitro and in vivo. Ifp12
can bind to viral DNA, thenp12
may function in the condensationand/or packaging
of the viral DNAintothepreformed capsids.
As stated
previously,
studies by Newcombe and Brown(32)
haveprovided
evidence thatp12 (referred
to as VP26)may be located at the vertices of the icosahedral HSV
capsids;
however,
it is not known whetherp12
actuallycomprises
the pentavalent capsomers or is located in closeproximity
tothe pentons.Itispossible
thatp12
mayinteract with both the viral DNA and the pentavalent capsomers. Suchanarrangementwould beanalogous
tothat found withadenovirus,
in whichabasic coreprotein (protein V)
inter-actswith the viral DNA andthe penton baseprotein (proteinIII) (24).
Ithas alsobeen suggestedthat adenovirusprotein V may have apositioning
function in the packaging of the virionDNA(24).
Whetherp12
has suchafunction during theassembly
andmaturation ofthe herpesvirusvirion remains to be determined. To date, no mutations within the geneencoding
p12
have been mapped; therefore, itis not known whetherp12
is essential forproductive viral infection. Thisproblem
canbe addressed byconstructing an HSV mutantcontaining
a null allele of UL35. Such a mutant would be valuable inaddressing
the function ofp12
in the assembly andmaturationofHSV.Wearecurrently performing studies to further characterizep12
in an attempt to understand its function in theassembly pathway
ofHSV.ACKNOWLEDGMENTS
We are grateful to Gary Cohen and Roselyn Eisenberg for providing the NC7 antiserum and toFeng Yaoforprovidingthetype Aand Bcapsids and virions used in some of these studies.
This investigation was supported in part bygrant A129026 from the National Institutesof Health.
REFERENCES
1. Baker, T. S., W. W. Newcomb, F. P. Booy, J. C. Brown, and A. C. Steven. 1990. Three-dimensional structuresofmaturable and abortive capsidsofequine herpesvirus 1fromcryoelectron microscopy.J. Virol. 64:563-573.
2. Bayliss, G. J., H. S. Marsden, and J. Hay. 1975.Herpessimplex virus proteins: DNA-binding proteins ininfected cells and the virus structure. Virology68:124-134.
3. Beemon, K., and T. Hunter. 1978. Characterization of Rous
sarcomavirussrcgeneproducts synthesized invitro. J. Virol. 28:551-566.
4. Blair, E. D., and R. W. Honess. 1983. DNA-binding proteins specified by herpesvirus saimiri. J. Gen. Virol. 64:2697-2715. 5. Bolivar, F.,R.L.Rodriquez, P. J.Greene, M. C. Betlach,H.L.
Heyneker, H. W. Boyer, J. H. Crosa, and S. Falkow. 1977. Construction andcharacterization ofnewcloningvehicles. II. A multipurpose cloning system.Gene2:95-113.
6. Bone, D. R., and R. J. Courtney. 1974. A temperaturesensitive mutantofherpes simplex virus type 1 defective in thesynthesis of the majorcapsid protein. J. Gen. Virol. 24:17-27.
7. Booy, F. P., W. W. Newcomb, B. L. Trus, J. C. Brown, T. S. Baker, and A. C. Steven. 1991. Liquid-crystalline, phage-like packing of encapsidated DNA in herpes simplex virus. Cell 64:1007-1015.
8. Braun, D. K., W. Batterson, and B.Roizman. 1984. Identifica-tion and genetic mapping of a herpes simplex virus capsid protein that binds DNA. J.Virol. 50:645-648.
9. Cassai, E. N., M. Sarmiento, and P. G. Spear. 1975.Comparison of the virionproteins specified by herpes simplex virus types 1 and 2.J. Virol. 16:1327-1331.
10. Cohen, G. H., D. Long, and R. J. Eisenberg. 1980. Synthesisand processing ofglycoproteins gD and gC of herpes simplex virus type 1. J. Virol. 36:429-439.
11. Cohen, G. H., M. Ponce de Leon, H. Diggelmann, W. C. Lawrence, S. K. Vernon, and R. J. Eisenberg. 1980. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J. Virol. 34:521-531.
12. Compton, T., and R. J. Courtney.1984. Virus-specific glycopro-teins associated with the nuclear fraction of herpes simplex virus type 1-infected cells. J. Virol. 49:594-597.
13. Costa, R. H., G. Cohen, R.Eisenberg, D. Long, and E. Wagner. 1984.Direct demonstration that the abundant 6-kilobase herpes simplex virus type 1 mRNA mapping between 0.23 and 0.27 map units encodes the major capsid proteinVP5. J. Virol. 49:287-292.
14. Eberle, R., and R. J. Courtney. 1980. Preparation and charac-terization of specific antisera to individual glycoproteinantigens comprising the major glycoprotein region of herpes simplex virus type 1. J. Virol. 35:902-917.
15. Flannery, V. L., R. J. Courtney, and P. A. Schaffer. 1977. Expression of an early, nonstructural antigen of herpes simplex virus in cells transformed in vitro by herpes simplex virus. J. Virol.21:284-291.
16. Gibson, W., and B. Roizman. 1972. Proteins specified by herpes simplex virus. VIII. Characterization and composition of mul-tiplecapsid forms of subtypes 1 and 2. J. Virol. 10:1044-1052. 17. Godowski, P. J., and D. M. Knipe.1986. Transcriptional control
of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc. Natl. Acad. Sci. USA 83:256-260.
18. Goodrich, L. D., F. J. Rixon, and D. S. Parris. 1989.Kinetics of expression of the gene encoding the 65-kilodaltonDNA-binding protein of herpes simplex virus type 1. J. Virol. 63:137-147. 19. Harlow, E., and D. Lane. 1988. Antibodies: a laboratory
man-ual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. 20. Heilman, C. J.,Jr., M. Zweig,J.R.Stephenson, and B. Hampar. J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
1979.Isolation of a nucleocapsid polypeptide of herpes simplex virus types 1 and 2 possessing immunologically type-specific and cross-reactive determinants. J. Virol. 29:34-42.
21. Heine, J. W., R. W. Honess, E. Cassai,andB.Roizman. 1974. Proteins specified by herpes simplex virus. XIII. The virion polypeptides oftype 1 strains. J. Virol. 14:640-651.
22. Homa, F. L., J. C. Glorioso, and M. Levine. 1988. A specific 15-bp TATA box promoter element is required for expression of aherpes simplex virus type 1 late gene. Genes Dev.2:40-53. 23. Homa, F. L., T. M. Otal, J. C. Glorioso, and M. Levine. 1986.
Transcriptional control signals of a herpes simplex virus type 1 late (Y2) gene lie within bases -34 to +124 relative to the 5' terminusofthe mRNA. Mol.Cell. Biol. 6:3652-3666. 24. Horwitz, M. S. 1986. Adenoviruses and their replication, p.
563-606. In B. N.Fields and D. M. Knipe (ed.), Fundamental virology. Raven Press, New York.
25. Knopf, K. W., and H. C. Kaerner. 1980. Virus-specific basic phosphoproteins associated with herpes simplex virus type 1 (HSV-1) particlesand the chromatin of HSV-1-infected cells. J. Gen. Virol.46:405-414.
26. Koerner, T. J., J. E. Hill, A. M. Myers, andA.Tiagoloff. 1991. High-expression vectors with multiple cloning sites for con-struction of trpE fusion genes: pATH vectors. Methods En-zymol. 194:477-490.
27. Lee, J. Y., A. Irmiere, and W. Gibson. 1988.Primate cytomeg-alovirusassembly: evidence that DNApackagingoccurs subse-quent to Bcapsid assembly. Virology 167:87-96.
28. McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M.C. Frame, D.McNab, L. J.Perry,J. E.Scott, and P. Taylor. 1988. ThecompleteDNA sequenceofthelong unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574.
29. McNabb, D. S.,andR.J. Courtney.Unpublisheddata. 30. Morrissey, J. H. 1981. Silver stain fromproteins in
polyacryl-amidegels:amodified procedurewith enhanced uniform sensi-tivity. Anal. Biochem. 117:307-310.
31. Newcombe, W.W.,andJ. C. Brown. 1989.Use of Ar+plasma etching to localize structural proteins in the capsid ofherpes simplex virustype 1.J. Virol. 63:4697-4702.
32. Newcombe, W. W., and J. C. Brown. 1991. Structure of the herpes simplex virus capsid:effectsofextraction withguanidine hydrochlorideandpartial reconstitution of extractedcapsids.J. Virol.65:613-620.
33. Powell, K. L., andR.J.Courtney. 1975.Polypeptides synthe-sized in herpes simplex virus type 1-infected HEp-2 cells. Virology66:217-228.
34. Powell, K. L., and D. H. Watson. 1975. Somestructuralantigens ofherpes simplexvirus type 1. J. Gen. Virol. 29:167-177.
35. Preston, V. G., J. A. V. Coates, and F. J. Rixon. 1983. Identifi-cation and characterization of a herpes simplex virus gene product required for encapsidation of virus DNA. J. Virol. 45:1056-1064.
36. Rixon, F. J., M. D. Davison, and A. J. Davison. 1990. Identifi-cation of the genes encoding two capsid proteins of herpes simplex virus type 1 by direct amino acid sequencing. J. Gen. Virol. 71:1211-1214.
37. Roizman, B., and W. Batterson. 1986. Herpesviruses and their replication, p. 607-636.In B. N. Fields and D. M. Knipe (ed.), Fundamental virology. Raven Press, New York.
38. Roizman?B., and D. Furlong. 1974. Thereplication of herpes-viruses. Compr. Virol.3:229-403.
39. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
40. Schrag, J. D., B. V. Venkataram Prasad, F. J. Rixon, and W. Chiu. 1989. Three-dimensional structure of the HSV-1 nucleo-capsid. Cell 56:651-660.
41. Shelton, L. S. G., M. N. Pensiero, and F. J. Jenkins. 1990. Identification andcharacterization of the herpes simplexvirus type 1 protein encoded by the UL37 open reading frame. J. Virol. 64:6101-6109.
42. Sherman,G., and S. L. Bachenheimer. 1988.Characterizationof intranuclear capsids made by ts morphogenic mutants of HSV-1. Virology163:471-480.
43. Smith, I. L., and R. M. Sandri-Goldin. 1988. Evidence that transcriptional control is themajormechanism ofregulationfor theglycoproteinD geneinherpes simplexvirus type 1-infected cells.J. Virol.62:1474-1477.
44. Spear, P.G.,and B. Roizman.1972.Proteinsspecified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion.J. Virol. 9:143-159.
45. Spear, P. G., and B. Roizman. 1980.Herpessimplex viruses, p. 615-745. In J. Tooze (ed.), DNA tumorviruses. ColdSpring HarborLaboratory, ColdSpring Harbor, N.Y.
46. Towbin, H., T.Staehelin,andJ.Gordon. 1979.Electrophoretic transfer ofproteinsfrompolyacrylamide gels tonitrocellulose sheets:procedureandsomeapplications.Proc. Natl. Acad.Sci. USA76:4350-4354.
47. Wagner, E. K. 1985. Individual HSVtranscripts. Characteriza-tion of specific genes, p. 45-104. In B. Roizman (ed.), The herpesviruses,vol. 3.PlenumPress,New York.
48. Wildy, P. 1986. Herpesvirus. Intervirology25:117-140. 49. Yie, S., S. I.Chowdhury,B. M.Bhat, A.J. Conley, W. S. M.
Wold,and W. Batterson. 1990. Identificationand characteriza-tion of the herpes simplex virus type 2 gene encoding the essentialcapsidprotein ICP32/VP19c. J. Virol.64:1124-1134.
![FIG. 3.werefectedHSV-infectedandUL35preparedphoresisinfectedinfectedimmunoprecipitatedextractslecular[35S]methionine Identification of the UL35 gene product in HSV-1 in- cells](https://thumb-us.123doks.com/thumbv2/123dok_us/1308465.84105/5.612.337.534.77.323/fig-werefectedhsv-infectedandul-preparedphoresisinfectedinfectedimmunoprecipitatedextractslecular-methionine-identification-product-cells.webp)